

Abstract
The ventromedial hypothalamic nucleus (VMN) is known to mediate autonomic responses in feeding and reproductive behaviors. To date, the most definitive molecular marker for the VMN is the orphan nuclear receptor steroidogenic factor-1 (SF-1). However, it is unclear whether SF-1 functions in the VMN as it does in peripheral endocrine organ development where loss of SF-1 results in organ agenesis due to apoptosis. Here, we provide evidence that SF-1 has a distinct role in later stages of VMN development by demonstrating the persistence of VMN precursors, the misexpression of an early marker (NKX2-1) concomitant with the absence of a late marker (BDNF neurotrophin), and the complete loss of projections to the bed nucleus of stria terminalis and the amygdala in sf-1 null mice. Our findings demonstrate that SF-1 is required for terminal differentiation of the VMN and suggest that transcriptional targets of SF-1 mediate normal circuitry between the hypothalamus and limbic structures in the telencephalon.
Introduction
Physical and chemical ablation studies of the hypothalamic ventromedial nucleus (VMN) suggest that this region of the hypothalamus controls a number of homeostatic and autonomic behavioral responses. For instance, the VMN has been implicated in regulating reproductive cycles, sexual and feeding behaviors, diurnal rhythm of glucocorticoid secretion, body temperature, and possibly locomotor behavior (Chateau et al., 1987; Egawa et al., 1991; Cohen and Pfaff, 1992; Choi et al., 1998; Flier and Maratos-Flier, 1998; Choi and Dallman, 1999; Majdic et al., 2002). The VMN is located in the mediobasal region of the diencephalon and is recognized morphologically as a dense bilateral aggregate of cell bodies surrounded by a cell-free neuropil zone. Based on different projections, possible functions, and histochemical properties, the VMN can be organized into dorsomedial, central, and ventrolateral regions (Altman and Bayer, 1986; Canteras et al., 1994; Canteras, 2002). VMN neurons send projections to adjacent hypothalamic regions and to other brain regions, including the bed nucleus of stria terminalis and the amygdala of limbic system (Altman and Bayer, 1986; Luiten et al., 1987; Swanson, 1987; Canteras et al., 1994). Although the precise molecular and chemical nature of the VMN circuitry remains to be defined, recent studies have proposed that the orphan nuclear receptor steroidogenic factor 1 (SF-1) is required for proper VMN development and function (Ikeda et al., 1995; Shinoda et al., 1995; Majdic et al., 2002).
SF-1 belongs to the hormone nuclear receptor gene family, and remains an orphan member because a cognate ligand has yet to be identified (Mangelsdorf et al., 1995). It is well established that SF-1 is an essential regulator in endocrine tissue and organ development. Indeed, targeted disruption of sf-1 in mice results in gonadal and adrenal agenesis, as well as the complete loss of pituitary gonadotropes (Ingraham et al., 1994; Luo et al., 1994; Sadovsky et al., 1995; Shinoda et al., 1995). SF-1 also regulates multiple targets that mediate normal adult endocrine physiology (reviewed in Parker, 1998; Hammer and Ingraham, 1999) and controls male sexual differentiation by regulating three important male hormones, including the Müllerian inhibiting substance, steroidogenic enzymes for testosterone synthesis and Insl-3, which is required for testicular descent (for review, see Roberts et al., 1999). Consistent with SF-1’s role in male development, sf-1 null (−/−) mice have female external genitalia regardless of their sex chromosomal complement (Luo et al., 1994; Sadovsky et al., 1995; Shinoda et al., 1995). More recently, phenotypic sex reversal of XY genotypes in humans has been associated with a partial loss of SF-1 function (Achermann et al., 1999, 2002). Although male sexual development in mice does not require a full dosage of SF-1, as shown in humans, two fully functional sf-1 alleles are required for proper adrenal development and function (Bland et al., 2000). The Sf-1 heterozygous (+/−) mice exhibit symptoms of adrenal insufficiency due to the hypoplastic and disorganized nature of the organ (Bland et al., 2000; Babu et al., 2002), consistent with adrenal insufficiency in humans due to partial loss of SF-1 function (Achermann et al., 1999; Biason-Lauber and Schoenle, 2000; Achermann et al., 2002). These studies suggest that a threshold level of SF-1 activity must be maintained for optimal growth and differentiation of the adrenogonadal primordium.
The precise role of SF-1 in VMN development is less clear, in part because SF-1 targets in the hypothalamus have not been identified and because of the early postnatal lethality in sf-1 −/− mice due to the complete lack of adrenal function. Previous analyses of sf-1 −/− mice report an apparent change in the cytoarchitecture of the VMN as assessed by the absence of a prominent condensed nucleus, an indistinct cell-free neuropil zone, and an increased number of neuroblasts in the paraventricular zone (Ikeda et al., 1995; Shinoda et al., 1995). Impaired VMN development in sf-1 −/− mice was explained as either a failure of VMN aggregation and organization or the regression of VMN neurons at late stages of development, analogous to the prominent cell death accounting for adrenal and gonadal agenesis in sf-1 −/− mice (Ikeda et al., 1995; Shinoda et al., 1995). More recently, detailed histological analyses reported an expansion of glutamic acid decarboxylase (GAD) 67 and estrogen receptor (ER)α-positive neurons in the presumptive embryonic sf-1 −/− VMN, leading to the suggestion that VMN precursors either fail to aggregate, survive, or differentiate properly (Dellovade et al., 2000). Collectively, these studies suggest that SF-1 could participate in multiple phases of VMN development; early phases would include the proliferation, survival, and migration of VMN precursors from the ventricular zone, while later stages would involve the aggregation and condensation of the VMN nucleus. At both early and late phases, SF-1 could also function to specify VMN cell fate.
Here, we investigated prenatal VMN development in the sf-1 +/+ and −/− mice using birth dating analysis, expression profiles of early and late VMN markers, and DiI neuronal tracing. Our collective results provide evidence that functional SF-1 protein is needed for terminal differentiation of VMN precursors.
Results
Cell proliferation is normal in the VMN of sf-1 −/− mice
VMN precursors are derived from neuronal stem cells located within the third ventricular zone of the diencephalon (Altman and Bayer, 1986). Consistent with this fact, we find that most bromo-deoxyuridine (BrdU) pulse-labeled cells were SF-1 negative and were localized to the neuroepithelial layer lining the third ventricle (Fig. 1A). Further labeling for mitotic cells with an anti-phospho-histone H3 antibody confirmed that cell proliferation is restricted to the third ventricle neuroepithelium (data not shown). By contrast, all SF-1-positive cells resided immediately outside of the ventricular zone (Fig. 1A). We conclude that SF-1 is expressed in postmitotic cells of the embryonic hypothalamus and is therefore unlikely to participate in the proliferative phase of VMN development. These findings contrast the role of SF-1 in adrenal development, where SF-1 is expressed in mitotically active cells and is required for normal proliferation (Bland and Ingraham, unpublished data, and Bland et al., 2000).
Fig. 1.

