

SUMMARY
Animals lacking neurotrophin-3 (NT-3) are born with deficits in almost all sensory ganglia. Among these, the trigeminal ganglion is missing 70% of the normal number of neurons, a deficit which develops during the major period of neurogenesis between embryonic stages (E) 10.5 and E13.5. In order to identify the mechanisms for this deficit, we used antisera specific for TrkA, TrkB, and TrkC to characterize and compare the expression patterns of each Trk receptor in trigeminal ganglia of wild type and NT-3 mutants between E10.5 and E15.5. Strikingly, TrkA, TrkB, and TrkC proteins appear to be exclusively associated with neurons, not precursors. While some neurons show limited co-expression of Trk receptors at E11.5, by E13.5 each neuron expresses only one Trk receptor. Neuronal birth dating and cell counts show that in wild-type animals all TrkB- and TrkC-expressing neurons are generated before E11.5, while the majority of TrkA-expressing neurons are generated between E11.5 and E13.5. In mice lacking NT-3, the initial formation of the ganglion, as assessed at E10.5, is similar to that in wild-type animals. At E11.5, however, the number of TrkC-expressing neurons is dramatically reduced and the number of TrkC-immunopositive apoptotic profiles is markedly elevated. By E13.5, TrkC-expressing neurons are virtually eliminated. At E11.5, compared to wild type, the number of TrkB-expressing neurons is also reduced and the number of TrkB immunoreactive apoptotic profiles is increased. TrkA neurons are also reduced in the NT-3 mutants, but the major deficit develops between E12.5 and E13.5 when elevated numbers of TrkA-immunoreactive apoptotic profiles are detected. Normal numbers of TrkA-and TrkB-expressing neurons are seen in a TrkC-deficient mutant. Therefore, our data provide evidence that NT-3 supports the survival of TrkA-, TrkB- and TrkC-expressing neurons in the trigeminal ganglion by activating directly each of these receptors in vivo.
Keywords: Neurotrophin-3, Knockout mouse, Trk receptors, Trigeminal ganglion, Neurogenesis
INTRODUCTION
The neurotrophins are a family of related polypeptides that regulate neuronal survival, differentiation and function. They promote neuronal survival primarily by binding to members of the Trk family of receptor tyrosine kinases. Individual neurotrophins interact specifically with members of the Trk receptor family: NGF activates TrkA; BDNF and NT-4/5 activate TrkB; and NT-3 activates primarily TrkC, but at higher concentrations can also activate other Trk receptors, in some, but not all cells (reviewed by Reichardt and Fariñas, 1997). Each neurotrophin additionally binds to another receptor, called p75NTR, a member of the TNF receptor family. While p75NTR can promote Trk-dependent survival of some developing neurons (see e.g. Davies et al., 1993; Stucky and Koltzenburg, 1997), neurotrophin-mediated signaling through p75NTR can also induce apoptosis of neurons when Trk receptor signaling is absent (Van de Zee et al., 1996; Bamji et al., 1998; Davey and Davies, 1998).
Experiments analyzing mice with targeted mutations have provided clear evidence for the importance of neurotrophins and Trk receptors in the developing nervous system. Elimination of any of the neurotrophins or Trk receptors results in mice with specific nervous system deficiencies (reviewed by Reichardt and Fariñas, 1997). Of particular interest, the trigeminal ganglia in NT-3 mutants lack about 70% of the wild-type number of neurons at birth (Fariñas et al., 1994; Wilkinson et al., 1996; ElShamy and Ernfors, 1996). This deficit is much more severe than the 6 to 22% deficit observed in the TrkC mutant (Silos-Santiago, personal communication; Piñon et al., 1996; Tessarollo et al., 1997), suggesting that NT-3 may have an effect on neurons expressing receptors other than TrkC. Consistent with this possibility, NT-3 has been shown to activate TrkA and TrkB when these receptors are expressed in fibroblasts and in PC12 cells (Ip et al., 1993). However, the concentration of NT-3 required to bind and activate TrkA or TrkB appears to be 10- to 100-fold higher than that required for activation by their cognate ligands, NGF and BDNF, respectively (Ip et al., 1993; Shelton et al., 1995). Furthermore, NT-3 has also been shown to promote survive of trigeminal, nodose, and sympathetic neurons derived from mice lacking TrkC, albeit at very high concentrations (Davies et al., 1995).
In the trigeminal ganglia of wild-type mice, neurogenesis occurs between E9.5 and E13.5 and this interval is associated with substantial changes in the expression of TrkA, TrkB and TrkC in vivo and in the responses of neurons to different neurotrophins in vitro. In addition, mRNA analyses indicate that expression of TrkB kinase isoforms decreases after E12.5, whereas expression of truncated isoforms increases progressively from E10.5 to E15.5 (Ninkina et al., 1996). In situ studies have shown that TrkB and TrkC mRNAs are prominently expressed in the rat trigeminal ganglion at E12, but are restricted to comparatively few cells by E16 and E18 (Arumae et al., 1993; Ernfors et al., 1992). In contrast, TrkA mRNA is expressed by increasing proportions of trigeminal neurons over the same period (Arumae et al., 1993; Wyatt and Davies, 1993). Interestingly, cultured trigeminal neurons have shown dramatic changes in neurotrophin responsiveness over this interval (Buchman and Davies, 1993; Paul and Davies, 1995). While BDNF or NT-3 promotes survival of most E11.5 trigeminal neurons, this responsiveness progressively declines and these neurotrophins promote survival of only a small proportion of neurons cultured from later stages. Over this same interval, NGF promotes survival of increasing numbers of trigeminal neurons cultured from E12.5 onwards. Taken together, the cell culture and in situ studies have suggested that trigeminal neurons switch Trk receptor expression and neurotrophin responsiveness during their maturation (Davies, 1997).
Consistent with the substantial changes in the expression of Trk receptors, trigeminal ganglia lacking individual Trk receptors show elevations in apoptotic cell death that peak at different embryonic stages, depending on which receptor is absent (Piñon et al., 1996). For example, a massive wave of cell death appears in the trigeminal ganglion of TrkA mutants at E13.5 and E14.5, whereas cell death of a smaller magnitude occurs in ganglia of TrkB mutants at E11.5 and E12.5 (Piñon et al., 1996). As expected decreases in neuronal number are seen immediately after these waves of apoptosis. The timing of neuronal loss in TrkC mutants, however, is less clear. Although it has been reported that a small wave of cell death occurs at E11.5 and E12.5, the decrease in neuron number reportedly does not develop until E17.5 (Piñon et al., 1996).
In our previous work, we have determined that the deficit in the trigeminal ganglion of NT-3 mutants occurs between E10.5 and E13.5 and is due to apoptotic death of neurons, not precursors (Wilkinson et al., 1996). However, there has been a conflicting report indicating precursors in the trigeminal ganglion are affected by the absence of NT-3 (ElShamy and Ernfors, 1996). To determine the expression patterns of Trk receptors in the sensory ganglia and to identify which subpopulations of cells in the trigeminal ganglion are affected in the NT-3 mutants, we prepared antisera specific for the extracellular domains of rat TrkB and rat TrkC, which are expected to recognize both kinase-containing and truncated isoforms of these receptors (reviewed by Barbacid, 1994; Ninkina et al., 1997). Together with a previously generated antiserum to rat TrkA (RTA; Clary et al., 1994), these were used to examine expression of TrkA, TrkB, and TrkC in wild-type and NT-3 mutant embryos. Our results show that, in the trigeminal ganglion, the expression of each receptor is confined to neurons, not precursors, at all stages examined. Furthermore, neuronal birth-dating experiments indicate that trigeminal neurons are generated in two waves, with TrkB- and TrkC-expressing neurons generated exclusively in the first wave and the majority of TrkA neurons generated in the second. Lack of NT-3 leads to virtual elimination of TrkC-expressing neurons and partial deficiencies in neurons expressing TrkA and TrkB. As these deficits in TrkA- and TrkB-expressing cells are not observed in TrkC mutants, NT-3 appears to promote survival of these neurons by directly activating each receptor.
