

SUMMARY
A key factor in the genetically programmed development of the nervous system is the death of massive numbers of neurons. Therefore, genetic mechanisms governing cell survival are of fundamental importance to developmental neuroscience. We report that inner ear sensory neurons are dependent on a basic helix-loop-helix transcription factor called NeuroD for survival during differentiation. Mice lacking NeuroD protein exhibit no auditory evoked potentials, reflecting a profound deafness. DiI fiber staining, immunostaining and cell death assays reveal that the deafness is due to the failure of inner ear sensory neuron survival during development. The affected inner ear sensory neurons fail to express neurotrophin receptors, TrkB and TrkC, suggesting that the ability of NeuroD to support neuronal survival may be directly mediated through regulation of responsiveness to the neurotrophins.
Keywords: Basic helix-loop-helix protein, NeuroD, Inner ear, Cell death, TrkC, TrkB, Deafness, Mouse
INTRODUCTION
As is the case in Drosophila neurogenesis, many basic helix-loop-helix (bHLH) transcription factors are implicated in vertebrate neurogenesis. NeuroD is a bHLH protein that is widely expressed in both the central and peripheral nervous systems (CNS and PNS) in vertebrates (Lee et al., 1995). NeuroD has been shown to be a potent neuronal differentiation factor, as evidenced by its gain-of-function phenotypes that include conversion of a non-neuronal cell fate into a neuronal fate (Lee et al., 1995). Other closely related NeuroD subfamily members, NeuroD2 and Math3 (Atoh – Mouse Genome Informatics), also display such fate conversion activities (McCormick et al., 1996; Takebayashi et al., 1997). Subsequently, neurogenins (Ngns) have been identified and shown to be the upstream activators of the NeuroD gene (Neurod – Mouse Genome Informatics). Ectopic expression of the Xenopus ortholog of Ngn (Neurod3 – Mouse Genome Informatics), X-ngnr-1, in frog embryos results in activation of the endogenous NeuroD and Xath3 genes (Ma et al., 1996; Perron et al., 1999), followed by formation of ectopic sensory neurons (Olson et al., 1998; Perron et al., 1999). Neurogenins are expressed in neuroblasts and are required for the activation of a cascade of downstream bHLH factors, including NeuroD, Math3 and Nscl1 (Nhlh – Mouse Genome Informatics), all of which are expressed in the differentiating neuronal population (Ma et al., 1998).
NeuroD is highly expressed in the nervous system (Lee et al., 1995). In the mouse CNS, a high level of NeuroD expression is found in differentiating neurons as well as in mature neurons such as the granule cells of the cerebellum and hippocampus and the neurons of the limbic system. In the PNS, NeuroD expression is found in developing and mature sensory neurons, but not in autonomic neurons. NeuroD is expressed in other peripheral endocrine cells including those in the pancreas, gut and stomach (Naya et al., 1997). Consequently, NeuroD-null mice die shortly after birth due to neonatal diabetes that results from developmental and functional defects of the pancreatic endocrine cells (Naya et al., 1997). By reintroducing NeuroD in the pancreatic endocrine cells of the NeuroD-null mutant mice, we have recently rescued neonatal lethality and shown that NeuroD is required for proper CNS development (Miyata et al., 1999). In the NeuroD-null brain, the granule cells in the hippocampus and cerebellum fail to survive during differentiation, which results in ataxic mice that lack most of the internal granule layer in the cerebellum, and completely lack the dentate gyrus of the hippocampus (Miyata et al., 1999). In addition to obvious motor dysfunction resulting from the cerebellar defects, we have observed that the rescued mice display an inability to balance themselves and, in some cases, severe circular head movements, suggestive of vestibular defects. Thus, we have examined NeuroD-null mice and find severe developmental defects in the inner ear, which is evident throughout the embryonic stages.
The neuronal cells emanate from the otic placode between embryonic day (E)9.5 and E15.5 to form the vestibulo-cochlear ganglia (VCG), which will later separate into the vestibular and spiral (or cochlear) ganglia. The basic inner ear structure and ganglia are settled by E14.5 after which maturation occurs (for a review, see Fritzsch et al., 1999). Inner ear development is critically dependent on neurogenin 1 (Ngn1; Neurod3 – Mouse Genome Informatics) which is also required for development of all neural crest-derived cranial sensory neurons (Ma et al., 1998; Ma et al., 2000). Development of the epibranchial placode derived sensory neurons, however, is dependent on Ngn2 (Atoh4 – Mouse Genome Informatics; Fode et al., 1998). All neurogenin proteins are expressed in dividing precursors and, thus, their phenotype can be attributed to defects in the precursor cells, such as a failure to proliferate. Consequently, all sensory neurons of the inner ear fail to develop in mice lacking Ngn1 and some defects also occur in sensory epithelia (Ma et al., 1998; Ma et al., 2000). Development of the inner ear hair cells in mice is crucially dependent on another bHLH protein, mammalian atonal homolog Math1 (Atoh1 – Mouse Genome Informatics), the absence of which results in complete loss of hair cells in an otherwise normal inner ear (Bermingham et al., 1999). Data on neurogenins and Math1 provide evidence that these proteins function during the early steps in generation of sensory neurons and sensory hair precursor cells of the inner ear, respectively, rather than during differentiation (Ma et al., 1998; Bermingham et al., 1999). The first transcription factor known to be specifically involved in late sensory cell differentiation of the inner ear is a POU-domain containing transcription factor, Brn3a (Pou4f1 – Mouse Genome Informatics), whose null mutant phenotype involves failure to maintain survival of sensory neurons after maturation (McEvilly et al., 1996). The inner ear phenotype in Brn3a-null mice resembles the phenotypes found in the mice carrying mutations in neurotrophins or their receptors (McEvilly et al., 1996; Fritzsch et al., 1999). However, a related protein, Brn3c (Pou4f3 – Mouse Genome Informatics), was shown to be required for hair cell development (Erkman et al., 1996). Here, we report that the inner ear sensory neurons are uniquely dependent on NeuroD for their survival during the early phases of differentiation. Migration of sensory neurons and the formation of correct synapses are also affected in the NeuroD-null inner ear, possibly due to failure of these neurons to fully differentiate.
Apart from these transcription factors, the survival of the inner ear sensory neurons is also dependent on TrkB and TrkC signaling (see Fritzsch et al., 1999 for a review). One of these neurotrophin receptor and ligand pairs, TrkB/BDNF (brain-derived neurotrophic factor), is important for the development of the vestibular sensory neurons, while the other pair, TrkC/NT3 (neurotrophin 3), is required for the development of the auditory sensory neurons. These two signaling pathways partially overlap and compensate functionally at the receptor and cellular levels. Thus, mice lacking components of the TrkB/BDNF or TrkC/NT3 signaling pathway lack a subset of sensory neurons in the inner ear at birth (Ernfors et al., 1995; Fritzsch et al., 1997). Other epibranchial placode-derived sensory neurons in the distal portions of cranial nerve ganglia VII (geniculate ganglion), IX (petrosal ganglion) and X (nodose ganglion) also show dependence on TrkB for their survival (Fritzsch et al., 1998). In contrast, the neural crest-derived cranial sensory ganglia do not display such crucial dependency on TrkB signaling, suggesting that the dependency on a specific neurotrophin signal relates to the origins of the cells. We show here that the inner ear sensory neurons lacking NeuroD fail to express TrkB and TrkC, and subsequently undergo cell death.