Normal cell proliferation is detected in sf-1 −/− mice. Coronal sections from embryonic day (E) 14.5 mouse embryos were stained with both anti-SF-1 and anti-BrdU antisera (A). SF-1 and BrdU signals were developed with the secondary goat anti-rabbit Alexa 488 conjugate (green) and secondary goat anti-rat Alexa 546 (red), respectively, using confocal microscopy. BrdU-labeled cells are mostly restricted to the third ventricle (3V) neuroepithelium (ne, arrowhead), whereas SF-1-positive cells are found outside of the neuroepithelium (arrow). Panel B shows the number of BrdU pulse-labeled neurons in the VMN plotted for different embryonic stages, spanning E10.5 to E15.5. The number of BrdU-labeled cells was determined from comparable coronal sections obtained from both sf-1 +/+ (yellow bars) and −/− (blue bars) mice. Equivalent sections were selected based on morphological landmarks surrounding the hypothalamus, including the fornix, the fasciculus retroplexus, the cerebral peduncle, and the arcuate nucleus. Four independent sections were counted for each developmental time point; data shown here represent the most medial VMN sections. A representative P0 section of each genotype is shown for BrdU-labeled cells (red) at stage E12.5 in panels C and D. The VMN outlined with a white dashed circle, based on SF-1 expression (green) in the +/+ mice. BrdU, bromo-deoxyuridine; VMN, ventromedial nucleus. Scale bars = 100 μm.
To assess the numbers and distribution of VMN precursors born at different ages in sf-1 −/− mice, BrdU birth dating analysis was carried out on +/+ and −/− littermates. Previous work established that in mice VMN neurons are born between embryonic day (E) 10 and E15, peaking at E13 (Shimada and Nakamura, 1973). BrdU-positive VMN neurons were quantified by colocalization with SF-1-positive cells in +/+ mice and in the comparable hypothalamic region for sf-1 −/− mice, as shown in Fig. 1C and D. Equivalent numbers of BrdU labeled neurons were observed in the mediobasal hypothalamus for both genotypes at all stages of labeling, with the peak number of VMN neurons born between E11.5 and E13 (Fig. 1B), suggesting that the birth rate of VMN precursors is unaffected by the loss of SF-1. We also noted that VMN neurons were born in a ventrolateral progression during development; for example, neurons born early (E11) reside in the ventrolateral VMN, while those born later (E14) reside in the dorsomedial VMN (data not shown). Furthermore, the distribution of BrdU-labeled cells appeared normal in mutant mice, implying that their migration is unaffected by the loss of SF-1. Taken together, these results show that SF-1 is not required for normal proliferation of VMN precursors, consistent with expression of SF-1 in postmitotic cells.
VMN precursors persist in sf-1 −/− neonates
Previous studies proposed that the altered hypothalamic cytoarchitecture observed in sf-1 −/− mice might arise from the complete loss of VMN neurons due to cell death (Ikeda et al., 1995; Dellovade et al., 2000). However, TUNEL analysis revealed no apparent cell death in the presumptive VMN of both +/+ and −/− mice from E11 through postnatal day (P) 0 despite our ability to detect TUNEL-positive cells in the developing olfactory epithelium, as previously reported (Pellier and Astic, 1994; Voyron et al., 1999; and data not shown). Based on these observations, we conclude that programmed cell death is unable to explain the apparent altered VMN morphology in sf-1 −/− mice.
These findings predict that VMN precursors may be present in sf-1 −/− mice. To study the fate of SF-1-positive cells, we analyzed expression of the sf-1 mutant transcript targeted in the sf-1 −/− mice, which includes the first two exons and a modified third exon that contains the selectable marker gene, neomycin (Ikeda et al., 1995). In situ hybridization studies revealed that sf-1 expression was maintained in the sf-1 −/− mice, and largely recapitulated the pattern observed in wild-type mice (Fig. 2). However, no SF-1 protein is observed in the sf-1 −/− mice due to disruption of third exon, which encodes the second zinc finger of the DNA binding domain (see insets). Although the boundaries of sf-1 transcript expression were maintained in sf-1 −/− mice, expression of the mutant transcripts decreased, especially in the ventrolateral VMN. Expression of the neuronal specific marker NeuN confirmed that the cells populating the presumptive VMN retain a neuronal identity. No labeling was detected with glial specific marker GFAP in either wild-type or mutant mice (data not shown). Thus, these data provide evidence that the absence of SF-1 does not preclude the birth, migration, and condensation of VMN neurons.
Fig. 2.
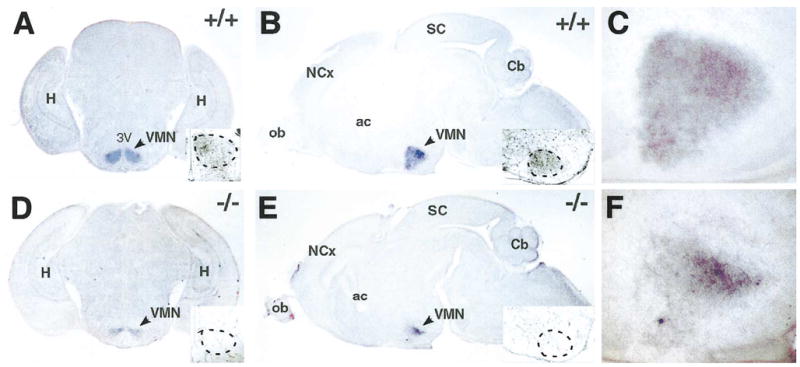
Mutant sf-1 transcripts are present in the ventromedial nucleus (VMN) of sf-1 −/− mice. In situ hybridization of sf-1 mRNA is shown for both coronal (A and D) and sagittal whole brain sections (B and E). Expression of sf-1 mRNA was restricted to the VMN of both wild-type (+/+) and sf-1 null (−/−) mice as indicated in whole brain sections or in enlarged sections of sagittal VMN regions (C and F). SF-1 protein staining is shown for corresponding sections as insets with the VMN region indicated (arrowhead). Abbreviations or anatomical landmarks are as follows: anterior commissure (ac), cerebellum (Cb), hippocampus (H), neocortex (NCx), olfactory bulb (ob), superior colliculus (SC), and the third ventricle (3V).
Altered expression of early and late VMN markers in sf-1 −/− mice
Previous reports suggested that GABAergic neurons are expanded into the VMN as early as E15 in sf-1 −/− mice (Dellovade et al., 2000). Therefore, to examine a potential change in cell fate of VMN neurons, we analyzed GAD67 expression by in situ hybridization in the VMN of sf-1 −/− mice. Indeed, we noted increased expression of GAD67 in the mutant VMN; however, this increased expression was still low relative to expression in neighboring parts of the hypothalamus (Fig. 3). Interestingly, the lowest expression of GAD67 was found in the dorsomedial VMN of mutant mice, where we also noted the strongest expression of the sf-1 mutant transcript (see above).
Fig. 3.