MATERIALS AND METHODS
Purification of rTrkB-ex and rTrkC-ex from COS cell supernatants
cDNA encoding the extracellular domain of rat TrkB (rTrkB-ex) or the extracellular domain of rat TrkC (rTrkC-ex) were obtained by PCR and amplified PCR products were cloned into pCR3 using the TA cloning kit (Invitrogen, Carlsbad, CA) for transfection in COS-7 cells. Sixty μl lipofectamine (Life Technologies, Gaithersburg, MD) and 15 μg DNA were added to COS-7 cells in 150 mm tissue culture dish. Cells were then transferred into expression medium (DME, 1% Nutridoma-HU [Boehringer Mannheim, Indianapolis, IN], penicillin/streptomycin). Supernatants were harvested at 48 and 96 hours, filter sterilized, and diluted with the following: PMSF (100 μg/ml), NaCl (400 mM), Tris-HC1 (pH 8.0, 20 mM), leupeptin (10 μg/ml) and pepstatin (5 μg/ml). Supernatants containing rTrkB-ex or rTrkC-ex were adsorbed using a wheat germ lectin column. After extensively washing with 20 mM Tris-HCl (pH 8.0) and 300 mM NaCl, lectin-binding proteins were eluted using 500 mM N-acetyl glucosamine in 200 mM NaCl, 20 mM Tris-HCl (pH 8.0), PMSF (100 μg/ml), sodium imidazole (10 mM, pH 8.0), β-mercaptoethanol (10 mM), glycerol (30%), and Tween-20 (0.1%). The eluates were then batch-adsorbed to NTA Nickel beads (Qiagen, Chatsworth, CA) for 3 hours at 4°C, and washed in buffer (10 mM Tris-HCl, pH 8.0, 300 mM NaCl, 0.1% Tween-20). Proteins were eluted using 10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 500 mM imidazole, concentrated on a Centricon-30 (Amicon Inc., Beverly, MA), and dialyzed extensively against TBS. A typical yield was 1.5–2 mg protein per 40 plates. Purified rTrkB-ex was used to inoculate two rabbits and two chickens, and purified rTrkC-ex was used to inoculate a goat.
Affinity purification of antisera to rTrkB-ex and rTrkC-ex
Purified rTrkB-ex was obtained using a Picchia pastoralis expression system (pPIC9K expression vector; Invitrogen, Carlsbad, CA) and recombinant rTrkC-ex protein was prepared using the pET bacterial expression system (Novagen, Madison, WI). Purified proteins were bound to cyanogen bromide-Sepharose CNBR-4MB beads (Pharmacia, Alameda, CA). For purification, antisera to rTrkB-ex and rTrkC-ex were incubated with appropriate affinity matrices using batch incubation. Beads were washed twice in 10 mM Tris-HCl, pH 7.5, 300 mM NaCl, once with TBS, and were then loaded onto a mini-column. Immunoglobulin was eluted using 150 mM glycine, pH 2.3, and was immediately neutralized by adding 1 M Tris-HCl, pH 8.0, to a final concentration of 100 mM followed by dialysis in TBS.
Western blot analysis of antisera specificity
COS-7 cells were transiently transfected with pCR3 containing full-length rat TrkA, TrkB or TrkC (Invitrogen, Carlsbad, CA) and each lysate was analyzed by western blot using affinity purified RTB (1 μg/ml), affinity purified RTC (1 μg/ml), or RTA IgG (1 μg/ml).
Immunohistochemistry
Wild-type and NT-3 mutant mice on a C57B/6 background (Fariñas et al., 1994) were obtained from our colony. Embryos were staged using the criteria of Theiler (1989), and placed into Carnoy’s fixative (10% glacial acetic acid: 30% chloroform: 60% ethanol). Embryos were dehydrated, embedded in paraffin, sectioned at 7 μm on a rotary microtome, and mounted in series. For immunohistochemistry, sections were rehydrated and endogenous peroxidases were quenched in TBS containing 10% methanol and 3% hydrogen peroxide. Sections were rinsed in TBS then blocked with TBS containing 0.4% Triton X-100, 1% glycine, 3% BSA (Fraction V) and 10% normal serum from the host species of the secondary antibody to be used (see below). Primary antibodies were added at the following concentrations or dilutions in blocking solution: rabbit anti-neurofilament medium molecular weight subunit (NF-M)(Chemicon, Temeluca, CA; 1:1000); rabbit anti-TrkA IgG (Clary et al., 1994, 1 μg/ml); affinity purified rabbit anti-TrkB (5 μg/ml); chicken anti-TrkB IgY, 1:100; affinity purified goat anti-TrkC (5 μg/ml); mouse anti-NF-M (Sigma, St. Louis, MO; 1:100); mouse anti-BrdU (Novocastra, Newcastle upon Tyne; 1:100). For analyses of BrdU incorporation, pregnant dams were injected intraperitoneally with 5-bromo-2-deoxyuridine (BrdU; Sigma, St. Louis, MO)(50 mg/kg body weight) 2 hours before being killed. Before staining with anti-BrdU antibody, sections were treated according to the manufacturer’s instructions. Detection of RTA immunoreactivity required microwave treatment prior to primary antibody application. The immunostaining patterns identified by chicken anti-TrkB IgY are very similar to those revealed by affinity purified rabbit RTB.
For detection of single antigens, biotinylated goat anti-rabbit IgG or rabbit anti-goat IgG (Vector Laboratories Inc., Burlingame, CA; 1:300) was used after which sections were developed with biotin-avidin-biotin peroxidase reagents from the Vectastain detection kit (Vector Laboratories Inc., Burlingame, CA). To assess co-expression of more than one antigen, TrkC immunoreactivity was detected using Cy3-coupled donkey anti-goat antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA; 1:100) except for the following experiments with E11.5 embryos: TrkC with TrkA, and TrkC with BrdU. These experiments were performed using the more sensitive Tyramide System Amplification kit (NEN, Boston, MA). For these experiments, TrkC immunoreactivity was detected using biotinylated horse anti-goat IgG (Vector Laboratories Inc., Burlingame, CA; 1:200) followed by Vectastain AB reagent, biotinyl tyramide, and FITC streptavidin from the TSA kit. To analyze co-expression of TrkA and TrkB, TrkB immunoreactivity was developed using biotinylated anti-chicken IgG, followed by Vectastatin AB reagent and FITC streptavidin.
Rabbit NF-M and TrkB immunoreactivity were detected using donkey anti-rabbit IgG (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, 1:300) coupled to either FITC or Cy3. Mouse NF-M immunoreactivity was detected using goat anti-mouse IgG (Cappel, Durham, NC; 1:100) coupled to either FITC or Texas Red. Fluorescent samples were photographed on a Biorad confocal microscope using a Zeiss Plan Neofluor 100× objective (n.a.=1.30), with an aperture of 3–4 mm and a zoom factor of 1–2. In double labeling studies with RTB and neurofilament or RTC and neurofilament, Z-series covering 5–8 μm were collected at Z-spacing of 0.68 μm. TrkA -neurofilament double label images were collected using a Zeiss Plan Neofluor 63× objective (n.a.=1.25).
Neuronal birth-dating experiments
To determine the time intervals during which neurons expressing each Trk receptor are generated, we designed an experimental paradigm in which pregnant dams were injected with a single dose of BrdU (50 mg/kg) at E9.5, E10.5, E11.5, or E12.5, and the embryos were collected at later stages for immunohistochemistry (see schematic diagram in Fig. 6A). By immunolabeling cells in trigeminal ganglia with antibodies to BrdU and each of the Trk receptors, it is possible to determine if neurons generated at early stages later express particular Trk receptors. For detection of BrdU and each of the Trk receptor antigen in neuronal birth-dating experiments, a sequential DAB-alkaline phosphatase method was used (Vector Laboratories Inc., Burlingame, CA). Data shown are representatives of at least two different embryos examined, and are presented as the percentage of neurons expressing a particular Trk receptor that are colabeled with antibodies to BrdU. The entire ganglion was counted in every instance. For data indicating injection and collection on the same date, the embryos were collected 10 hours after BrdU injection.
Fig. 6.