MATERIALS AND METHODS
Surgical procedure
All procedures were performed in accordance with IACUC guidelines for the humane use of laboratory animals in biological research. Adult ND−/−Tg and sibling control (9 months old) mice were anesthetized to surgical levels using interperitoneal injections of ketamine hydrochloride (85 mg/kg) and xylazine (17 mg/kg) and secured in a stereotaxic frame. A unilateral craniotomy extending from bregma to lambda and lateral to the temporal bone was performed over the right hemisphere, exposing a wide region of parietotemporal cortex where the dura was reflected. The exposed cortical surface was regularly bathed with physiological saline, and body temperature was maintained using a regulated heating pad. Additional ketamine and xylazine was administered at approximately 40 minute intervals during subsequent recording, as required, to maintain a level of anesthesia such that the corneal reflex could barely be elicited.
Stimulation
Auditory evoked potentials (AEP) were obtained using click stimuli (0.1 ms monophasic pulses) presented contralaterally using a piezo-electric speaker positioned approximately 10 cm lateral to the animal’s head. Somatosensory evoked potentials (SEP) were obtained by tying together the 25 large facial vibrissae of the contralateral (left) mystacial pad and displacing them simultaneously at approximately 0.5 cm from their base using a laboratory-built apparatus which converts computer-generated square-wave pulses (duration 0.4 ms) into silent dorsolateral displacements (~0.5 mm) of the stimulator arm. In the ND−/−Tg mouse, many facial vibrissae were missing, and it was necessary to stimulate both the remaining vibrissae and the underlying mystacial pad to obtain a response.
Surface recording
Epipial maps of the click-evoked AEP complex and the vibrissa-evoked SEP complex were recorded using a flat multi-channel electrode array consisting of 64 silver wires arranged in an 8 × 8 grid (tip diameter, ~100 μm; interelectrode spacing, 0.5 mm) covering a 3.5 × 3.5 mm area of the cortical surface in a single placement (see Fig. 1A). This placement covered the primary auditory (A1) cortex as well as the vibrissal representation of primary somatosensory cortex (‘vibrissa/barrel field’ or S1) and secondary somatosensory cortex (S2). Epipial potentials were referenced to a silver ball electrode secured over the contralateral frontal bone, and were simultaneously amplified (×2000), analog filtered (band-pass cut-off, −6dB at 0.001 to 5000 Hz; roll-off, 5 dB/octave) and digitized at 10 kHz. AEP and SEP were averaged over 100 stimulus presentations.
Fig. 1.
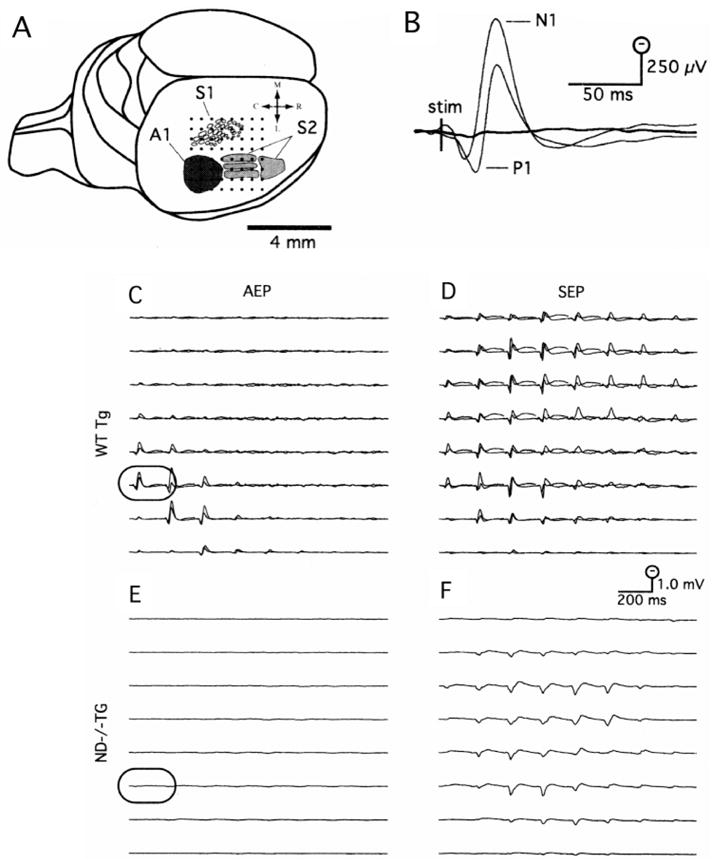
Surface mapping the AEP and SEP from ND−/−Tg mouse and sibling controls. (A) Placement of the 64 channel epipial recording array (dots) covered a 3.5 × 3.5 mm area of the exposed right hemisphere. This placement included primary auditory cortex (A1; black region), as well as the vibrissal representation of primary and secondary somatosensory cortex (S1 and S2, respectively), the approximate borders of which were adapted from Wallace (Wallace, 1987). (B) Enlargements of the AEP from and electrode over auditory cortex (C and E; outlined traces) in ND−/−Tg mouse (thick trace) and controls (thin traces). The AEP was composed of a biphasic positive/negative fast wave labeled P1/N1 to indicate the polarity and sequence of occurrence. (C) Superimposed AEP from two siblings had a similar amplitude, morphology and spatial distribution, centered over auditory cortex in the caudolateral region of the array. (D) The SEP complex in the controls was more widespread, covering both somatosensory cortex in the caudolateral electrodes of the array, as well as motor cortex more medially. (E) In ND−/−Tg mouse, the AEP complex was absent. (F) The SEP complex in ND−/−Tg mouse was comparable in amplitude and spatial distribution to the controls.
Mice and genotyping
Generation of the NeuroD mutant mice and genotyping were performed as described previously (Miyata et al., 1999). Briefly, NeuroD−/− mice were generated by replacing the entire NeuroD coding region with a cytoplasmic lacZ reporter gene (Miyata et al., 1999). These mice die shortly after birth due to neonatal diabetes (Naya et al., 1997). The ND−/−Tg mice refer to the NeuroD−/− mice carrying a transgene encoding Myc-tagged NeuroD under the control of the insulin gene promoter. These mice are rescued from neonatal lethality and survive to adulthood, but their nervous systems remain devoid of NeuroD (Miyata et al., 1999).