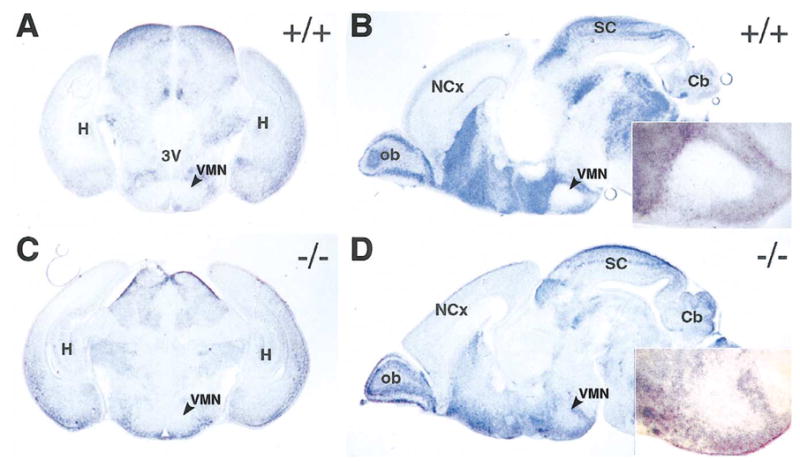
Increased GAD67 expression in the ventromedial nucleus (VMN) of sf-1 −/− mice. In situ hybridization of GAD67 mRNA is shown for 100-μm coronal (A and C) and sagittal sections (B and D) of P0 mice. The VMN is indicated (arrow) in all panels of sections obtained from either wild-type (+/+) or sf-1 null (−/−) mice. Enlargements of the VMN area for both genotypes are shown as insets in B and D. Abbreviations for anatomical landmarks are as follows: cerebellum (Cb), hippocampus (H), neocortex (NCx), olfactory bulb (ob), superior colliculus (SC), and third ventricle (3V).
To examine more closely VMN differentiation in the sf-1 mutant we used two molecular markers; the first is the homeobox gene Nkx2-1, which is expressed in basal telencephalic and ventral hypothalamic neurons (Kimura et al., 1996; Sussel et al., 1999; Marin et al., 2002). The second marker is brain-derived neurotrophic factor (BDNF), which was found previously to mark ventrolateral VMN neurons in adult mice (Kernie et al., 2000). Using mutant mice that harbor lacZ in the BDNF locus (BDNFlacZ), we observed prominent expression of BDNFlacZ (β-galactosidase) in the ventrolateral region of the anterior and medial VMN of adult mice that overlapped significantly with SF-1, especially in the anterior VMN (Fig. 4). During prenatal development, BDNFlacZ expression followed an anterior to posterior progression in the presumptive VMN and was not apparent until E17.5 (Fig. 5A). BDNFlacZ expression was markedly decreased in the VMN of sf-1 −/− mice, as shown by the loss of β-galactosidase expression (Fig. 5B). However, BDNFlacZ was maintained in the substantia nigra in sf-1 −/− mice (data not shown), implying that BDNF expression in the VMN is dependent on SF-1.
Fig. 4.
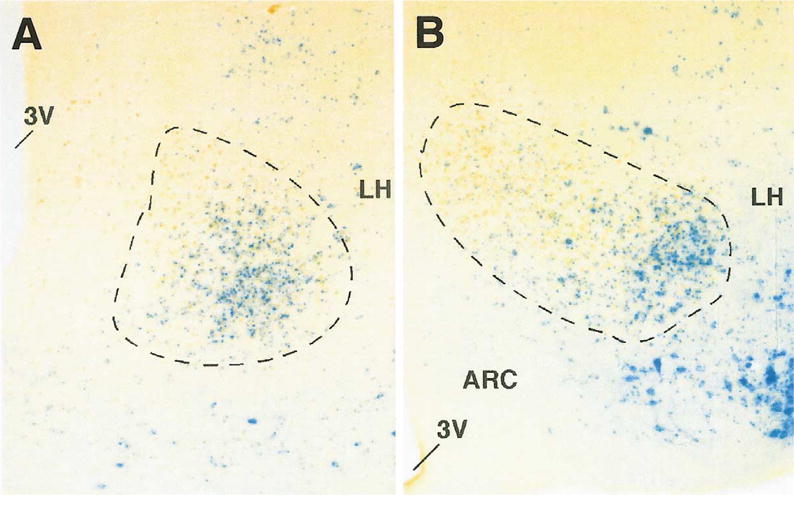
Brain-derived neurotrophic factor (BDNF) expression overlaps with SF-1 in the adult ventromedial nucleus (VMN). Anterior (A) and posterior sections (B) of adult mouse mediobasal hypothalamus show both SF-1 (brown) and BDNFlacZ (blue), as described in Experimental methods. A dashed line outlines the area of SF-1-positive cells. Other hypothalamic regions indicated include the third ventricle (3V), the lateral hypothalamus (LH), and arcuate nucleus (ARC).
Fig. 5.
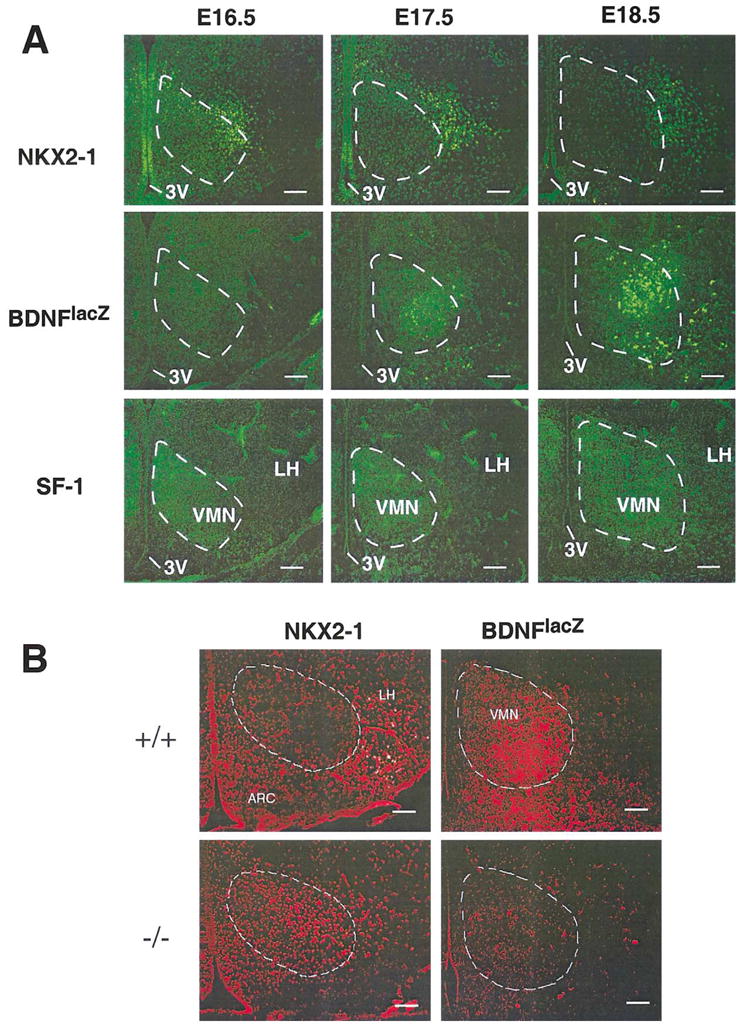
NKX2-1 and brain-derived neurotrophic factor (BDNF) mark early and late ventromedial nucleus (VMN) development, respectively. NKX2-1 staining was carried out on coronal brain sections using rabbit anti-mouse NKX2-1 antibody (A). Staining is shown for wild-type (+/+) mice at different stages of development (top panels). BDNF expression was examined using mice carrying a lacZ insertion at the BDNF locus (see Experimental methods). Expression of β-galactosidase (β-gal) was detected in BDNFlacZ +/− mice using rabbit anti-β-gal and developed with the secondary goat anti-rabbit Alexa 488 at different stages of development, beginning at E16.5 (middle panels). β-gal expression was found in the ventrolateral portion of both anterior and medial VMN as indicated by the dashed line in A and B. SF-1 staining in wild-type embryos is shown for adjacent sections at each stage (bottom panels). Panel B shows NKX2-1 and BDNFlacZ staining (red) for sf-1 null (−/−) neonates (P0) and wild-type littermates. A similar boundary for the VMN is assumed in both the wild-type and mutant mice. By P0 NKX2-1 is completely excluded from the VMN in +/+ mice, while persisted in the VMN of −/− mice. In contrast, BDNFlacZ is expressed in +/+ VMN, but is markedly lost in sf-1 −/− VMN. For all panels, the relative position of the VMN (dashed circle), as judged by SF-1 expression, and the arcuate nucleus (ARC) are indicated. Scale bar = 100 μm.