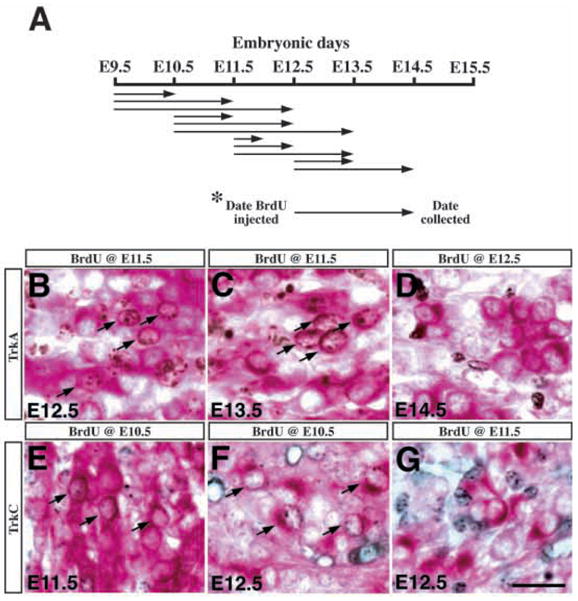
Neurons expressing different Trk receptors are generated at distinct time intervals during embryogenesis. (A) A schematic diagram indicating the experimental paradigm for birth-dating Trk-expressing neurons. The arrows designate the embryonic days when BrdU was injected intraperitoneally into the pregnant dams and the days when the embryos were collected for immunohistochemistry. (B–D) When pulse labeled with BrdU at E11.5, a significant number of TrkA-expressing neurons (red cytoplasmic staining) at E12.5 and E13.5 were immunoreactive for BrdU (brown nuclear staining) (arrows in B,C). However, when embryos received BrdU injections at E12.5 and were collected at either E13.5 or E14.5, there was no colabeling of BrdU and TrkA in the trigeminal neurons (D) (notice the complete separation of nuclear brown staining and red cytoplasmic staining). (E–G) Embryos labeled with BrdU at E10.5 showed colabeling of BrdU (black nuclear staining) and TrkC (red cytoplasmic staining) at E11.5 or E12.5 in the trigeminal neurons (arrows in E,F). However, in embryos injected with BrdU at E11.5 or E12.5, there was no colabeling of BrdU and TrkC in the trigeminal neurons (G). Scale bar, 20 μm.
RESULTS
Specificity of Trk receptor antisera
In order to characterize Trk receptor expression patterns in sensory ganglia, we developed antisera that specifically recognize the extracellular domains of rat TrkB or TrkC to supplement a similar serum specific for TrkA, developed previously in this laboratory. Results in Fig. 1 document the specificity of the antibodies to each of the Trk receptors prepared using extracellular domains of rat TrkA (Clary et al., 1994), TrkB, or TrkC as immunogens (see Materials and Methods). Hereafter, we will refer to rabbit anti-rat TrkA-ex IgG as anti-TrkA (RTA; Clary et al., 1994), affinity purified rabbit anti-TrkB-ex as anti-TrkB (RTB), and affinity purified goat anti-rat TrkC-ex as anti-TrkC (RTC). In Fig. 1A, western blots of lysates of COS cells transiently transfected with full-length rat TrkA, TrkB and TrkC, or of untransfected COS cells, were probed with each antibody. Each recognized protein bands of specific molecular weights which correspond to previously reported sizes of the different Trk receptors (Martin-Zanca et al., 1990; Klein et al., 1990; Tsoulfas et al., 1993). Of particular importance, RTA (left panel) recognized protein bands only in lysates of TrkA transfected cells; RTB (middle panel) recognized protein bands only in lysates of TrkB transfected cells; and RTC (right panel) recognized protein bands only in lysates of TrkC transfected cells. Thus each antibody appears to recognize only the Trk receptor corresponding to its antigen and does not cross-react with other proteins.
Fig. 1.
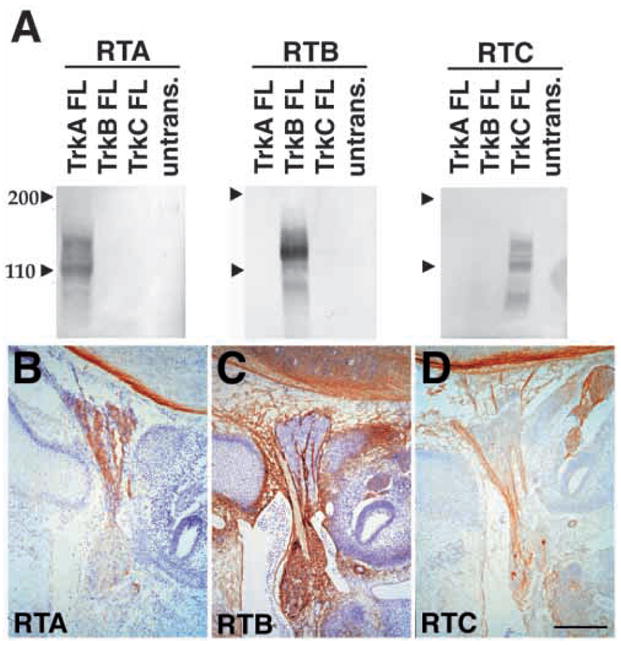
Specificity of Trk receptor antisera. (A) Western blot analysis of lysates of COS cells transiently transfected with full length rat TrkA, TrkB or TrkC, or of untransfected COS cells. Lysates were subjected to western blot analysis with RTA IgG (1 μg/ml; left panel), affinity purified RTB (1 μg/ml; center panel), or affinity purified RTC (1 μg/ml; right panel). Each antiserum recognizes protein bands of specific molecular weights in lysates of COS cells transfected with corresponding Trk receptor cDNA.
(B–D) Immunohistochemical staining of E13.5 IX-X ganglion complex. The intracranial superior jugular ganglion is at the top in these images and the extracranial nodose-petrosal complex is toward the bottom. RTA (B) identifies neurons in the superior jugular ganglion, but not the nodose-petrosal complex. RTB (C) recognizes neurons in the nodose-petrosal complex but not in the superior jugular ganglion and RTC (D) recognizes a small number of neurons in the nodose-petrosal complex. Scale bars (B–D) 200 μm.
The specificity of each Trk receptor antibody is further demonstrated in staining patterns of E13.5 cranial ganglion complex IX-X, which is composed of the intracranial superior jugular ganglion (toward the top of these images) and the extracranial nodose-petrosal complex (toward the bottom) (Fig. 1B–D). RTA recognizes a majority of neurons within the superior jugular ganglion, but does not recognize cells of the nodose-petrosal complex, similar to in situ analyses of TrkA mRNA distribution (Fig. 1B; Martin-Zanca et al., 1990). RTB recognizes a majority of cells within the nodose-petrosal complex, but not cells within the superior jugular ganglion (Fig. 1C); in contrast, RTC recognizes a minor population within the nodose-petrosal ganglion, and does not bind cell bodies within the superior jugular ganglion (Fig. 1D). These findings are consistent with in situ studies of the distributions of Trk mRNAs in these ganglia (Ernfors et al., 1992).
In E14.5 lumbar dorsal root ganglion (DRG), RTA recognizes numerous neurons with small cell bodies, whereas RTB and RTC recognize less abundant populations of neurons with larger cell bodies (Fariñas et al., 1998; data not shown). RTB additionally recognizes the mesenchyme surrounding the DRGs, which has been reported to express an isoform of TrkB lacking a functional kinase domain (reviewed by Barbacid, 1994). These results are consistent with in situ studies on the distributions of Trk receptor mRNAs in developing DRGs and surrounding tissues (Martin-Zanca et al., 1990; Mu et al., 1993; Klein et al., 1990; Tessarollo et al., 1993).
Trk receptor proteins are expressed exclusively in neurons, not in proliferating precursors
We examined which cell populations express TrkA, TrkB and TrkC proteins in the developing trigeminal ganglion, beginning at E10.5, the first time at which the trigeminal ganglion is well-coalesced. Using double labeling experiments, we compared the distributions of TrkA, TrkB, and TrkC proteins with those of neurofilament-medium weight subunit (NF-M), a neuronal marker, or of cells able to incorporate bromo-deoxyuridine (BrdU), a marker for proliferating precursor cells.
Fig. 2 shows confocal images of E11.5-E13.5 wild-type trigeminal ganglia double labeled with RTA, RTB or RTC plus anti-neurofilament or anti-BrdU antibody. Results of these colabeling experiments are quantitated in Table 1. At E11.5, TrkB and TrkC (red) were always associated with neurofilament-positive profiles (green; Fig. 2A,C). Although a few cells appeared to express TrkC or TrkB, but not neurofilament (Fig. 2A,C; arrowheads), these cells invariably proved to be neurofilament-positive in other confocal planes (arrowheads; inset). The integral membrane Trk receptors and neurofilaments are expected to have distinct distributions, which explains why the antigens are not superimposed in the same confocal planes. Furthermore, at E11.5, the nuclei of TrkC or TrkB immunopositive cells (red) did not incorporate detectable amounts of BrdU, a marker for proliferating precursors (green; Fig. 2B,D; Table 1). Finally, at E13.5 TrkC and TrkB immunoreactive cells (red) were always associated with neurofilament immunoreactivity (green; Fig. 2F). We conclude that during stages E11.5-E13.5, TrkC and TrkB are expressed at significant levels exclusively in trigeminal neurons; detectable amounts of these proteins are not present in proliferating precursors.
Fig. 2.