In situ hybridization
Nonradioactive in situ hybridization using the antisense RNA probes labeled with digoxigenin was performed as described elsewhere (Lee et al., 1995). Briefly, the plasmids containing the mouse cDNAs for NeuroD (1.7 kb; Lee et al., 1995), NT3 (0.8 kb; Hohn et al., 1990) and BDNF (0.8 kb; Hofer et al., 1990) were used to generate the antisense RNA probes by in vitro transcription in the presence of digoxigenin-labeled UTPs. The embryos were fixed for 1–3 hours with 4% paraformaldehyde at 4°C, immersed in 20% sucrose and frozen in OCT freezing compound. The frozen blocks were sectioned at 10 μm and hybridized overnight at 60°C to the riboprobes in hybridization solution containing 50% formamide. The probes were detected by an anti-digoxigenin-antibodies conjugated with alkaline phosphatase (Roche, IN), which were reacted with NBT (nitro blue tetrazolium chloride) and BCIP (5-bromo-4-chloro-3-indolyl phosphate) substrates for color reaction.
X-gal staining
Dissected tissues were briefly fixed with 3% formaldehyde and 0.25% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3), rinsed in wash solution (0.1 M sodium phosphate, 0.1% deoxycholic acid, 0.2% NP40, 2 mM magnesium chloride) and stained in wash solution containing 1 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside). The stained whole-mount samples were either further dissected for microstructure examination or were embedded in sucrose and sectioned at 16 μm.
Immunohistochemistry and cell death assay
For TrkB and TrkC staining, embryos were fixed in Carnoy’s fixative (10% acetic acid; 30% chloroform; 60% ethanol), dehydrated with ethanol and embedded in paraffin. 7 μm sections were collected, defatted and rehydrated, and the endogeneous peroxidase activity was quenched with 3% peroxidase solution. The sections were equilibrated in TBS (Tris-buffered saline, pH 7.4) and blocked with TBS containing 0.4% Triton X-100, 3% BSA (Fraction V, Sigma, NE) and 10% normal serum from the host species of the secondary antibody used (goat serum for TrkB and rabbit serum for TrkC). Primary antibodies, chicken anti-TrkB (1:1000) or goat anti-TrkC (1:500), and biotinylated secondary antibodies (Vector Lab, CA) were applied followed by treatment with Vectastatin AB complex (1:500, Avidin-Biotin and peroxidase complex), which was visualized with DAB (3,3′ diaminobenzidine tetrahydrochloride).
To reveal the fiber projection patterns, whole-mount immunostaining using an antibody to acetylated-tubulin was performed as described elsewhere (Fritzsch et al., 1997). Briefly, the X-gal stained, and defatted inner ears were reacted with primary antibody (6-11B1, 1:500, Sigma, MO) for 24 hours. The specimen was incubated with HRP-conjugated secondary antibody and reacted with DAB until the fibers could be identified.
The dying cells were detected by a modified TUNEL assay (ApopTag, Intergeon, NY) according to the manufacturer’s instructions, followed by neuronal staining using monoclonal anti-neuronal tubulin antibody, TUJ-1 (1:1500 Babco, CA) and rhodamine-labeled goat anti-mouse secondary antibody. Sections were washed and mounted after DAPI staining of the nuclei. The digital images of the Fluorescein-labeled dying cells and Rhodamine-labeled neurons were captured using a fluorescence microscope, Nikon Eclipse E8000 (Nikon, Tokyo), and analyzed with Slidebook software (Intelligent Imaging Innovation, CO).
DiI labeling
To label the afferent fibers, the heads from the mouse or embryos were cut sagittally at the midline, and DiI-soaked filter strips were applied onto the alar plate of the brainstem to label the eighth cranial nerve fibers coming from the inner ear. Efferent fibers were labeled by applying DiI-soaked filter strips into the olivo-cochlear efferent bundle of the contralateral side near the floor plate (Fritzsch and Nichols, 1993). After an appropriate diffusion time, the ears were dissected and examined with a compound epifluorescence microscope. The images were captured with cooled CCD camera and processed using a deconvolution technique (Vaytek and ImagePro software, IA)
Transmission electron microscopic analysis
The ears dissected from the mouse were osmicated in 1% OsO4 for 1 hour, dehydrated and embedded in epoxy resin. Semi-thin and ultra-thin sections were viewed by light microscope and electron microscope (Phillips CM10), respectively.
Cell count
Every fourth 7 μm sagittal section of the paraffin-embedded embryo heads were stained with antibody to TUJ1 or with Cresyl Violet. Only the cells containing a clear, round nucleus were counted from the vestibulo-cochlear ganglia using at least two independent samples of the same litter.
RESULTS
NeuroD-null mice are deaf
To measure the extent of the inner ear sensory deficit in the rescued adult NeuroD-null mice (ND−/−Tg) that are severely ataxic (Miyata et al., 1999), we measured auditory evoked potentials (AEP) and somatosensory evoked potentials (SEP) by cortical surface recording using 64 channel-electrodes routinely applied to rats. Fig. 1A depicts a schematic of the electrode array placement covering both the auditory and somatosensory cortex of the right hemisphere. In sibling control animals, the AEP complex was prominent in the lateral and caudal electrodes of the array (Fig. 1C), covering the estimated region of primary auditory cortex (A1) of the mouse. Enlarged traces from an electrode directly above the auditory cortex (Fig. 1C, framed trace) are shown in Fig. 1B (thin traces). The recording shows that the AEP, which was composed of a typical positive/negative fast wave (peaking at 12 and 32 ms post-stimulus for the positive and negative waves, respectively), labeled the P1/N1, reflecting the polarity and sequence of occurrence of these temporal components. The SEP complex in sibling controls was quite similar across animals (Fig. 1D), and was also composed of a prominent P1/N1 component that could be recorded at electrode sites adjacent to the primary auditory response (the approximate location of S2) and also at more medial electrode sites covering the vibrissa/barrel field or S1. The waveform morphology and relative spatial distributions of the AEP and SEP complex in these animals were quite similar to those mapped in previous studies of the rat (Barth and Di, 1991; Barth et al., 1993; Di and Barth, 1991; Di and Barth 1992). However, mice lacking NeuroD protein exhibit no AEP while maintaining the SEP. SEP responses with a similar amplitude and spatial distribution in ND−/−Tg mice can be seen in Fig. 1F. The latency of the SEP was slightly longer (~5 mseconds to P1 peak) than that in controls, probably due to stimulation of the mystacial pad along with the remaining vibrissae. However, as shown in Fig. 1E (enlarged in Fig. 1B – thick trace), the AEP response in ND−/−Tg animals was grossly attenuated when compared with sibling controls.