In contrast to the late onset of BDNF expression, NKX2-1 expression was observed early in the ventral hypothalamus and overlapped with SF-1 expression until E18.5, at which time its expression was greatly reduced in the VMN (Fig. 5A). At this stage, NKX2-1 expression was restricted to the ventral neuroepithelium and the ventrolateral area of lateral hypothalamus (vlLH) (Fig. 5A). Therefore, NKX2-1 is an early marker of VMN development that is downregulated as the VMN precursors differentiate into a morphologically distinct nucleus. In sf-1 mutant mice, NKX2-1 expression persisted in the presumptive VMN concomitant with reduced expression in the vlLH (Fig. 5B). Interestingly, SF-1-positive cells persist in Nkx2-1 null embryos (Marin, O., unpublished data). These preliminary results could imply that some and perhaps all VMN precursors arise from a distinct lineage other than NKX2-1. Alternatively, the presence of SF-1-positive cells in Nkx2-1 null mice could also result from possible compensation activities of other NKX homeobox proteins, such as NKX2-2 and NKX2-4. Whether NKX2-1 and SF-1 function in the same or separate genetic pathways that govern VMN development remains to be determined. In summary, the expansion of GAD67, the persistence of NKX2-1, and the corresponding loss of BDNF in sf-1 −/− mice suggest that SF-1 participates in terminal differentiation of VMN neurons.
VMN projections are lost in sf-1 −/− neonates
The hypothesis that VMN neurons in sf-1 −/− mice fail to differentiate properly was further tested by tracing afferent projections of this nucleus to other regions of the brain; these include projections to the paraventricular hypothalamus (PVN), bed nucleus of stria terminalis (BST), and amygdala (Swanson, 1987). Consistent with previous reports, all of these known projections were identified in wild-type mice using the neuronal tracer DiI (Fig. 6A–C) with a large number of fibers extending through the stria terminalis to innervate the BST and the amygdala (Fig. 6A–C). In contrast, sf-1 −/− littermates showed a complete loss of VMN projections to both the BST and amygdala, but maintained limited projections to the anterior hypothalamus (Fig. 6D–F). Closer examination of these projections in mutant mice showed that these are bona fide afferent fibers that terminate at axonal junctions (Fig. 6G–I). Collectively, these data provide strong evidence that SF-1 is required for VMN precursors to establish their respective neuronal connections.
Fig. 6.
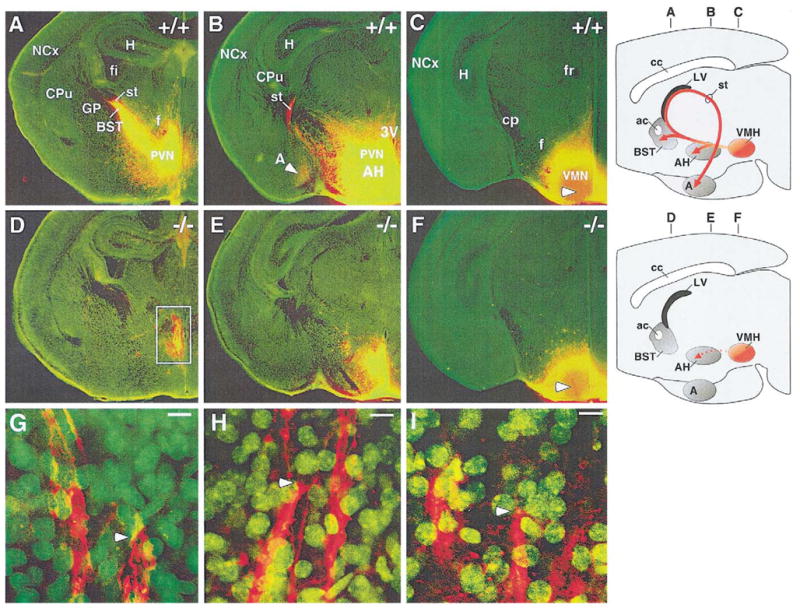
Loss of ventromedial nucleus (VMN) projections in sf-1 −/− neonates. Coronal sections of brains implanted with DiI crystals are shown for either wild-type mice (+/+; A–C) or sf-1 null mice (−/−; D–F). Sections are shown in an anterior (left) to posterior (right) arrangement with nuclei stained with Cytox Green (green; Molecular Probes), and the DiI neuronal tracer (red). Stained projections from the ventromedial nucleus (VMN) to the amygdala are indicated with arrows. Panels C and F show the central site where DiI crystals were implanted in the VMN (white arrowhead). Schematics illustrating the afferent projections emanating from the VMN in wild-type and SF-1 mutant brains are shown on the far-right-hand side of panels A–F; the corresponding planes of sectioning for panels A–F are indicated above each schematic. Abbreviations for anatomical landmarks include the amygdala (A), anterior hypothalamus (AH), bed nucleus of stria terminalis (BST), caudoputamen (CPu), cerebral peduncle (cp), fasciculus retroflexus (fr), fimbria (fi), fornix (f), globus pallidus (GP), hippocampus (H), neocortex (NCx), paraventricular hypothalamus (PVN), and stria terminalis (st). Afferent fibers terminating on axons within the anterior hypothalamus are shown in panels G–I (white arrowheads); each panel represents a higher magnification of the white-boxed area from panel D. Scale bar = 10 μm or 100× magnification.
Discussion
SF-1 regulates multiple genes in the endocrine system and is essential for peripheral endocrine organ development. Although the function of SF-1 in adrenal, gonadal, and pituitary function has been studied extensively, the role of SF-1 in VMN development is less well understood. Here, unexpectedly, our studies provide evidence that early stages of VMN development, which commence when neuronal precursors are born and migrate from the third ventricle neuroepithelium, are independent of SF-1 function. The following two additional observations are consistent with this hypothesis: first, SF-1 expression is restricted to postmitotic cells, and, second, neurons in the presumptive VMN of sf-1 −/− mice continue to express sf-1 mutant transcripts. Instead, our findings showing misexpression of early and late molecular markers and a loss of neuronal projections in the sf-1 −/− VMN suggest strongly that SF-1 is required in late stages of VMN development. Moreover, no overt differences were noted between wild-type and heterozygous sf-1 mice for all parameters examined, including cell proliferation, cell death, expression of molecular markers, and neuronal projections (data not shown). Thus, the function of SF-1 in VMN development appears to be independent of gene dosage and is therefore distinct from its role in gonadal and adrenal development, where cell survival and cell proliferation required a full complement of SF-1 activity.