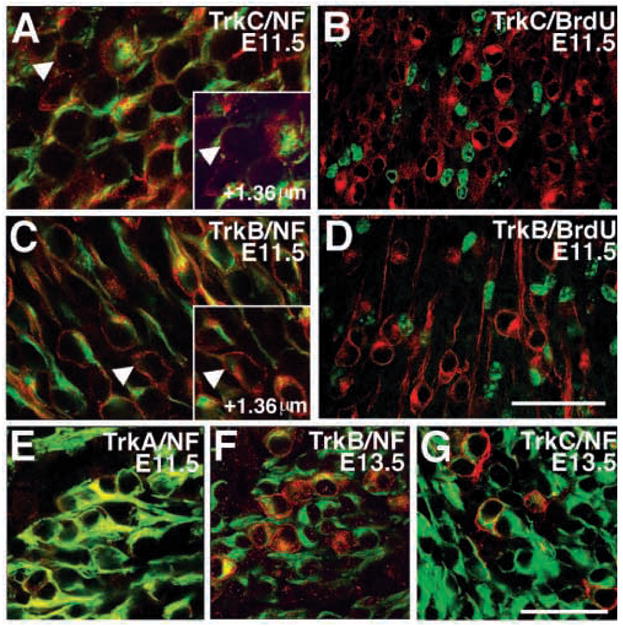
Trk receptor proteins are expressed exclusively in neurons, not precursors. (A,C) Confocal images showing (A) RTC (red) and neurofilament (green) and (C) RTB (red) and neurofilament (green) colocalization in E11.5 trigeminal ganglion. Cells that are immunoreactive for RTC or RTB, but not neurofilament (arrowheads), in some confocal plane almost always show colocalization with neurofilament in another confocal planes (inset) (B,D). Confocal images showing RTC or RTB (both in red) and BrdU (green) immunoreactivity in E11.5 trigeminal ganglion. The nuclei of (B) RTC- and (D) RTB-immunopositive cells do not incorporate BrdU. (E) Merged confocal image showing RTA and NF- 160 colocalization (yellow) in E11.5 trigeminal ganglion. (FG) Confocal images of E13.5 trigeminal ganglion stained with neurofilament (green) and RTC (F) or RTB (G) (red for both). RTB and RTC immunoreactivity is confined to neurons. Scale bars (A,C) 50 μm; (E) 50 μm; (B,D,F) and (G) is 100 μm.
Table 1.
Trk receptor expression in neurons and in precursors
| Embryonic stage | Antigens Green/Red (n) | Green only | Double | Red only |
|---|---|---|---|---|
| E11.5 | NF/TrkA (6) | 182 | 365 | 0 |
| NF/TrkB (6) | 17 | 146 | 0 | |
| BrdU/TrkB (5) | 86 | 0 | 106 | |
| NF/TrkC (2) | 126 | 156 | 0 | |
| BrdU/TrkC (5) | 53 | 0 | 77 | |
| E12.5 | NF/TrkA (4) | 83 | 261 | 0 |
| E13.5 | NF/TrkB (1) | 68 | 11 | 0 |
| NF/TrkC (3) | 185 | 53 | 0 |
Experiments compare the distribution of antigen pairs using two separate fluorophores. Results of double labeling experiments are reported as the number of profiles scored as immunopositive for the green label only (Green only); immunopositive for both labels (Double); or immunopositive for the red label only (Red Only). Total numbers from at least 3 different confocal fields (n) from two different experiments are listed. The fields selected for analysis were chosen to contain many profiles immunoreactive for each fluorophore. Thus the total counts may not reflect the true abundance of these antigens throughout the ganglion. Therefore, TrkB which is not randomly distributed throughout the ganglia is over-represented as a percentage of NF-positive profiles. Abbreviations used: NF, neurofilament-M; BrdU, bromodeoxyuridine.
TrkA mRNA is largely confined to neuronal structures, indicating that this receptor is largely expressed by neurons (reviewed by Barbacid, 1994). However, some nonneuronal tissues also express TrkA (Barker et al., 1993; Shibayama et al., 1996). We therefore examined whether TrkA expression is confined to neurofilament-positive profiles in the developing trigeminal ganglion. Double labeling experiments with TrkA and NF-M at stages E11.5 and E12.5 confirmed that TrkA protein is confined to neurons at these stages (Fig. 2E; Table 1).
Quantitation of neurons expressing different Trk receptors
At E11.5 or E13.5, RTB recognizes a subpopulation of neurons within the trigeminal ganglion (Fig. 3A,C). When viewed at high magnification (Fig. 3E,F), these cells are seen to have morphological hallmarks of neurons, including an asymmetric cytoplasm and a labeled axonal process. The neuronal profiles in the E11.5-E13.5 trigeminal ganglion show a skewed spatial distribution, occurring with greater frequency toward the anterior of the trigeminal ganglion (to the right in Fig. 3A,C). RTB also intensely labels axons in the peripheral (not shown) and central branches of the E11.5-E13.5 trigeminal nerve (Fig. 3A,C). RTB also labels nonneuronal structures near the trigeminal ganglion such as the mesenchyme surrounding the ganglion and, at E13.5, blood vessels within the ganglion. In situs have detected mRNAs encoding TrkB isoforms lacking a kinase domain in numerous non-neuronal tissues in murine embryos, so these isoforms are almost certainly the antigen(s) detected within the mesenchyme and vasculature by RTB (reviewed by Barbacid, 1994).
Fig. 3.

Dynamic changes in the expression patterns of TrkB- and TrkC-expressing neurons between E11.5-E13.5. (A) TrkB-expressing neurons in E11.5 trigeminal ganglion appear to cluster toward the anterior of the ganglion (to the right in this image). (B) TrkC-expressing neurons in E11.5 trigeminal ganglion are distributed throughout the entire ganglion. (C) In E13.5 trigeminal ganglion, RTB recognizes a smaller proportion of neurons, vascular structures, and the surrounding mesenchyme. (D) Similar to RTB, RTC recognizes only a small fraction of neurons at E13.5. (E–H) High magnification of TrkB at E11.5 (E) and E13.5 (F); TrkC at E11.5 (G) and E13.5 (H). Scale bars: (A–D) 200 μm; (E–H) 50 μm.
Examinations of E11.5-E13.5 embryos stained with RTC show striking developmental changes in TrkC expression in the trigeminal ganglion (Fig. 3B,D,G,H). RTC immunoreactive cells are abundant at E11.5 (Fig. 3B,G). When viewed at higher magnification, these immunoreactive cells show the morphological hallmarks of neurons (Fig. 3G). At E13.5, the relative extent of labeling has decreased, such that RTC immunoreactive profiles represent a minor population of trigeminal neurons (Fig. 3D,H). The reduction in TrkC-expressing cells correlates with similar changes in mRNA expression observed over the same developmental interval (Arumae et al., 1993; Ernfors et al., 1992). At all stages examined, RTC intensely labels axons in the central and peripheral branches of the trigeminal nerve (Fig. 3B,D). Examinations of E11.5-E13.5 embryos stained with RTA showed expression of this antigen within large numbers of neurons (data not shown). Similar to the profiles described above for RTB and RTC, staining was not restricted to cell bodies but included axons in both the central and peripheral processes.
In order to provide quantitative analyses of the dynamic changes in neuronal populations expressing different Trk receptors, we determined the total number of trigeminal neurons and the number of neurons expressing each Trk receptor between E10.5 to E15.5. The total number of neurons (Fig. 4A black squares), counted as neurofilament-positive profiles, increased 7-fold between E10.5 and E12.5, peaked at E13.5, and declined slightly over the next 2 days (Table 2). Neurons expressing TrkB or TrkC appeared to be generated at earlier stages than those expressing TrkA. TrkC-expressing cells, corresponding to approximately 35% of neurons, were detected at E10.5, increased approximately 5-fold in number by E11.5 and declined thereafter (Fig. 4A, white circles; Table 2). TrkB-expressing cells were similarly present at E10.5, representing approximately 15% of neurons, increased approximately 8-fold in number by E11.5, and also decreased at later times (Fig. 4A, white diamonds). TrkA expression was very weak and diffuse at E10.5 and the number of cells expressing this antigen could not be quantified reliably at this stage. At E11.5, however, TrkA (Fig. 4A, white squares) was expressed by approximately 40% of neurons. TrkA-expressing neurons increased 2.5-fold in number by E12.5 when approximately 75% of neurons expressed this antigen. They declined in number significantly only after E13.5. Interestingly, the reductions in the numbers of neurons expressing TrkA, TrkB, or TrkC at E13.5-E15.5, E11.5-E13.5, and E11.5-E12.5, respectively, were associated with increases in the numbers of apoptotic profiles immunoreactive for each Trk receptor (Fig. 7G).
Fig. 4.