NeuroD is expressed in developing inner ear sensory neurons
To understand the basis of deafness in NeuroD-null mice, we first analyzed the NeuroD expression pattern during inner ear development. We determined the expression patterns of the NeuroD mRNA as well as that of the lacZ reporter gene, which was inserted in place of the NeuroD coding region. Heterozygous mouse embryos, which show no discernible phenotype, were used as positive controls for the expression analysis. The X-gal staining, which reflects the endogenous NeuroD mRNA expression, was restricted to the neuronal cells in the inner ear (Fig. 2A,B). In situ hybridization analysis confirmed neuronal-specific expression of NeuroD throughout inner ear development in embryos between E9.5 and E12.5. At E9.5, when a few postmitotic neurons are known to leave the otic placode (see Ma et al., 1998; Fritzsch et al., 1999 for reviews) and form the VCG anterior to the ear, NeuroD is already expressed in the cells within the otic epithelium. Stronger expression both within the otic epithelium and among the delaminating cells migrating away from the ear to form sensory neurons can be seen at E10.5 (Fig. 2A,B). Vestibular sensory neurons become postmitotic between E9.5 and E12.5, and spiral sensory neurons between E10.5 and E15.5 in a base to apex progression (Ruben, 1967). Throughout that period the number of neurons increases within the vestibular and spiral ganglia (VIII) most of which express NeuroD.
Fig. 2.

NeuroD is expressed in developing inner ear and is required for proper development of the inner ear sensory neurons. (A) In situ hybridization of E10.5 wild-type mouse embryo section using antisense NeuroD RNA as a probe and alkaline phosphatase reaction for visualization. (B) E10.5 NeuroD+/− mouse embryo section stained with X-gal. (C,D) Inner ear dissected from newborn (P0) NeuroD+/− (C) and NeuroD−/− mice (D), and stained with X-gal as whole mount. Anterior vertical canal (AVC), horizontal canal (HC), posterior vertical canal (PVC), saccule (S), utricle (U), vestibular ganglion (vgl), geniculate ganglion (ggl), spiral ganglion (sgl), modiolus (M) and cochlear hair cells (CHC) are indicated. In NeuroD−/− inner ear, the spiral ganglia (sgl) staining is absent and that of vestibular ganglia (vgl) is reduced and displaced compared with the control. (E,F) The apical and middle turn of the P0 cochleae are dissected and mounted after whole-mount staining with X-gal. The absence of spiral ganglia staining in NeuroD−/− inner ear is evident. Hair cells expressing lacZ are present in the NeuroD−/− cochleae. (G,H) Part of vestibular organ containing the vestibular ganglia, utricle and saccule is dissected out and mounted after whole-mount staining with X-gal to reveal the reduction in the size of the vestibular ganglia in NeuroD−/− mice. Scale bars: 100 μm.
The inner ear sensory neurons are depleted in NeuroD−/− mice
To determine the developmental time at which the inner ear phenotype first occurs, we analyzed the inner ear of NeuroD−/− embryos at various developmental stages. During the initial phases of sensory neuron formation at E9.5, no difference can be observed between control and NeuroD−/− mice at the X-gal staining level. However, starting at E10.5, a noticeable reduction in the number of lacZ-expressing cells is evident in the vestibulocochlear ganglion (VCG) of NeuroD−/− mice. By E14.5, the spiral (cochlear) ganglion of NeuroD−/− mutants is almost absent based on X-gal staining, while the spiral ganglion in control heterozygous mice is composed of many cells strongly expressing lacZ. This phenotype in NeuroD−/− mice persists until birth (P0) (Fig. 2C–H) when a small number of lacZ-positive spiral sensory neurons is found scattered predominantly in the center of the cochlear turn near the fiber bundle of the modiolus, the central axis of the cochlea (Fig. 2D). The volume of the modiolus and the radius of the cochleae in NeuroD−/− mice appeared drastically reduced compared with those in control siblings (Fig. 2E,F). In addition, an empty space in the middle of the cochlea is pronounced in NeuroD−/− mice (Fig. 2F). This phenotype persists until at least 9 months of age, which is the latest age examined using the rescued ND−/−Tg mice (Miyata et al., 1999). The density of the few remaining spiral neurons and the number of radial fibers extending toward the cochleae showed variation among individuals. In the majority of cases, only about 30 spiral neurons that express lacZ were found, as compared with thousands of neurons in the control inner ear at P0. In two cases, we found only two labeled neurons with fibers extending toward the middle turn. The distribution of the few remaining spiral sensory neurons was always in the middle turn, with no neurons near the apex or the base. In fact, most of the surviving spiral ganglion cells in NeuroD−/− mice were not in the typical spiral ganglion position but in or near the modiolus, a position where only a few neurons are encountered in control animals (Fig. 2C,D).
At E14, the vestibular sensory neurons of both heterozygous control and NeuroD−/− mice show a diffuse distribution of lacZ-positive and -negative cells, whereas at P0, most vestibular sensory neurons express lacZ. However, there is a topological difference in the position of the vestibular ganglia between control and NeuroD−/− mice. In heterozygous and wild-type control mice, the vestibular ganglion is in a small medial depression outside the otocyst, whereas the vestibular ganglion of NeuroD−/− mice is inside the otic capsule, adjacent to the lacZ-positive geniculate ganglion. In addition, the volume of the vestibular ganglia is considerably decreased (Fig. 2G,H). This decrease is comparable with that found in TrkB- or BDNF-null mice (Bianchi et al., 1996).
Tracing afferent and efferent fibers with DiI and tubulin immunohistochemistry
To assess the extent and patterns of the axon projection in the affected inner ear in NeuroD−/− mice, we labeled the afferent and efferent fibers using DiI implantation and acetylated tubulin immunohistochemistry. The eighth nerve afferent fibers were labeled from the alar plate of the brainstem where their fibers terminate centrally, and the olivo-cochlear efferent fibers from rhombomere 4 where they cross (Cowan et al., 2000).
In the cochleae of NeuroD−/− mice, there was an almost complete loss of all afferent and most efferent innervation to the basal and apical turns as early as E14.5, while the greatly reduced radial fibers extended toward the middle turn of the cochlea. This phenotype persists until birth. Interestingly, the radial fibers interdigitated upon approaching the cochlea, forming a candelabra of fibers next to the inner hair cells. However, only a few afferent fibers could be traced to the three rows of outer hair cells in the NeuroD−/− cochleae (Fig. 3A–D). There was a pronounced elongation of inner spiral bundle fibers along the inner hair cells from the middle turn to the tip of the base (Fig. 3B).
Fig. 3.

Innervation defects of the spiral ganglia in the NeuroD−/− mice revealed by DiI labeling. DiI labeling of the afferent nerve fibers (A–D) and efferent nerve fibers (E,F) innervating cochlear sensory epithelium at P0. The spiral ganglion tightly innervates the cochlear hair cells in sibling control animals (A). (B) In NeuroD−/− cochleae, only a few afferent fibers are present in the middle turn of the cochleae. (C,D) The higher magnification images showing the innervation to the inner and outer hair cells (ihc and ohc, respectively). (E) At P0, efferent fibers of sibling control animal reach the inner hair cells of the cochleae through out the entire epithelium. (F) In contrast, efferent fibers in the inner ear of NeuroD−/− mice are restricted to few fibers to the middle turn of the cochleae. NeuroD−/− efferent fibers fail to branch on their way from the modiolus to the cochlea. Scale bars: 100 μm.