While our data suggest that VMN precursors fail to undergo terminal differentiation, it is unclear whether they are blocked from undergoing full differentiation or adopt an alternative neuronal cell fate. In wild-type mice, normal repression of NKX2-1 and the subsequent expression of BDNF coincide with initial stages of VMN condensation. Therefore, the persistence of NKX2-1 and the loss of BDNF expression in sf-1 −/− mice may indirectly imply that the final phases of VMN development are blocked. The loss of VMN afferent projections in sf-1 −/− mice also implies that establishing the definitive VMN neuronal phenotype requires SF-1 activity. However, the presence of limited afferent projections from the mutant VMN area suggests that either terminal differentiation of a limited number of VMN neurons is not entirely dependent on SF-1 function, or alternatively, VMN precursors in mutant mice adopt neuronal fates of neighboring hypothalamic nuclei that also send axonal projections to the anterior hypothalamus. In this regard, Tobet and colleagues noted an expansion of neurons expressing ERα, galanin, and neuropeptide Y in the presumptive VMN of sf-1 −/− neonates (Dellovade et al., 2000). Normally, these peptides are selectively expressed in the neurons of lateral hypothalamus, dorsomedial nucleus, and arcuate nucleus, respectively. Identification of additional markers for later stages of VMN differentiation should help to clarify the molecular events and regional specification underlying VMN development.
SF-1 and BDNF in feeding and locomotor behaviors
Our findings suggest that the normal BDNF expression in mature vlVMN neurons is dependent on SF-1. Whether SF-1 actively participates in regulating BDNF expression is not known at this time. Elucidating the exact mechanisms governing BDNF expression has proven difficult given that four distinct promoter regions are found in the BDNF upstream genomic sequences (Timmusk et al., 1993; Nakayama et al., 1994) and it is not clear which of the four BDNF transcripts participates in the differentiation of vlVMN neurons. However, we have noted two potential SF-1 binding sites within the 5′ regulatory region of transcript 4 (Liu et al., 2001). BDNF has been established to promote neuronal differentiation and survival both in vitro and in vivo (Jones et al., 1994; Schwartz et al., 1997), and thus it is possible that BDNF functions to promote the maturation of VMN neurons.
Previous ablation studies implicate the VMN as one of the homeostatic centers of energy expenditure. Although the precise molecular circuitry for this function has not been clarified as for other centers, such as the arcuate and paraventricular nuclei, it is known that reduced locomotor activity leading to an obesity phenotype is observed in rats with reversible chemical VMN lesions (Choi et al., 1998; Choi and Dallman, 1999). Parker and colleagues observed a similar phenotype in adrenal rescued sf-1 −/− mice; these animals exhibit late onset obesity associated with a significant reduction in locomotor activity and normal overall food intake (Majdic et al., 2002). Similarly, BDNF heterozygous mice also display abnormal locomotor activity and late onset obesity. However, the obesity phenotype observed in BDNF +/− mice results from hyperphagia and shows incomplete penetrance. Obese BDNF +/− mice exhibit normal locomotor activity, whereas nonobese BDNF +/− mice are thought to counteract this hyperphagia through hyperactivity (Kernie et al., 2000). More recently, conditional inactivation of BDNF in adult mice also resulted in hyperactivity associated with increased anxiety-like behavior (Rios et al., 2001). BDNF is not expressed in the arcuate nucleus (Fig. 4A), suggesting that this hypothalamic center is unlikely to be involved in BDNF regulation of feeding and locomotive behavior. Although the functional significance of an altered BDNF expression pattern in the vlVMN of sf-1 −/− mice remains to be established, the obvious links between the VMN to energy homeostasis and to conditioned responses is intriguing. Further studies are needed to decipher the possible interplay in the VMN between SF-1 and the other molecular determinants of energy homeostasis found in the VMN, such as BDNF, the leptin receptor, the tubby peptide, and the ghrelin receptor (Kleyn et al., 1996; Mercer et al., 1996; Guan et al., 1997; Wren et al., 2000).
Orphan nuclear receptors in CNS development
Based on our findings presented here, we can now add SF-1 to the subset of orphan nuclear receptors that control specific aspects of CNS development. For example, Nurr1, a member of the Nurr1/Nur77/Nor1 subfamily of receptors, appears to be essential for the migration, differentiation, and survival of dopaminergic neurons of the ventral mesencephon (Zetterstrom et al., 1997; Saucedo-Cardenas et al., 1998; Le et al., 1999; Wallen et al., 1999). Similarly, the orphan nuclear receptor RORα is crucial for maturation and survival of Purkinje cells in the cerebellum (Dussault et al., 1998; Chu and Zingg, 1999; Vogel et al., 2000). Finally, the inhibitory orphan receptor, COUP-TFI, is known to mediate neocortex identity as suggested by the loss of regional organization and specific gene expression in the absence of COUP-TFI function (Zhou et al., 2001). In the absence of bona fide ligands, potential mechanisms for regulating these orphan receptors include posttranslational phosphorylation and selective recruitment of coactivator proteins, as shown for SF-1 (Hammer et al., 1999; Ito et al., 2000; Desclozeaux et al., 2002). Further insights into SF-1’s function in the hypothalamus should be directly relevant to these other orphan nuclear receptors that function in neural development. In addition, our findings are complementary to the known role of the bHLH-PAS transcription factor, SIM1 in the development of several hypothalamic nuclei, including the PVN, supraoptic nucleus (SON), and anterior periventricular nucleus (aPV). All of these nuclei share a common precursor and SIM1 function is required for the terminal differentiation of PVN/SON/aPV precursors (Michaud et al., 1998; Michaud, 2001). In the absence of SIM1, PVN/SON/aPV common precursors persist initially, but are subsequently lost upon terminal differentiation. Whether they undergo embryonic cell death or fail to fully differentiate has yet to be established. Nonetheless, these data suggest that the morphological identification of distinct hypothalamic nuclei will arise from distinct developmental genetic pathways in a cell autonomous manner.
In summary, we provide evidence that SF-1 regulates late stages of VMN development by showing that terminal differentiation and establishment of normal afferent projections in the VMN are dependent on SF-1 activity. Our findings in the VMN illustrate the versatility of this orphan nuclear receptor in regulating distinct developmental programs in multiple tissues of the neuroendocrine reproductive and stress axes. Future studies aimed at delineating the circuitry of the VMN by genetic tracing, and at eliminating the VMN by conditional ablation to avoid the early postnatal lethality, should help to clarify how this hypothalamic region regulates complex homeostatic behavioral responses.