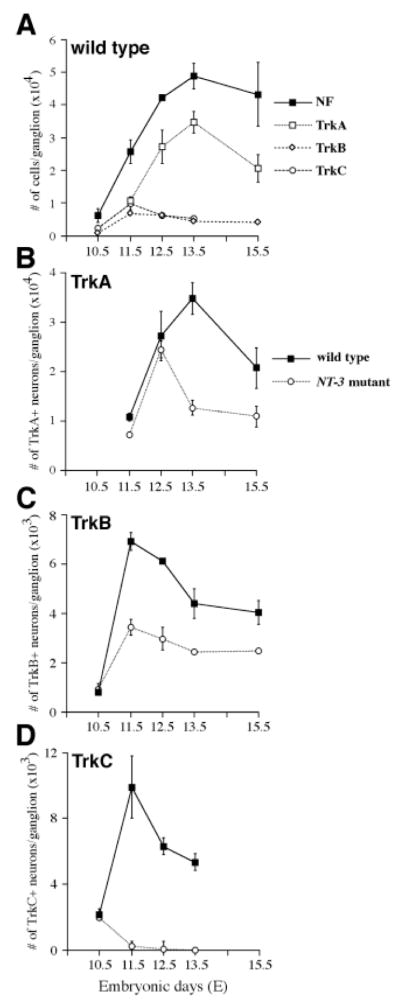
Quantitation of neurons and of Trk-immunoreactive cells in E10.5-E15.5 trigeminal ganglia of wild type and NT-3 mutants. (A) Total numbers of neurons (black squares) and of neurons expressing TrkA (white squares), TrkB (white diamonds), and TrkC (white circles) in wild-type trigeminal ganglion. (B) Numbers of TrkA-expressing neurons in trigeminal ganglion of wild type (black squares) and NT-3 mutants (white squares). (C) Numbers of TrkB-expressing neurons in trigeminal ganglion of wild type (black squares) and NT-3 mutants (white squares). (D) Numbers of TrkC-expressing neurons in trigeminal ganglion of wild type (black squares) and NT-3 mutants (white squares). Counts from at least three animals are shown as mean ± s.e.m. for each time point.
Table 2.
Numbers of neurons and cells immunoreactive for RTA, RTB, and RTC in the trigeminal ganglion of wild type and NT-3 mutant from E10.5 to E15.5
| Embryonic stages | Immumoreactant | Wild type | NT-3 mutant | % of wild type |
|---|---|---|---|---|
| E10.5 | Neurofilament‡ | 6093±2065 | 5375±1738+ | n.s. |
| RTB | 792±83 (3) | 911±215(3)+ | n.s. | |
| RTC | 2152±316 (4) | 1943±302 (3)+ | n.s. | |
| E11.5 | Neurofilament | 25545±3562 | 14743±5513* | 58 |
| RTA | 10733±831 (3) | 7117±542(3)** | 66 | |
| RTB | 6325±926 (3) | 3418±315 (3)*** | 54 | |
| RTC | 9893±1874 (3) | 210±207 (3)*** | 2 | |
| E12.5 | Neurofilament | 42170±960 | 31353±2181** | 74 |
| RTA | 27088±5067 (4) | 24357±1669 (3)+ | n.s. | |
| RTB | 6120±70 (3) | 2946±464 (3)*** | 48 | |
| RTC | 6273±510 (3) | 50±10 (2)*** | <1 | |
| E13.5 | Neurofilament | 48755±3943 | 15217±1023*** | 31 |
| RTA | 34667±3207 (3) | 12560±1555 (3)*** | 36 | |
| RTB | 4390±602 (3) | 2425±76 (3)** | 55 | |
| RTC | 5313±510 (3) | 0 | 0 | |
| E15.5 | Neurofilament | 43137±9794 | 13032±4614*** | 30 |
| RTA | 20630±4142 (4) | 10805±2075 (4)* | 52 | |
| RTB | 4022±469 (3) | 2465±75 (3)* | 61 | |
| Embryonic stages | Immunoreactant | wild type | TrkC mutant | % of wild type |
|
| ||||
| E13.5 | RTA | 36423±1147(3) | 32343±712 (3)+ | n.s. |
| RTB | 5300±623 (3) | 4400±265 (3)+ | n.s. | |
Numbers are from at least three embryos from each genotype and embryonic stage, and are presented as mean±s.e.m. The actual numbers of embryos examined are indicated in parentheses.
Neurofilament counts of wild type and mutants at E10.5, E11.5 and E13.5, neuron number (determined by Nissl stain) at E15.5 are reprinted from Wilkinson et al., 1996.
P < 0.05,
P < 0.01
P < 0.001,
not significant (n.s.) (Student’s two-tailed t-test).
Fig. 7.

Stage-dependent apoptotic cell death of TrkA-, TrkB- and TrkC-expressing neurons in NT-3 mutant embryos. (A,D) RTA immunoreactivity in wild-type (A) and mutant (D) embryos at E13.5. While most TrkA-expressing neurons in NT-3 mutants show normal morphology, there is increase in apoptotic figures associated with TrkA immunoreactivity (arrows). (B,E) RTB immunoreactivity in wild-type (B) and mutant (E) embryos. In E11.5 NT-3 mutant trigeminal ganglion, the number of TrkB-expressing neurons is reduced and the number of apoptosis associated with TrkB immunoreactivity (arrows) is increased. (C,F) RTC immunoreactivity in wild-type (C) and mutant (F) embryos. Numerous TrkC-immunoreactive apoptoses are identified in NT-3 mutant ganglion at E11.5. Scale bar, 20 μm. (G) Quantitation of Trk receptor immunoreactive apoptosis at different embryonic stages in wild type and NT-3 mutants. Note that the numbers for TrkA-, TrkB- and TrkC-immunoreactive apoptosis peaks at E13.5, E11.5 and E11.5, respectively. Counts are derived from at least two animals and are presented as mean ± s.e.m.
Limited co-expression of Trk receptors in early trigeminal neurons
The total numbers of cells in the trigeminal ganglion expressing TrkA, TrkB and TrkC slightly exceed those neurons at E11.5, indicating that some neurons may express more than one Trk receptor. To determine the extent of Trk receptor co-expression, we performed double labeling experiments using combinations of Trk receptor antibodies. Double labeling with RTC (red) and RTA (green) revealed some overlap between these antigens in the E11.5 trigeminal ganglion (Fig. 5A). Examining the approximately 9% of neurons co-expressing detectable amounts of both antigens, RTC immunoreactivity was often associated with profiles which appeared to be only weakly immunopositive for RTA (Fig. 5A). However, at E12.5 only 5% of neurons co-expressed detectable amounts of both antigens, and no such cells were observed a day later at E13.5 (Fig. 5D,G). Results using RTC (red) and RTB (green) demonstrated that at E11.5 about 10% of neurons co-expressed detectable amounts of both receptors (Fig. 5B). The number of these neurons decreased progressively and by E13.5 RTB and RTC labeled completely distinct sets of neurons (Fig. 5E,H; Table 3). Double labeling with RTB (green, chicken anti-TrkB Ab) and RTA (red) showed similar results at E13.5, in which each antibody labeled a completely different neuronal population (Fig. 5F,I). However, at E11.5 there appeared to be a higher percentage (24%) of neurons co-expressing both receptors (Fig. 5C; Table 3), although the TrkA immunoreactivity was much weaker than that of TrkB. Thus there appears to be limited co-expression of Trk receptors at E11.5 which is reduced at E12.5 and is not observed at later stages.
Fig. 5.
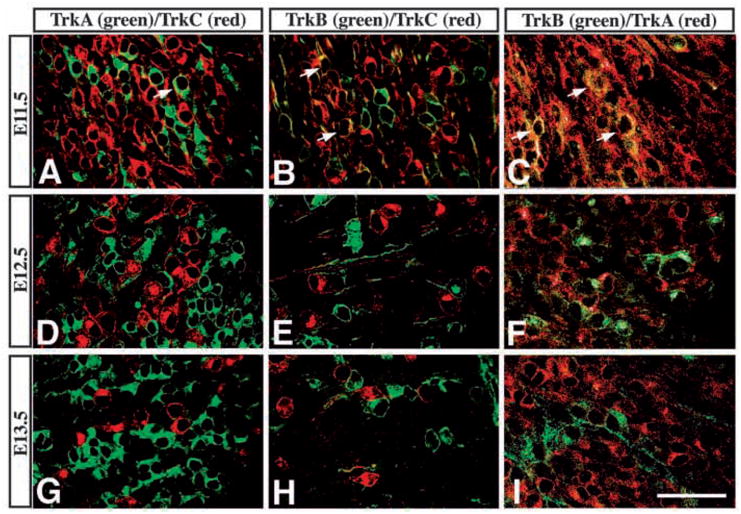
Limited coexpression of Trk receptor proteins in E11.5, but not in E13.5, trigeminal neurons. (A–C) Confocal images showing coexpression of (A) TrkA (green) and TrkC (red), (B) TrkB (green) and TrkC (red), and (C) TrkA (red) and TrkB (green) in E11.5 trigeminal ganglion. Cells coexpressing both antigens are present as yellow and indicated by arrows. (D–I) At E12.5 (D–F) and E13.5 (G–I), each Trk receptor antiserum labels separate cell populations. Scale bar, 100 μm.