As with the afferent fibers, the efferent fibers to the cochleae were also greatly reduced in NeuroD−/− mice. The pattern of efferent innervation closely followed the afferent pattern, suggesting that efferents somehow navigate along afferents as previously recognized (Fritzsch et al., 1999). As with afferents, efferents did not reach the apex or the base of the cochleae in radial bundles (Fig. 3E,F). Nevertheless, some efferents extended as inner spiral bundles along inner hair cells toward the apex or the base in many NeuroD−/− mice. In some cases, the afferent and efferent fibers were so greatly reduced in number that large parts of the cochleae appeared denervated.
We also analyzed the pattern of innervation in the vestibular sensory epithelia. As mentioned above, the vestibular ganglia in NeuroD−/− mice were in an unusual position, inside the otic capsule and near the utricle and saccule. DiI tracing and tubulin immunostaining showed that all vestibular sensory epithelia received a variable degree of innervation by both afferent and efferent fibers. However, there were dramatic differences in the density of fibers innervating the semi-circular canal epithelia. For example, innervation density to the anterior vertical canal in NeuroD−/− mice was indistinguishable from that in control littermates, as assessed by DiI labeling of neuronal fiber. The innervation in the horizontal canal was also mildly reduced in NeuroD−/− mice. The density of fibers in the saccule and utricle was only moderately reduced in comparison with those in control siblings. In contrast, innervation density to the posterior vertical canal of NeuroD−/− mutants was drastically reduced (Fig. 4H). Occasionally, a single afferent or efferent fiber extended to the posterior vertical canal. Overall, the vestibular sensory epithelium showed an anterior to posterior gradient of fiber loss with the posterior vertical canal being the most affected and the anterior vertical and horizontal canal being the least affected.
Fig. 4.

The pattern of innervation and of lacZ-expressing hair cells (HC: blue in A–F) in the P0 vestibular epithelia. (A–F) Dense innervation of all sensory epithelia in the NeuroD+/− animals (A,C,E) and the reduced density and partial absence of nerve fibers to parts of the sensory epithelium in NeuroD−/− mice (B,D,F) are shown in black. Also evident is disorganized fiber projection towards the sensory epithelia, as indicated by acetylated tubulin staining of the fibers. (G,H) DiI labeling of the afferent fibers to the PVC in control sibling (G) and NeuroD−/− (H) ears at P0 shows significant reduction in innervation into the PVC in NeuroD−/− mice. (I,J) DiI-labeling of the extending fibers at E11.5 indicates that fiber projection fails to initiate towards the PVC in NeuroD−/− mice. Scale bar in G, 100 μm for G,H; in I, 100 μm for I,J.
In addition to such a quantitative difference in the fiber projection, there was also a qualitative difference among the fibers that extended toward the vestibular sensory organ of NeuroD−/− mice. Tubulin immunostaining clearly indicated that the patterns of the fiber projection throughout the epithelium was highly disorganized (Fig. 4). Even in the places where a substantial amount of the fibers remained, such as in the horizontal and anterior vertical canals, the fibers seemed to project into random directions and failed to form large bundles. We next addressed whether absence of posterior vertical canal innervation, which represents the most severe fiber reduction phenotype, resulted from neurite outgrowth problems or from a rapid loss of fibers after a normal initial outgrowth. To distinguish between these two possibilities, we examined the posterior vertical canal (PVC) at E11.5 when the fibers begin to extend toward their targets. As shown in Fig. 4I,J, the fibers failed to project to the PVC in NeuroD−/− mice, indicating that the problem lies in failure to initiate fiber projection. Therefore, in addition to extensive neuronal loss, failure in both initiation and/or guidance of fiber projection can be observed in NeuroD−/− inner ear.
The inner ear sensory neurons of NeuroD-null mice die early during development
We examined if the loss of the inner ear sensory neurons is due to cell death during development, as we have found to be the case with the NeuroD−/− neurons in the cerebellum and hippocampus (Miyata et al., 1999). Starting at E10.5, reduction in volume and cell number in the VCG is observed in NeuroD−/− mice (Fig. 5A). By E11.5, the cell number in the VCG of NeuroD−/− mice is only about half of that found in control mice, indicating that extensive cell loss occurs between E10.5 and E11.5 (Fig. 5A). We performed a TUNEL assay and observed that numerous TUNEL-labeled cells are detected at E11.5 in the VCG of NeuroD−/− embryos, while few TUNEL-positive cells are detected in the VCG of the control animals (Fig. 5B,C). By E12.5, the VCG has separated into the vestibular and spiral ganglia, and the cell loss in the spiral ganglia is nearly complete by E13.5 (Fig. 5D,E). The timing of cell death in the NeuroD−/− VCG corresponds to the time during which they normally exit cell cycle and differentiate (Ruben, 1967).
Fig. 5.
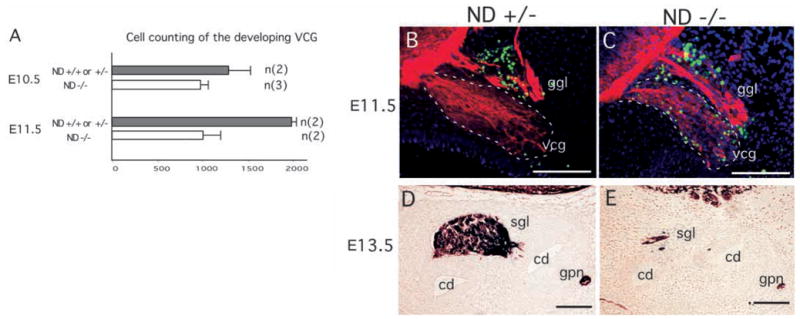
Cell loss in the developing vestibulo-cochlear ganglia (vcg) of NeuroD−/− mice. (A) Cell counting was performed in the vcg at E10.5 and E11.5. By E11.5, a significant reduction in vcg cell number is evident in NeuroD−/− embryos. Error bars indicate standard deviations. (B,C) Cell death as detected by a TUNEL assay in the vcg at E11.5. Green indicates the fluorescein-labeled TUNEL-positive cells. Dark blue indicates nuclear staining by DAPI. Red indicates anti-β-tubulin staining (TUJ1), as visualized with rhodamine-conjugated secondary antibody. The TUNEL-positive profiles near the geniculate ganglion layer (ggl) are present in both the control (B) and NeuroD−/− (C) inner ears. The vcg of NeuroD−/− mice display an increased number of TUNEL-positive cells. (D,E) The spiral ganglia of E13.5 NeuroD+/− and NeuroD−/− embryos are stained using the TUJ1 antibody and visualized by DAB (3,3′ diaminobenzidene). The spiral ganglia (sgl), cochlear ducts (cd) and great petrosal nerve (gpn) are indicated. Scale bars:100 μm.