Experimental methods
Animals
The sf-1 +/− mice (obtained from the Jackson Laboratory) were maintained on a C57BL/6J × FVB background and kept on a 12-h light-dark cycle. sf-1 +/− mice were bred to generate +/+, +/−, and −/− embryos and were designated E0.5 on the morning when sperm plug was found. Newly born pups were collected immediately after birth and were designated P0. Embryos, P0, and adult mice were genotyped by using genomic DNA isolated from tail tissue. Genotyping was determined by polymerase chain reaction using the following oligos: sf-1For (ACAAGCATTACACGTGCACC), sf-1Rev (TGACTAGCAACCACCTTGCC), and neoRev (AGGTGAGATGACAGGAGATC). BDNFlacZ mice were maintained on an FVB background. The BDNFlacZneo (BDNFlacZ) mouse strain was constructed by replacing the BDNF coding region, beginning at the initial methionine codon, with the Escherichia coli lacZ gene and the PGKneo selectable marker, as previously described (Bennett et al., 1999). Transcription from the targeted genomic locus is predicted to produce active β-galactosidase (β-gal) protein rather than the neurotrophin. Genotyping of BDNFlacZ mice was determined by using primers GTGCTGCAAGGCGATTAAGT (lacZN5-For) and GTGGAGTTCTGCTAATGAGA (MBDSA 10-Rev) to detect the presence of LacZ insertion. BDNFlacZ heterozygous (+/−) mice were mated to sf-1 +/− mice to generate compound heterozygous, which are viable and fertile. These mice were mated to sf-1 +/− to generate both BDNFlacZ +/−; sf-1 +/+ and BDNFlacZ +/−; sf-1 −/− mice. Newly born pups were collected and perfused with 4% paraformaldehyde (PFA) fixative for all subsequent analyses. All research with animals was performed according to guidelines of the UCSF Committee on Animal Research.
Histology and immunohistochemistry
Dissected postnatal brains and embryonic heads were fixed in 4% PFA overnight at 4°C. For cryosectioning, fixed tissues were cryoprotected by infusion with 15% phosphate-buffered saline (PBS)-sucrose and followed by 30% PBS-sucrose overnight at 4°C. Tissues were embedded in OCT compound (Tissue-tek, Sakura Finetek USA, Inc., Torrance, CA) and sectioned at 12 or 20 μm. For all vibratome sections, fixed tissues were rinsed once in PBS and embedded in 4% low-melt agarose and sectioned at 100 μm by using a Leica VT1000S vibratome.
Immunohistochemistry analyses were performed on sections equilibrated at room temperature, rinsed in PBS for 10 min, and permeabilized in PBT (PBS + 0.2% Triton X-100) for 1 h. All sections were first incubated in blocking solution (10 mg/ml bovine serum albumin [BSA] in PBT) for 30 min, followed by primary antibody incubation overnight at 4 °C. Sections were rinsed in PBT (3×) for 10 min and then incubated with fluorescence-conjugated secondary antibody for 1 h at room temperature. Excess antibody was removed by rinsing sections in PBT for 10 min (3×), following by DNA staining with DAPI for 5 min. Finally, sections were rinsed again in PBS for 10 min (3×) and mounted. The following antibodies were used: rabbit antimouse SF-1 (kindly provided by Dr. K. Morohashi; 1:1000), rabbit antimouse NKX2-1 (1:1000; Biopat Biotechnologies), rat anti-BrdU (1:50; Harlan Sera-lab), rabbit anti-β-galactosidase (1:1000; ICN Pharmaceuticals), mouse anti-neuronal nuclei NeuN (1:500; Chemicon International), and mouse monoclonal GFAP (1:10,000; Cymbus Biotech). Secondary antibodies used in this study include Alexa 488-conjugated goat anti-rabbit (1:200) and Alexa 546-conjugated goat anti-rat (1:200; Molecular Probes).
TUNEL-positive cells were detected by using a modified version of a method originally developed by (Gavrieli et al., 1992). Briefly, brain sections (12 μm) were equilibrated to room temperature and rehydrated in PBS. Sections were permeabilized in PBT for 1 h, rinsed twice in TdT buffer (30 mM Tris, pH 7.6, 0.024% CoCl2, 30 mg/ml sodium cacodylate), and labeled with biotinylated dUTP using terminal transferase for 1 h at 37°C. The labeling reaction was stopped by incubation in termination buffer (300 mM NaCl, 30 mM sodium citrate). Tissues were rinsed twice in PBT and incubated in blocking solution (10 mg/ml BSA in PBS) for 10 min. Biotin-labeled DNA was detected by incubating tissue with fluorescence-conjugated streptavidin (Molecular Probes) diluted in PBS containing 1 mg/ml BSA for 30 min. Slides were then rinsed three times in PBT and mounted. All data were collected using a Bio-Rad confocal microscope.
BrdU labeling and neuronal birth-dating
For bromo-deoxyuridine (BrdU) labeling, BrdU solution (10 mg/ml in H2O; Sigma) was injected into the peritoneal cavity of pregnant mice (40 μg per gram of body weight). For birth-dating, injected pregnant mice were allowed to give birth and newly born pups were collected immediately and perfused with 4% PFA fixative. For shorter pulse-labeling experiments, pregnant mice were killed 1 h after BrdU injections, and embryos were collected and fixed in 4% PFA. Fixed tissues were sectioned and treated with 2 N HCl at 37°C for 20 min to denature DNA prior to staining with BrdU antisera. BrdU-labeled cells were counted by using NIH Image software and inverted images. The number of digitally counted positive cells was confirmed by visual assessment to ensure appropriate parameter settings.
In situ hybridization
In situ hybridization was performed as described (Erskine et al., 2000). Briefly, PFA-fixed newborn brains were dissected from the cranium and sectioned by using a vibratome. Sections were mounted (Superfrost plus, Fisher Scientific, Houston, TX, USA), dried overnight, and rehydrated in PBT. Sections were dehydrated followed by rehydration in a series of methanol-PBT (25%, 50%, and 100%) and rinsed in PBT followed by bleaching in 6% H2O2-PBT for 1 h. Hybridization conditions followed exactly the protocol by Erskine et al. (2000) using DIG-labeled probes (Roche Chemical, Indianapolis, IN, USA) made from the full-length cDNAs encoding mouse sf-1 and rat GAD67.
DiI implantation and labeling
Experiments were performed as described (Marin et al., 2002) with minor modifications. In brief, sf-1 +/+ and −/− P0 littermates were cold anesthetized and perfused with 4% PFA. Brains were dissected and stored in 4% PFA. Crystals of the axonal tracer 1,1′-dioctodecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, Molecular Probes) were inserted into the VMN of fixed brains by light dissecting microscopy using visual landmarks. Brains were kept in fixative at room temperature for 6 or 7 weeks to allow sufficient diffusion of the DiI tracer. Brains were then embedded into 4% low-melt agarose and sectioned at 100 μm using a vibratome. All sections were counterstained with Cytox Green (Molecular Probes). Data were collected and analyzed by using a Nikon compound microscope equipped with a CCD camera.