Table 3.
Detection of neurons coexpressing more than one Trk receptors
| Embryonic stages | Antigens Green/Red (n) | Green only | Double | Red only | Percentage coexpression |
|---|---|---|---|---|---|
| E11.5 | TrkA/TrkC (6) | 93 | 24 | 162 | 9 |
| TrkB/TrkC (5) | 85 | 21 | 95 | 10 | |
| TrkB/TrkA (6) | 34 | 43 | 104 | 24 | |
| E12.5 | TrkA/TrkC (4) | 185 | 13 | 51 | 5 |
| TrkB/TrkC (3) | 47 | 7 | 58 | 6 | |
| TrkB/TrkA (6) | 60 | 4 | 122 | 2 | |
| E13.5 | TrkA/TrkC (4) | 311 | 0 | 41 | 0 |
| TrkB/TrkC (3) | 76 | 0 | 87 | 0 | |
| TrkB/TrkA (3) | 22 | 0 | 126 | 0 |
Experiments compare the distribution of antigen pairs using two separate fluorophores. Results of double labeling experiments are reported as number of profiles immunopositive for the green label only (Green only), immunopositive for both labels (Double), or immunopositive for the red label only (Red Only). Total numbers from at least 3 different confocal fields (number given in parenthesis) from two different experiments are listed. The percentage of cells coexpressing both antigens is reported as the percentage of double labeled profiles per total cells. The fields selected for analysis were chosen to contain many profiles immunoreactive for each fluorophore.
Neurons expressing different Trk receptors are generated at distinct embryonic stages
The observation that neurons expressing different Trk receptors reach maximal numbers at different times during embryogenesis strongly suggests that the generation of these different neuronal populations follows an orderly schedule (Fig. 4). Based on this model, precursors in the trigeminal ganglia differentiate into neurons expressing predominantly TrkB and TrkC before E11.5, while after E11.5 they generate almost exclusively TrkA-expressing neurons. To test this hypothesis, we injected pregnant dams with single doses of BrdU (50 mg/kg) at E9.5, E10.5, E11.5, or E12.5 and collected the embryos at later stages for immunohistochemistry (Fig. 6A). In the E11.5 DRG, this protocol labels approximately 50% of proliferating precursors (I. Fariñas, unpublished observations). By colabeling cells in trigeminal ganglia with antibodies to BrdU and to each of the Trk receptors, it is possible to determine when proliferating precursors generate the different types of neurons.
Consistent with the predicted model, we found that many descendants of precursors, labeled with BrdU at E9.5, differentiated into neurons expressing TrkB or TrkC by E10.5 and E11.5 (Table 4). Similarly, descendants of precursors in S-phase at E10.5 also differentiated into neurons expressing TrkB or TrkC at E11.5 or E12.5. Few E11.5 precursors and no E12.5 precursors, however, differentiated subsequently into neurons expressing TrkB or TrkC. Consistent with the observation that maximal numbers of neurons expressing either of these Trk receptors are present at E11.5 (Table 2), the BrdU-labeling data indicate that essentially all neurons expressing either of these Trk receptors are generated from precursors that have exited S-phase before E11.5. As very few neurons isolated the same day (10 hours) after a BrdU pulse express either Trk receptor, it appears that more time is required for precursors to differentiate sufficiently to express these receptors.
Table 4.
Birth-dating of trigeminal neurons expressing different Trk receptor
| TrkA |
TrkB |
TrkC |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Stage for BrdU injection | % BrdU+ |
% BrdU+ |
% BrdU+ |
|||||||||
| E11.5 | E12.5 | E13.5 | E14.5 | E10.5 | E11.5 | E12.5 | E13.5 | E10.5 | E11.5 | E12.5 | E13.5 | |
| E9.5 | 16.8 | 48 | 16 | 38 | 20 | |||||||
| E10.5 | 4.3 | 30.3 | 25.6 | 4.5 | 3.8 | 0.7 | 7.5 | 7.3 | ||||
| El 1.5 | 12.7 | 33.3 | 0.3 | 0.1 | 1.6 | 0.8 | 0 | 0 | ||||
| E12.5 | 1.5 | 0.6 | 0 | 0 | ||||||||
| Total no. | 10,733 | 27,088 | 34,667 | n.d. | 792 | 6,325 | 6,120 | 4,390 | 2,152 | 9,893 | 6,273 | 5,313 |
Pregnant mice were injected with a single dose of BrdU (50 mg/kg) at various embryonic stages (E9.5, E10.5, E11.5 and E12.5) and embryos were collected 1 to 3 days after the injection. For data indicating injection and collection on the same date, the embryos were collected 10 hours after BrdU injection. Data shown are means of at least two different embryos examined, and are presented as the percentage of neurons expressing a particular Trk receptor that were colabeled with antibodies to BrdU. The entire ganglion was counted in every instance. The mean number of neurons expressing each Trk receptor at each stage is indicated in the bottom row. These numbers are derived from Table 2. (n.d., not done.)
Similar to neurons expressing the other Trk receptors, TrkA-expressing neurons were generated from precursors labeled with BrdU at E9.5 and E10.5. In addition, precursors in S-phase at E11.5 also generated large numbers of TrkA-expressing neurons by 1 or 2 days later. Precursors in S-phase at E12.5 no longer generated neurons expressing TrkA. As summarized in Table 2, approximately 70% of TrkA expressing neurons accumulate between E11.5 and E13.5, so it is not surprising that they are generated from precursors still in S-phase at E11.5. While some neurons expressing TrkA are derived from precursors in S-phase 24 hours earlier, more extensive colabeling is seen 48 hours after a BrdU pulse (Table 4). This suggests that it requires approximately 24 hours for precursors in S-phase to differentiate into neurons that express TrkA, a somewhat longer time than appears to be required for similar precursors to generate TrkB or TrkC-expressing neurons. The major conclusion from these experiments is that neurons expressing TrkB or TrkC are generated from precursors at an earlier embryonic stage than the vast majority of neurons expressing TrkA.
Stage-dependent losses of TrkA, TrkB, and TrkC-expressing neurons in NT-3 mutant embryos
In a previous study (Wilkinson et al., 1996), we reported that the deficit in neuronal numbers in the trigeminal ganglia of animals lacking NT-3 emerges after E10.5 and appears to be complete by stage E13.5. In order to evaluate the effects of NT-3 deficiency on neurons expressing each Trk receptor, we compared TrkA, TrkB, and TrkC expression in trigeminal ganglia of wild-type and NT-3 mutant embryos between E11.5-E15.5 (Table 2; Fig. 7). Consistent with our previous observations, no deficits in either number or morphology of TrkB- or TrkC-expressing neurons were detected at E10.5. As described above, TrkA expression is very weak at this stage, but there was no obvious difference between control and mutant embryos (Table 2 and data not shown). At E11.5, however, absence of NT-3 affects all populations of neurons. The most dramatic consequence of the loss of NT-3 is abnormal apoptotic elimination of TrkC-expressing neurons (Fig. 7C,F,G). As quantified in Table 2, approximately 40% of neurons in the trigeminal ganglia of E11.5 wild-type embryos express TrkC. In E11.5 NT-3 mutants, however, the number of RTC immunopositive profiles is severely reduced to about 2% of the number in control embryos (Figs 4D, 7F). RTC immunoreactive apoptotic profiles are frequently seen throughout the mutant ganglion (arrow, Fig. 7F), and their numbers are at least 6-fold higher than those in wild-type embryos (Fig. 7G), indicating that these neurons are eliminated by apoptosis. At E12.5, an even smaller number of TrkC expressing neurons remains in the mutant ganglion (Table 2; Fig. 4D). At E13.5, TrkC neurons have been essentially eliminated.