Expression of TrkC and TrkB is absent during early stages of inner ear development in NeuroD-null mice
The loss of neurons in both the spiral and vestibular ganglia observed in NeuroD−/− mice is reminiscent of the phenotypes found in TrkC and TrkB mutant mice, respectively. TrkC-null mice display a severe loss of neurons in the spiral ganglia, whereas TrkB-null mice show severe neuronal loss within the vestibular ganglia (Ernfors et al., 1995). To determine whether expression of these Trk receptors is affected in NeuroD−/− mice, we examined their inner ear using antibodies specific to each TrkB and TrkC receptor (Huang et al., 1999). At E10.5, control mice displayed high levels of TrkC and TrkB expression in the developing VCG (Fig. 6A,C). However, in NeuroD−/− mice, expression of TrkC was completely and selectively absent in the VCG at E10.5 (Fig. 6B), shortly before extensive neuronal loss was observed (Fig. 5C). The expression of TrkB was maintained in a few cells of the vestibular-cochlear ganglia (Fig. 6D). In contrast, we did not observe any changes in TrkB or TrkC expression in the nearby trigeminal ganglia (Fig. 6A–D, inset). Likewise, the geniculate sensory neurons, which are located near the VCG and strongly express TrkB but not TrkC at E11.5, did not display any change in Trk expression in NeuroD−/− embryos (data not shown).
Fig. 6.

Expression analysis of the neurotrophin receptors, TrkB and TrkC, and their respective ligands, BDNF and NT3, in E10.5 inner ear. (A–D) Immunohistochemistry was performed on E10.5 vcg of control sibling (A,C) and NeuroD−/− (B,D) mice with anti-TrkC (A,B) and anti-TrkB (C,D) antibodies followed by visualization using a DAB. The arrowheads in D indicate a small number of TrkB-positive cells remaining in the NeuroD−/− vcg. The insets show unaltered TrkB/C staining in the trigeminal ganglion (tgg) from the same embryos. (E–H) In situ hybridization using antisense NT3 (E,F) and antisense BDNF (G,H) reveals that both ligands are expressed in the E10.5 inner ear of NeuroD−/− mice (F,H). Scale bar: 100 μm.
To determine whether the expression of the ligands to these receptors is also affected, we examined the expression patterns of NT3 and BDNF. As shown in Fig. 6E–H, the ligand expression in NeuroD−/− mice was comparable with that found in control mice, suggesting that the lack of Trk expression in the inner ear of NeuroD−/− mice is a specific cell-autonomous event that is independent of the expression of the ligands. These results indicate that the inner ear sensory neurons are uniquely dependent on NeuroD for expression of neurotrophin receptors, TrkB and TrkC.
Hair cell development in the sensory epithelium
Finally, we examined the sensory hair cells in the inner ear epithelia. Vestibular sensory epithelia show lacZ-positive hair cells as early as E13.5, whereas the cochlear sensory epithelia do not display such lacZ-positive hair cells prior to E14.5. The distribution of lacZ-positive hair cells is not uniform throughout the sensory epithelia. For example, in the utricle and saccule, lacZ-positive hair cells are found more frequently in the striola region, whereas the extrastriola region shows fewer lacZ-positive hair cells. In the semi-circular canal sensory epithelia, there is only a slight aggregation of lacZ-positive hair cells in the abneuronal side. At P0, both inner and outer hair cells of the cochleae show lacZ positive hair cells (Fig. 7A). However, many more inner than outer hair cells are lacZ-positive. Interestingly, a high number of lacZ-positive hair cells are found in NeuroD−/− mutant mice, indicating that hair cell development does not require NeuroD.
Fig. 7.

The inner ear hair cells develop and are maintained in the absence of innervation in NeuroD−/− mice. (A) Normally organized cochlear hair cells in NeuroD−/− mice are shown by X-gal staining. Many of the inner hair cells (IHC) and less of the outer hair cells (OHC) express various levels of lacZ. (B) Cochlear hair cells develop normally, even in 9-month-old animals in which the apical turn never received any innervation. (C–F) Electron micrographs of the posterior vertical canal (PVC) of a 9-month-old control (C) and NeuroD−/− mouse (D–F) show the absence of large calyxes, afferent innervation shown as empty space, around the Type I hair cells in NeuroD−/− mouse. The smaller size of the sensory epithelium and the absence of nerve fibers underneath the sensory epithelium are also evident. Despite the complete lack of innervation throughout embryonic and adult stages, hair cells are rather normal (D–F). Closer examination shows no nerve endings inside the sensory epithelium (D) but the presence of two types of hair cells (F): one type with stereocilia larger than kinocilia (asterisks), and the other with stereocilia the same diameter as kinocilia (# and circles). The Type I (I) and Type II (II) hair cells are marked in E. Note that the nuclei of the Type I cells are positioned deeper from the surface. In addition, the cuticular plate underneath the cilia coming out at the apex is thicker in the type I hair cells. The presence of two different types of hair cells suggests that development of the hair cells is independent of innervation. Scale bars: 100μm in A; 10 μm in B–E; 1 μm in F.
The apparent absence of innervation in the posterior vertical canal and in the basal and apical regions of the cochleae presented an opportunity to study the long-term differentiation of hair cells in the absence of innervation. Given the large variation in cochlear innervation, we focused here on the development of hair cells in posterior vertical canal of ND−/−Tg mice that were 9 months of age. Remarkably, hair cells are rather normal in their overall development in ND−/−Tg mice (Fig. 7C–F). Both light microscopic and transmission electron microscopic data showed complete absence of any innervation in these two cases. Nevertheless, two types of hair cells were present in the posterior vertical canal sensory epithelia based on the diameter of the stereocilia compared with the kinocilia consistent with suggestions of different hair cell types (Eatock and Rüsch, 1997) and the aggregation of heterochromatin (Fig. 7D,E). Likewise, the cochleae of the adult animals showed differentiated hair cells in the basal and apical turns that did not receive any afferent or efferent innervation at this stage of development (Fig. 7B). To our knowledge, this is the first time that this degree of autonomy of hair cell development and maintenance has been analyzed in vivo over such a long period.
DISCUSSION
Patterns of sensory neuronal loss in NeuroD−/− mice
The present data demonstrate that NeuroD is required for proper development of inner ear sensory neurons in mice. Electrophysiological analysis reveals that while somatosensory function remains in NeuroD−/− mice, they are deaf, as reflected by a complete absence of the AEP. The phenotypic analysis reveals an extensive sensory neuronal deficit in the inner ear of NeuroD−/− mice.
The pattern of remaining sensory neurons and afferent innervation shows an early onset of developmental loss that has reached its final pattern by E14.5. However, in contrast to the Ngn1-null mutants, where a complete loss of sensory neurons is observed (Ma et al., 1998; Ma et al., 2001), some sensory neurons still remain in NeuroD−/− mice. Thus, a factor whose function can compensate for the loss of NeuroD must exist for the survival of those remaining vestibular and spiral sensory neurons that persist up to 9 months of age. We have recently generated NeuroD−/−;Math3−/− and NeuroD−/−; NeuroD2−/− double-null mice and found that they do not display additional phenotype in the inner ear (our unpublished data), suggesting that NeuroD2 and Math3 are not likely to be such compensatory factors in the inner ear.