Acknowledgments
We wish to thank Dr. Ken Morohashi (Okazaki, Japan) for his generosity in providing the SF-1 antisera and Drs. M. Dallman and U. Greishammer for advice on technical aspects of this project, and members of the Ingraham lab, especially M. Bland, for meaningful discussions and review of the manuscript. This study was supported by an NRSA F32HD41327 (to P.V.T.), the National University of Singapore Scholarship (to M.B.-H.L), and research grants from: Nina Ireland, NIDA (R01DA12462), and NIMH K02 MH01046-01 (to J.R.R.), NARSAD Young Investigator Award and MIND Institute (to O.M.), and HHMI and NIH-NS P01-16033 (to L.F.R., an investigator of the Howard Hughes Medical Institute), and the UCSF Sandler Award for Basic Research, Brook Byers Award, and NICHD RO1 support (to H.A.I.).
References
- Achermann JC, Ito M, Hindmarsh PC, Jameson JL. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999;22:125–126. doi: 10.1038/9629. [DOI] [PubMed] [Google Scholar]
- Achermann JC, Ozisik G, Ito M, Orun UA, Harmanci K, Gurakan B, Jameson JL. Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1, in a dose-dependent manner. J Clin Endocrinol Metab. 2002;87:1829–1833. doi: 10.1210/jcem.87.4.8376. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. The development of the rat hypothalamus. Adv Anat Embryol Cell Biol. 1986;100:1–178. [PubMed] [Google Scholar]
- Babu PS, Bavers DL, Beuschlein F, Shah S, Jeffs B, Jameson JL, Hammer GD. Interaction between Dax-1 and steroidogenic factor-1 in vivo: increased adrenal responsiveness to ACTH in the absence of Dax-1. Endocrinology. 2002;143:665–673. doi: 10.1210/endo.143.2.8658. [DOI] [PubMed] [Google Scholar]
- Bennett JL, Zeiler SR, Jones KR. Patterned expression of BDNF and NT-3 in the retina and anterior segment of the developing mammalian eye. Invest Ophthalmol Vis Sci. 1999;40:2996–3005. [PubMed] [Google Scholar]
- Biason-Lauber A, Schoenle EJ. Apparently normal ovarian differentiation in a prepubertal girl with transcriptionally inactive steroidogenic factor 1 (NR5A1/SF-1) and adrenocortical insufficiency. Am J Hum Genet. 2000;67:1563–1568. doi: 10.1086/316893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland ML, Jamieson CA, Akana SF, Bornstein SR, Eisenhofer G, Dallman MF, Ingraham HA. Haploinsufficiency of steroidogenic factor-1 in mice disrupts adrenal development leading to an impaired stress response. Proc Natl Acad Sci USA. 2000;97:14488–14493. doi: 10.1073/pnas.97.26.14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canteras NS. The medial hypothalamic defensive system: hodological organization and functional implications. Pharmacol Biochem Behav. 2002;71:481–491. doi: 10.1016/s0091-3057(01)00685-2. [DOI] [PubMed] [Google Scholar]
- Canteras NS, Simerly RB, Swanson LW. Organization of projections from the ventromedial nucleus of the hypothalamus: a Phaseolus vulgaris-leucoagglutinin study in the rat. J Comp Neurol. 1994;348:41–79. doi: 10.1002/cne.903480103. [DOI] [PubMed] [Google Scholar]
- Chateau D, Chabli A, Aron C. Effects of ventromedial nucleus lesions on the display of lordosis behavior in the male rat. Interactions with facilitory effects of male urine. Physiol Behav. 1987;39:341–345. doi: 10.1016/0031-9384(87)90232-0. [DOI] [PubMed] [Google Scholar]
- Choi S, Dallman MF. Hypothalamic obesity: multiple routes mediated by loss of function in medial cell groups. Endocrinology. 1999;140:4081–4088. doi: 10.1210/endo.140.9.6964. [DOI] [PubMed] [Google Scholar]
- Choi S, Wong LS, Yamat C, Dallman MF. Hypothalamic ventromedial nuclei amplify circadian rhythms: do they contain a food-entrained endogenous oscillator? J Neurosci. 1998;18:3843–3852. doi: 10.1523/JNEUROSCI.18-10-03843.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu K, Zingg HH. Activation of the mouse oxytocin promoter by the orphan receptor RORalpha. J Mol Endocrinol. 1999;23:337–346. doi: 10.1677/jme.0.0230337. [DOI] [PubMed] [Google Scholar]
- Cohen RS, Pfaff DW. Ventromedial hypothalamic neurons in the mediation of long-lasting effects of estrogen on lordosis behavior. Prog Neurobiol. 1992;38:423–453. doi: 10.1016/0301-0082(92)90045-g. [DOI] [PubMed] [Google Scholar]
- Dellovade TL, Young M, Ross EP, Henderson R, Caron K, Parker K, Tobet SA. Disruption of the gene encoding SF-1 alters the distribution of hypothalamic neuronal phenotypes. J Comp Neurol. 2000;423:579–589. doi: 10.1002/1096-9861(20000807)423:4<579::aid-cne4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Desclozeaux M, Krylova IN, Horn F, Fletterick RJ, Ingraham HA. Phosphorylation and intramolecular stabilization of the ligand binding domain in the nuclear receptor steroidosenic factor 1. Mol Cell Biol. 2002;22:7193–7203. doi: 10.1128/MCB.22.20.7193-7203.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussault I, Fawcett D, Matthyssen A, Bader JA, Giguere V. Orphan nuclear receptor ROR alpha-deficient mice display the cerebellar defects of staggerer. Mech Dev. 1998;70:147–153. doi: 10.1016/s0925-4773(97)00187-1. [DOI] [PubMed] [Google Scholar]
- Egawa M, Inoue S, Sato S, Takamura Y, Murakami N, Takahashi K. Restoration of circadian corticosterone rhythm in ventromedial hypothalamic lesioned rats. Neuroendocrinology. 1991;53:543–548. doi: 10.1159/000125772. [DOI] [PubMed] [Google Scholar]
- Erskine L, Williams SE, Brose K, Kidd T, Rachel RA, Goodman CS, Tessier-Lavigne M, Mason CA. Retinal ganglion cell axon guidance in the mouse optic chiasm: expression and function of robos and slits. J Neurosci. 2000;20:4975–4982. doi: 10.1523/JNEUROSCI.20-13-04975.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flier JS, Maratos-Flier E. Obesity and the hypothalamus: novel peptides for new pathways. Cell. 1998;92:437–440. doi: 10.1016/s0092-8674(00)80937-x. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinath-singhji DJ, Smith RG, Van der Ploeg LH, Howard AD. Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res. 1997;48:23–29. doi: 10.1016/s0169-328x(97)00071-5. [DOI] [PubMed] [Google Scholar]
- Hammer GD, Ingraham HA. Steroidogenic factor-1: its role in endocrine organ development and differentiation. Front Neuroendocrinol. 1999;20:199–223. doi: 10.1006/frne.1999.0182. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Luo X, Abbud R, Nilson JH, Parker KL. The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol Endocrinol. 1995;9:478–486. doi: 10.1210/mend.9.4.7659091. [DOI] [PubMed] [Google Scholar]
- Ingraham HA, Lala DS, Ikeda Y, Luo X, Shen WH, Nachtigal MW, Abbud R, Nilson JH, Parker KL. The nuclear receptor steroidogenic factor 1 acts at multiple levels of the reproductive axis. Genes Dev. 1994;8:2302–2312. doi: 10.1101/gad.8.19.2302. [DOI] [PubMed] [Google Scholar]
- Ito M, Park Y, Weck J, Mayo KE, Jameson JL. Synergistic activation of the inhibin alpha-promoter by steroidogenic factor-1 and cyclic adenosine 3′,5′-monophosphate. Mol Endocrinol. 2000;14:66–81. doi: 10.1210/mend.14.1.0410. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 1996;10:60–69. doi: 10.1101/gad.10.1.60. [DOI] [PubMed] [Google Scholar]
- Kleyn PW, Fan W, Kovats SG, Lee JJ, Pulido JC, Wu Y, Berkemeier LR, Misumi DJ, Holmgren L, Charlat O, et al. Identification and characterization of the mouse obesity gene tubby: a member of a novel gene family. Cell. 1996;85:281–290. doi: 10.1016/s0092-8674(00)81104-6. [DOI] [PubMed] [Google Scholar]
- Krylova I, Zhang Y, Darimont BD, Simpson K, Weigel NL, Ingraham HA. Phosphorylation of the nuclear receptor SF-1 modulates cofactor recruitment: integration of hormone signaling in reproduction and stress. Mol Cell. 1999;3:521–526. doi: 10.1016/s1097-2765(00)80480-3. [DOI] [PubMed] [Google Scholar]
- Le W, Conneely OM, Zou L, He Y, Saucedo-Cardenas O, Jankovic J, Mosier DR, Appel SH. Selective agenesis of mesencephalic dopaminergic neurons in Nurr1-deficient mice. Exp Neurol. 1999;159:451–458. doi: 10.1006/exnr.1999.7191. [DOI] [PubMed] [Google Scholar]
- Liu, Q.-R., Walther, D., Uhl, G.R., 2001. Human BDNF gene: variation, expression and haplotypes associated with altered substance abuse vulnerability, Molecular Neurobiology Branch, NIDA-IRP, NIH, 5500 Nathan Shock Drive, Baltimore, MD 21224, USA.