Comparison of numbers of TrkB neurons in control and NT-3-deficient embryos shows that mutant ganglia contain only approximately 50% of the wild-type number of TrkB neurons at E11.5 (Table 2; Fig. 4C). Similar to the findings for TrkC neurons, we observed a 2- to 3-fold elevation in numbers of TrkB-immunoreactive apoptotic profiles in mutants at this stage (arrows, Fig. 7B,E,G). Elevated apoptosis did not persist at E12.5 or E13.5, and the remaining TrkB-expressing neurons showed apparently normal morphology (Fig. 7E). Compared to controls, the deficit in RTB immunopositive neurons in NT-3-deficient embryos actually becomes smaller at E12.5 and E13.5 (Table 2; Fig. 4C). The number of neurons expressing TrkB in mutant animals remain essentially constant while the number declines in wild-type embryos. Finally, at E13.5 and E15.5, mutant trigeminal ganglia contain between 50 and 60% the normal number of RTB immunopositive neurons (Table 2).
Quantification of TrkA-expressing cells in wild-type and NT-3 mutant embryos reveals slightly diminished numbers of TrkA-expressing neurons in mutants at E11.5 (66%). However, similar to wild-type embryos, the number of TrkA neurons in NT-3 mutants increases about 2-fold from E11.5 to E12.5, and did not appear to be significantly different from that of wild type at E12.5 (Table 2; Fig. 4B). The number of TrkA neurons in wild-type embryos reaches its maximum between E12.5 and E13.5, and declines approximately 30% by E15.5. The most dramatic effect on the TrkA neuron numbers in NT-3 mutants occurs between E12.5 and E13.5 when approximately 50% of TrkA-expressing neurons are lost. While apoptosis was not elevated in mutants at E11.5 or E12.5, at E13.5 a 2- to 3-fold increase in TrkA-positive apoptotic profiles was observed (Fig. 7A,D,G).
Results presented above suggest that in vivo NT-3 promotes survival of trigeminal neurons expressing TrkB or TrkA by directly activating these receptors. Alternatively, as some neurons co-express TrkC with TrkB or with TrkA, it seemed possible that apoptosis of TrkB and/or TrkA-expressing cells only reflects a requirement for activation of TrkC within neurons expressing another receptor. To investigate whether deficiencies in TrkC activation account for losses in either the TrkB or TrkA populations, we determined whether survival of either population was reduced in the TrkC mutant. Compared to wild-type littermates, however, there were not significantly fewer neurons expressing TrkB or TrkA in TrkC mutants (Table 2). Therefore, losses of TrkB and TrkA-expressing neurons in the NT-3 mutant are not caused by disruption of the NT-3 to TrkC signaling pathway.
DISCUSSION
Using antisera specific for each of the Trk receptors, we have characterized their expression patterns during the major period of neurogenesis in the trigeminal ganglion. Since the deficit in trigeminal ganglion develops in NT-3 mutants during this same period (Wilkinson et al., 1996), we have also used these antisera to examine effects of NT-3 deficiency on development and survival of each of these three subpopulations during this interval. Several conclusions can be drawn from our study. First, in the developing trigeminal ganglion, detectable quantities of Trk receptor antigens are present in neurons, but not precursors. Secondly, BrdU colabeling studies have shown that the generation of Trk-expressing neurons follows a dynamic pattern, with most TrkB- and TrkC-expressing neurons generated between E10.5 and E11.5, while vast majority of TrkA-expressing neurons is generated between E11.5 and E13.5. Thirdly, neurons co-expressing detectable amounts of more than one Trk receptor are present at early stages, but are always a minority. Although there are limited numbers of neurons with co-expression of TrkA and TrkB, TrkB and TrkC, or TrkA and TrkC at E11.5, the numbers of these neurons decrease progressively, and by E13.5 essentially all neurons express just one Trk receptor. Finally, loss of NT-3 results in apoptotic cell death of all neurons that express TrkC and substantial proportions of the neurons that express TrkA or TrkB. The loss of the latter is not due to death of neurons that co-express TrkA or TrkB with TrkC in earlier stages, but reflects a requirement for direct activation by NT-3 of TrkA and TrkB for survival of neurons expressing these receptors in vivo.
One of the most important observations of the present work is the failure to observe detectable expression of Trk receptor proteins in precursor cells within the trigeminal ganglion (Fig. 2; Table 1). In particular, previous reports on the expression of TrkC mRNA in very early embryos, and observations in vitro of NT-3 effects on neural crest and neuronal precursor populations, have strongly suggested that neuronal precursor cells express TrkC (reviewed by Lewin and Barde, 1996). Consistent with this expectation, TrkC mRNA has been observed in vivo in what appear to be migrating neural crest cells in the vicinity of the aorta in murine embryos (Tessarollo et al., 1993), and within trigeminal placodal cells at the time of commitment to neuronal fate in chick embryos (Williams et al., 1995). Moreover, lack of NT-3 has also been reported to cause apoptotic cell death in precursors of the trigeminal ganglion, suggesting that receptors for NT-3 are present on proliferating precursor cells in vivo (ElShamy and Ernfors, 1996). In our previous work, however, we found that there are normal numbers of precursor cells present in the trigeminal ganglion of NT-3 mutants during the period of neurogenesis (Wilkinson et al., 1996). In agreement with our previous work, double labeling experiments comparing RTA, RTB, or RTC immunoreactivity with neurofilament staining show that TrkA, TrkB, and TrkC proteins colocalize tightly with neuronal profiles in the developing trigeminal ganglion. Furthermore, detectable TrkB and TrkC proteins are not present within proliferating trigeminal precursors, identified by incorporation of BrdU (Table 1; Fig. 2). Similar results have been obtained in parallel studies of TrkC protein expression in embryonic trunk neural crest and dorsal root ganglia (Fariñas et al., 1998). Interestingly, our counts at E10.5 indicate that there are likely to be neurons not expressing detectable amounts of any Trk receptor, suggesting that there may be a short interval during which these neurons survive in absence of neurotrophin-mediated signaling (Table 2; see also Davies, 1997).
Another important observation from this study is the different waves of neurogenesis for neurons expressing Trk receptors. The numbers of neurons expressing TrkA, TrkB, or TrkC peak at different times (Fig. 4). In addition, results from neuronal birth-dating experiments indicate that proliferating precursors from E9.5 and E10.5, but not those from E11.5, contribute to the generation of TrkB- and TrkC-expressing neurons (Fig. 6; Table 4). No TrkB or TrkC neurons are born after this stage. In contrast, precursors from E10.5 and E11.5, and to a lesser extent E9.5, contribute to the generation of TrkA-expressing neurons, but most of these appear during the 2-day interval after E11.5 (Table 4). Interestingly, the birth-dating experiments also indicate that it takes more than 10 hours after S-phase for precursors to differentiate into TrkB or TrkC neurons, and that a longer time interval of approximately 24 hours after S-phase is required for precursors to fully mature into TrkA-expressing neurons. This suggests that there may be intrinsic differences in the precursors that give rise to neurons expressing different Trk receptors. Alternatively, unknown regulatory mechanisms may delay expression of TrkA in precursors committed to that differentiation pathway. Taken together, our data strongly suggest a model in which generation of neurons occurs in two waves with the first wave generating neurons expressing TrkA, TrkB, or TrkC, while the second wave produces only neurons expressing TrkA. In each case, neurogenesis is followed by apoptotic cell death, most likely due to competition for neurotrophins (reviewed by Reichardt and Fariñas, 1997).
The expression patterns of Trk receptors in the mouse trigeminal ganglion determined by these specific antibodies are in agreement with those from the in situ studies (Arumae et al., 1993; Ernfors et al., 1992) in which TrkB and TrkC mRNAs are prominent in rat trigeminal ganglion at E12, but become restricted to few cells at E16 and E18. Using antibodies to Trk receptors, our double labeling experiments have clearly shown that after E12.5, these receptors are present in distinct, non-overlapping populations of trigeminal neurons. However, it has been shown by RT-PCR that, after dissociated culture in vitro, individual E16 rat trigeminal neurons express detectable levels of mRNAs encoding more than a single Trk receptor (Moshnyakov et al., 1996). These mRNA assays may well be more sensitive than our protein assays, so neurons with detectable immunoreactivity for one Trk receptor may contain low levels of others. Alternatively, it has been shown by Barde and colleagues that the presumptive TrkA and TrkC-expressing chick DRG neurons, as result of dissociated culture in vitro, exhibit changes in gene expression, including changes in expression of genes encoding TrkA and TrkC (Friedel et al., 1997). Thus, Trk receptor expression data obtained from trigeminal neurons in dissociated culture may not be a perfect reflection of expression in vivo.