Loss of Trk expression as a potential cause of cell death
Interestingly, the neuronal loss and patterns of the remaining fibers observed in NeuroD−/− mice are similar to what has been published for compound Trk mutations (Fritzsch et al., 1998). Specifically, in the cochleae, the loss of basal turn innervation and spiraling of afferent and efferent fibers along the inner hair cells toward the basal tip of the cochlea is reminiscent of what has been described for NT3 (Fritzsch et al., 1997) and TrkC-null mutants (Fritzsch et al., 1998). However, the pattern of the neuronal loss in NeuroD−/− mice resembles most closely the phenotype found in the TrkC-null mutation combined with TrkB heterozygosity (Fritzsch et al., 1998). As in those mutants, there is a high degree of interindividual variation in the density of remaining afferent and efferent fibers. This evidence strongly supports the idea, derived from our TrkC and TrkB immunocytochemical data, that NeuroD functions upstream of TrkC and, to a lesser extent, of TrkB in the inner ear.
Although the inner ear sensory neurons die in both NeuroD and single Trk mutant mice, the timing of neuronal loss is different. Neuronal death in the inner ear of NeuroD−/− mice occurs early during development, before the sensory neurons make contact with their target cells, while that in the single Trk mutant mice occurs after target contacts have been made (see Fritzsch et al., 1999 for a review). These observations lead us to propose that in certain neuronal population, such as those affected in NeuroD−/− mice, the TrkC and TrkB receptors play compensatory roles for supporting the survival of young developing neurons before any target contact is made. Once the target contact is made, the TrkB and TrkC receptors each play specific roles in neuronal maintenance, presumably due to target specific expression of neurotrophins (Fritzsch et al., 1999). According to this model, we would predict that the onset of sensory neuron loss in TrkB/TrkC or BDNF/NT3 double-null mutants would occur early during development, as in NeuroD-null mice, but such study has not been conducted (see Fritzsch et al., 1999 for a review). In support of this model, TrkB−/−; TrkC+/− or TrkB+/−; TrkC−/− mice, but not single Trk mutant mice, display granule cell loss in the cerebellum and hippocampus of the CNS early during development (Minichiello, 1996) similar to what has been reported for in the NeuroD-null brain (Miyata et al., 1999).
Migration defect and disorganized fiber projection among the surviving vestibular sensory neurons
In addition to the simple loss of sensory neurons and accompanying fiber loss, we also observed misdirected migration of the remaining sensory neurons in NeuroD−/− mice, which was not previously reported for neurotrophin or Trk-null mutants. This finding suggests that NeuroD not only regulates survival, but also plays a role in placing neurons in proper topological positions. Migration defect is particularly strong for the vestibular ganglia that are mislocated inside the otic capsule. Such a phenotype is reminiscent of the defect found among the NeuroD-null pancreatic endocrine cells, which cluster near the ductal epithelium, rather than migrating and forming islets (Naya, 1997). From these phenotypes, we postulate that NeuroD controls some factors involved in cell migration. Such developmental defect, however, could also be due to incomplete differentiation resulting in arrested or defective migration.
In the vestibular organ, the remaining vestibular sensory neurons project poorly to the PVC, which is normally among the first vestibular sensory epithelia to be innervated around E11.5. The AVC, which is normally innervated at around the same time, is moderately innervated at E11.5, suggesting that the innervation defect is not due to a developmental delay. Rather, the defect may be due to the topological reorganization of the vestibular sensory ganglia, which are situated further away from the PVC in the NeuroD−/− mice. In addition to the poor innervation to the PVC, projection to nearly all the vestibular sensory epithelia is highly disorganized, indicating that NeuroD is involved in guiding fibers to correct targets during inner ear development. Owing to this highly irregular pattern of fiber projection, we suspect that the vestibular function is greatly disturbed, despite the presence of some surviving sensory neurons.
Hair cells are independent of afferent and efferent innervation
We also examined the involvement of NeuroD in inner ear hair cell development. Interestingly, a significant population of hair cells expresses NeuroD in all three semicircular canals, in the utricle and saccule of the vestibular system, and in the organ of Corti. Nevertheless, we can find normally differentiated hair cells in all the sensory epithelium under light microscopy. Electron microscopy of semicircular canal hair cells failed to reveal any phenotype at the ultra structural level, indicating that NeuroD is not required for hair cell formation.
However, we have noticed shortening of the cochlear sensory epithelium, possibly due to the absence of the mechanical stretching effect normally imposed by the radial fiber extension. We also find more X-gal-stained cells in the NeuroD−/− cochleae compared with the number in NeuroD+/− mice. Since the density of the hair cells in the cochleae does not seem to be different in NeuroD−/− mice that contain two copies of the lacZ gene, the increased level of X-gal staining may be due to a detection above a certain threshold level by X-gal staining. Indeed, X-gal staining of the sensory epithelia has always yielded a non-uniform level of staining among the hair cells, supporting this idea. Such differences in expression were also noted in a recent study visualizing the α9 acetylcholine receptor (Zuo et al., 1999).
In summary, we have identified NeuroD as a critical transcription factor involved in survival of differentiating inner ear sensory neurons. At the present time, the molecular and physiological basis of such neuronal death at an early developmental stage is unknown. However, our finding that the Trk genes fail to be activated suggests an intriguing possibility that Trk signaling, which is involved in the survival of mature neurons, may also play an important role for the survival of young differentiating neurons.
Acknowledgments
We thank J. Eby and C. Dufton for valuable comments on our manuscript, and K. Jones for providing the probes for NT3 and BDNF. This work was supported by the NIH (NIDCD: 2P01 DC00215 to B. F.), (NINDS: R01NS35118 to J. E. L.) and the Juvenile Diabetes Foundation International (to J. E. L.). L. F. R. is an investigator of the Howard Hughes Medical Institute.