- Luiten PG, ter Horst GJ, Steffens AB. The hypothalamus, intrinsic connections and outflow pathways to the endocrine system in relation to the control of feeding and metabolism. Prog Neurobiol. 1987;28:1–54. doi: 10.1016/0301-0082(87)90004-9. [DOI] [PubMed] [Google Scholar]
- Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, McGarry JD, Parker KL. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology. 2002;143:607–614. doi: 10.1210/endo.143.2.8652. [DOI] [PubMed] [Google Scholar]
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Baker J, Puelles L, Rubenstein JL. Patterning of the basal telencephalon and hypothalamus is essential for guidance of cortical projections. Development. 2002;129:761–773. doi: 10.1242/dev.129.3.761. [DOI] [PubMed] [Google Scholar]
- Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 1996;387:113–116. doi: 10.1016/0014-5793(96)00473-5. [DOI] [PubMed] [Google Scholar]
- Michaud JL. The developmental program of the hypothalamus and its disorders. Clin Genet. 2001;60:255–263. doi: 10.1034/j.1399-0004.2001.600402.x. [DOI] [PubMed] [Google Scholar]
- Michaud JL, Rosenquist T, May NR, Fan CM. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998;12:3264–3275. doi: 10.1101/gad.12.20.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M, Gahara Y, Kitamura T, Ohara O. Distinctive four promoters collectively direct expression of brain-derived neurotrophic factor gene. Brain Res Mol Brain Res. 1994;21:206–218. doi: 10.1016/0169-328x(94)90251-8. [DOI] [PubMed] [Google Scholar]
- Parker KL. The roles of steroidogenic factor 1 in endocrine development and function. Mol Cell Endocrinol. 1998;140:59–63. doi: 10.1016/s0303-7207(98)00030-6. [DOI] [PubMed] [Google Scholar]
- Pellier V, Astic L. Cell death in the developing olfactory epithelium of rat embryos. Brain Res Dev Brain Res. 1994;79:307–315. doi: 10.1016/0165-3806(94)90137-6. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Roberts LM, Shen J, Ingraham HA. New solutions to an ancient riddle: defining the differences between Adam and Eve. Am J Hum Genet. 1999;65:933–942. doi: 10.1086/302601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadovsky Y, Crawford PA, Woodson KG, Polish JA, Clements MA, Tourtellotte LM, Simburger K, Milbrandt J. Mice deficient in the orphan receptor steroidogenic factor 1 lack adrenal glands and gonads but express P450 side-chain-cleavage enzyme in the placenta and have normal embryonic serum levels of corticosteroids. Proc Natl Acad Sci USA. 1995;92:10939–10943. doi: 10.1073/pnas.92.24.10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucedo-Cardenas O, Quintana-Hau JD, Le WD, Smidt MP, Cox JJ, De Mayo F, Burbach JP, Conneely OM. Nurr1 is essential for the induction of the dopaminergic phenotype and the survival of ventral mesencephalic late dopaminergic precursor neurons. Proc Natl Acad Sci USA. 1998;95:4013–4018. doi: 10.1073/pnas.95.7.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron. 1997;19:269–281. doi: 10.1016/s0896-6273(00)80938-1. [DOI] [PubMed] [Google Scholar]
- Shimada M, Nakamura T. Time of neuron origin in mouse hypothalamic nuclei. Exp Neurol. 1973;41:163–173. doi: 10.1016/0014-4886(73)90187-8. [DOI] [PubMed] [Google Scholar]
- Shinoda K, Lei H, Yoshii H, Nomura M, Nagano M, Shiba H, Sasaki H, Osawa Y, Ninomiya Y, Niwa O, et al. Developmental defects of the ventromedial hypothalamic nucleus and pituitary gonadotroph in the Ftz-F1 disrupted mice. Dev Dyn. 1995;204:22–29. doi: 10.1002/aja.1002040104. [DOI] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- Swanson LW. The Hypothalamus. Elsevier Science Publishers B.V.; Amsterdam, New York, Oxford: 1987. [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Vogel MW, Sinclair M, Qiu D, Fan H. Purkinje cell fate in staggerer mutants: agenesis versus cell death. J Neurobiol. 2000;42:323–337. doi: 10.1002/(sici)1097-4695(20000215)42:3<323::aid-neu4>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Voyron S, Giacobini P, Tarozzo G, Cappello P, Perroteau I, Fasolo A. Apoptosis in the development of the mouse olfactory epithelium. Brain Res Dev Brain Res. 1999;115:49–55. doi: 10.1016/s0165-3806(99)00055-3. [DOI] [PubMed] [Google Scholar]
- Wallen A, Zetterstrom RH, Solomin L, Arvidsson M, Olson L, Perlmann T. Fate of mesencephalic AHD2-expressing dopamine progenitor cells in NURR1 mutant mice. Exp Cell Res. 1999;253:737–746. doi: 10.1006/excr.1999.4691. [DOI] [PubMed] [Google Scholar]
- Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DG, Ghatei MA, et al. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology. 2000;141:4325–4328. doi: 10.1210/endo.141.11.7873. [DOI] [PubMed] [Google Scholar]
- Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Zhou C, Tsai SY, Tsai MJ. COUP-TFI: an intrinsic factor for early regionalization of the neocortex. Genes Dev. 2001;15:2054–2059. doi: 10.1101/gad.913601. [DOI] [PMC free article] [PubMed] [Google Scholar]