References
- Barth DS, Di S. The functional anatomy of auditory evoked potentials in rat neocortex. Brain Res. 1991;565:109–115. doi: 10.1016/0006-8993(91)91741-i. [DOI] [PubMed] [Google Scholar]
- Barth DS, Kithas J, Di S. Anatomic organization of evoked potentials in rat parietotemporal cortex: Somatosensory and auditory responses. J Neurophysiol. 1993;69:1837–1849. doi: 10.1152/jn.1993.69.6.1837. [DOI] [PubMed] [Google Scholar]
- Bermingham NA, Hassan BA, Pricem SD, Vollrath MA, Ben-Arie N, Eatock RA, Bellen HJ, Lysakowski A, Zoghbi HY. Math1: an essential gene for the generation of inner ear hair cells. Science. 1999;284:1837–1841. doi: 10.1126/science.284.5421.1837. [DOI] [PubMed] [Google Scholar]
- Bianchi LM, Conover JC, Fritzsch B, DeChiara T, Lindsay RM, Yancopoulos GD. Degeneration of vestibular neurons in late embryogenesis of both heterozygous and homozygous BDNF null mutant mice. Development. 1996;122:1965–1973. doi: 10.1242/dev.122.6.1965. [DOI] [PubMed] [Google Scholar]
- Cowan CA, Yokoyama N, Bianchi LM, Henkemeyer M, Fritzsch B. EphB2 guides axons at the midline and is necessary for normal vestibular function. Neuron. 2000;26:417–430. doi: 10.1016/s0896-6273(00)81174-5. [DOI] [PubMed] [Google Scholar]
- Di S, Barth DS. Topographic analysis of field potentials in rat vibrissa/barrel cortex. Brain Res. 1991;546:106–112. doi: 10.1016/0006-8993(91)91164-v. [DOI] [PubMed] [Google Scholar]
- Di S, Barth DS. The functional anatomy of middle latency auditory evoked potentials: Thalamocortical connections. J Neurophysiol. 1992;68:425–431. doi: 10.1152/jn.1992.68.2.425. [DOI] [PubMed] [Google Scholar]
- Eatock RA, Rüsch A. Developmental changes in the physiology of hair cells. Semin Cell Dev Biol. 1997;8:265–275. doi: 10.1006/scdb.1997.0142. [DOI] [PubMed] [Google Scholar]
- Erkman L, McEvilly RJ, Luo L, Ryan AK, Hooshmand F, O’Connell SM, Keithley EM, Rapaport DH, Ryan AF, Rosenfeld MG. Role of transcription factors Brn-3.1 and Brn-3.2 in auditory and visual system development. Nature. 1996;381:603–606. doi: 10.1038/381603a0. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Van De Water T, Loring J, Jaenisch R. Complementary roles of BDNF and NT-3 in vestibular and auditory development. Neuron. 1995;14:1153–1164. doi: 10.1016/0896-6273(95)90263-5. [DOI] [PubMed] [Google Scholar]
- Fode C, Gradwohl G, Morin X, Dierich A, LeMeur M, Goridis C, Guillemot F. The bHLH protein NEUROGENIN 2 is a determination factor for epibranchial placode-derived sensory neurons. Neuron. 1998;20:483–494. doi: 10.1016/s0896-6273(00)80989-7. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Nichols DH. DiI reveals a prenatal arrival of efferents at the differentiating otocyst of mice. Hear Res. 1993;65:51–60. doi: 10.1016/0378-5955(93)90200-k. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Farinas I, Reichardt LF. Lack of neurotrophin 3 causes losses of both classes of spiral ganglion neurons in the cochlea in a region-specific fashion. J Neurosci. 1997;17:6213–6225. doi: 10.1523/JNEUROSCI.17-16-06213.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzsch B, Barbacid M, Silos-Santiago I. The combined effects of trkB and trkC mutations on the innervation of the inner ear. Int J Dev Neurosci. 1998;16:493–505. doi: 10.1016/s0736-5748(98)00043-4. [DOI] [PubMed] [Google Scholar]
- Fritzsch B, Pirvola U, Ylikoski J. Making and breaking the innervation of the ear: neurotrophic support during ear development and its clinical implications. Cell Tissue Res. 1999;295:369–382. doi: 10.1007/s004410051244. [DOI] [PubMed] [Google Scholar]
- Hohn A, Leibrock J, Bailey K, Barde YA. Identification and characterization of a novel member of the growth factor/brain-derived neurotrophic factor family. Nature. 1990;344:339–341. doi: 10.1038/344339a0. [DOI] [PubMed] [Google Scholar]
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990;9:2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Wilkinson GA, Farinas I, Backus C, Zang K, Wong SL, Reichardt LF. Expression of Trk receptors in the developing mouse trigeminal ganglion: in vivo evidence for NT-3 activation of TrkA and TrkB in addition to TrkC. Development. 1999;126:2191–2203. doi: 10.1242/dev.126.10.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- Ma Q, Kintner C, Anderson DJ. Identification of neurogenin, a vertebrate neuronal determination gene. Cell. 1996;87:43–52. doi: 10.1016/s0092-8674(00)81321-5. [DOI] [PubMed] [Google Scholar]
- Ma Q, Chen Z, del Barco Barrantes I, de la Pompa JL, Anderson DJ. Neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron. 1998;20:469–482. doi: 10.1016/s0896-6273(00)80988-5. [DOI] [PubMed] [Google Scholar]
- Ma Q, Anderson DJ, Fritzsch B. Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. J Assoc Res Otolaryngol. 2000;1:129–143. doi: 10.1007/s101620010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick MB, Tamimi RM, Snider L, Asakura A, Bergstrom D, Tapscott SJ. NeuroD2 and neuroD3: distinct expression patterns and transcriptional activation potentials within the neuroD gene family. Mol Cell Biol. 1996;16:5792–5800. doi: 10.1128/mcb.16.10.5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvilly RJ, Erkman L, Luo L, Sawchenko PE, Ryan AF, Rosenfeld MG. Requirement for Brn-3.0 in differentiation and survival of sensory and motor neurons. Nature. 1996;384:574–577. doi: 10.1038/384574a0. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Klein R. TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev. 1996;10:2849–2858. doi: 10.1101/gad.10.22.2849. [DOI] [PubMed] [Google Scholar]
- Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EC, Schinder AF, Dantzker JL, Marcus EA, Spitzer NC, Harris WA. Properties of ectopic neurons induced by Xenopus neurogenin1 misexpression. Mol Cell Neurosci. 1998;12:281–299. doi: 10.1006/mcne.1998.0712. [DOI] [PubMed] [Google Scholar]
- Perron M, Opdecamp K, Butler K, Harris WA, Bellefroid EJ. X-ngnr-1 and Xath3 promote ectopic expression of sensory neuron markers in the neurula ectoderm and have distinct inducing properties in the retina. Proc Natl Acad Sci USA. 1999;96:14996–5001. doi: 10.1073/pnas.96.26.14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben RJ. Development of the inner ear of the mouse: a radioautograhic study of terminal mitosis. Acta Otolaryngol. 1967;220(Suppl):1–44. [PubMed] [Google Scholar]
- Takebayashi K, Takahashi S, Yokota C, Tsuda H, Nakanishi S, Asashima M, Kageyama R. Conversion of ectoderm into a neural fate by ATH-3, a vertebrate basic helix-loop-helix gene homologous to Drosophila proneural gene atonal. EMBO J. 1997;16:384–395. doi: 10.1093/emboj/16.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MN. Histochemical demonstration of sensory maps in the rat and mouse cerebral cortex. Brain Res. 1987;418:178–182. doi: 10.1016/0006-8993(87)90977-2. [DOI] [PubMed] [Google Scholar]
- Zuo J, Treadaway J, Buckner TW, Fritzsch B. Visualization of α9 acetylcholine receptor expression in hair cells of transgenic mice containing a modified bacterial artificial chromosome. Proc Natl Acad Sci USA. 1999;96:14100–14105. doi: 10.1073/pnas.96.24.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]