Assays in vitro have shown that developing trigeminal neurons require different neurotrophins for survival, depending upon time of isolation for culture. When cultured at E10.5 and E11.5, neuronal survival appears complete in the presence of NT-3 or BDNF, but few survive in the presence of NGF. When cultured after E12.5, most neurons survive in the presence of NGF, but not NT-3 or BDNF (Buchman and Davies, 1993). Our data suggest that these differences in responsiveness to neurotrophins by trigeminal neurons cultured at different stages reflect primarily differences in the proportions of neurons generated by two waves of neurogenesis. The early generation of neurons expressing TrkB and TrkC correlates with the ability of E11.5 trigeminal neurons to survive in BDNF or NT-3 in vitro. The almost exclusive generation of TrkA-expressing neurons at stages after E11.5 reduces the proportion of neurons expressing TrkB or TrkC in vivo to below 30% after E12.5. Thus it is not surprising that a much lower percentage of these neurons survive in either BDNF or NT-3 in cell culture. Although about 40% of E11.5 trigeminal neurons express detectable levels of TrkA, trigeminal neurons at this stage did not respond efficiently to low concentrations of NGF in culture (Buchman and Davies, 1993). As TrkA receptor expression appears weak in vivo (e.g. Fig. s 2E,5C), low levels of TrkA receptor may explain their poor responsiveness. After E12.5, TrkA receptor expression becomes intense and is present in more than 70% of all neurons; thus it is not surprising that most trigeminal neurons cultured at E12.5 and later stages survive in the presence of NGF. In conclusion, the changes in Trk receptor expression in vivo seem generally consistent with the reported changes in acute survival responses to different neurotrophins of cultured trigeminal neurons (Buchman and Davies, 1993).
The most compelling evidence that neurons switch Trk receptor expression is data indicating that neurons can acquire the ability to respond to a neurotrophin with time in cell culture. For example, it has been shown that trigeminal neurons, cultured at E12.5 for 2 days in BDNF or NT-4/5, become responsive to NGF (Paul and Davies, 1995; see also Davies, 1997). In other words, there is an almost complete switch of neurotrophin dependence in this neuronal population in vitro. In contrast, our birth-dating analyses and our failure to detect significant number of neurons coexpressing multiple Trk receptors suggest that comparatively few trigeminal neurons switch Trk receptor expression between E11.5 and E13.5 in vivo. Our data also indicate that TrkA-expressing neurons generated in the second major wave of neurogenesis have not switched from expression of another Trk receptor. Specifically, E11.5 precursors labeled with BrdU were shown to generate large numbers of TrkA-expressing neurons over the following 2 days, but the progeny of these precursors never included significant numbers of neurons expressing TrkB or TrkC (Table 4). However, our data indicate that a minority of neurons do coexpress TrkA and TrkB at E11.5. Thus, similar to in vitro observations (Paul and Davies, 1995), it is possible that some, but clearly not all neurons expressing TrkB immediately after neurogenesis switch to expression of TrkA at later times. It seems also possible that more extensive switching may occur in vitro because long-term culture of dissociated neurons results in changes in their Trk receptor expression not normally seen in vivo (see e.g. Friedel et al., 1997).
In a previous paper (Wilkinson et al., 1996), we characterized the development of the trigeminal ganglion in wild type and NT-3 mutants between E10.5 and P0. We demonstrated that both neurons and precursors are present in normal numbers at E10.5; that in the NT-3 mutant there is an approximately 40% neuronal deficit, but no significant deficit in precursor number or proliferation rate at E11.5; and that the deficit in neuronal numbers in the NT-3 mutant remains approximately constant after E13.5. In the present paper, we have extended our earlier observations utilizing antibodies specific for each Trk receptor. The absence of detectable quantities of any Trk receptor on precursors is certainly consistent our observation that the defects in the NT-3 mutants is due to apoptotic death of neurons, not precursors. The antibodies have additionally enabled us to evaluate the consequences of the lack of NT-3 on the subpopulations of trigeminal ganglion neurons expressing each of these receptors. At E10.5, similar numbers of all neuronal subpopulations are seen in wild-type and NT-3-deficient embryos, whereas at E11.5, the first time at which we observed a reduction in neuronal number in the NT-3 mutant (Wilkinson et al., 1996), approximately 40% of trigeminal neurons express TrkC in wild-type embryos, but only 2% of this number express this protein in the NT-3 mutant. Moreover, most TrkC-immunopositive cells in mutant embryos have abnormal morphologies and there are widespread TrkC-immunoreactive apoptotic profiles. In E12.5 and older ganglia, almost no TrkC-immunoreactive neurons remain. These findings document that the entire population of TrkC expressing neurons requires NT- 3 for survival. Although present in normal numbers at E10.5, in E11.5 embryos lacking NT-3, the population of TrkB-expressing neurons is reduced by approximately 60% (Table 2; Fig. 4C). A 3-fold increase in TrkB-immunoreactive apoptotic profiles in NT-3 mutants at E11.5 indicates that this deficit is also caused by abnormal cell death (Fig. 7G). Remaining TrkB-expressing neurons appeared normal as assessed by morphological criteria. Thus, only a fraction of the TrkB-expressing neuronal population requires NT-3 to survive between E10.5 and E11.5. Similar to the TrkB-expressing neurons, there is about a 50% reduction in the TrkA-expressing neurons between E12.5 and E13.5, and the number of TrkA-immunoreactive apoptotic profiles increase by 2–3 fold at E13.5. In the end, total elimination of TrkC neurons and 40–50% losses of TrkA and TrkB neurons account for the final loss of 70% neurons in the trigeminal ganglion in the NT-3 mutant.
Our interpretation of the dramatic loss of TrkC neurons in NT-3 mutants is that NT-3 is absolutely required for the survival of TrkC-expressing neurons. While remotely possible, our data provide no evidence for a regulatory circuit in which absence of NT-3 reduces TrkC expression to below detectable levels in neurons that continue to survive. First, there is no difference in the intensity of TrkC expression and the number of TrkC-positive neurons between wild type and NT-3 mutants at E10.5. Thus NT-3 does not appear to regulate TrkC expression at this early stage (Table 2). Second, if NT-3 regulated TrkC expression, one would anticipate that the number of neurofilament-positive cells would exceed the sum of neurons expressing TrkA, TrkB, and TrkC in NT-3 mutants at E11.5. However, our data indicate that the sum of neurons expressing Trk receptors is very similar to the number of neurofilament-positive cells at this stage (Table 2). Finally and most importantly, the overwhelming presence of apoptosis argues that cell death is the major, if not the only, contributor to loss of TrkC neurons (Fig. 7).
Loss of TrkA neurons at E13.5 and TrkB neurons at E11.5 is almost certainly due to a direct effect of NT-3 on subsets of TrkA and TrkB neurons because the numbers of TrkA and TrkB neurons are not reduced by the loss of TrkC (Table 2). Further, since the timing of cell death differs between the TrkB and TrkA populations, TrkB neurons can not be dying because of disruption of an NT-3 to TrkA signaling pathway and vice versa. Indeed, activation of TrkB by NT-3 in vivo has been shown in the developing dorsal root ganglion (Fariñas et al., 1998) and in vitro, high concentrations of NT-3 also have been shown to support the survival of trigeminal neurons lacking TrkC (Davies et al., 1995). Although much more NT-3 is required to activate TrkA or TrkB in vitro (Ip et al., 1993), our data imply that there must be high enough local concentrations of NT-3 in vivo to support neurons expressing these receptors.
Survival of the remaining TrkB- and TrkA-expressing neurons almost certainly depends on the availability of other neurotrophins in target tissues to which these neurons project their axons. Indeed, by examining the expression of the lacZ gene which is integrated into the NT-3 or BDNF loci, we have identified distinct regions in the developing branchial arches where only NT-3 is expressed between E11.5 and E12.5 (E. J. H., unpublished data). Presumably, TrkB-expressing neurons that project to these regions will require NT-3 for their survival, while those projecting to regions expressing BDNF will not. Hence, the spatial and temporal distributions of neurotrophins may dictate the requirement for different neurotrophins for TrkA and TrkB neurons.
In conclusion, the isolation of antibodies specific for TrkA, TrkB and TrkC has allowed us to examine Trk expression at the cellular level in the mouse trigeminal ganglion during neurogenesis. We have shown that Trk receptors are present in neurons, not precursors, and that there is only limited co-expression of Trk receptors in the neurons. After E13.5, trigeminal neurons express detectable levels of only one Trk receptor. Between E10.5 and E13.5, the number of neurons increases rapidly. There are also dramatic changes in the percentages of neurons that express particular Trk receptors. Our data indicate that it is different waves of neurogenesis that explain these changes in Trk receptor expression and neurotrophin dependence. Finally, by analyzing the trigeminal ganglion in NT-3 and TrkC mutants, we have shown that, in addition to supporting TrkC-expressing neurons, NT-3 can also support the survival of significant numbers of TrkA- and TrkB-expressing neurons in vivo.
Acknowledgments
We thank Andrea Schmidt for help with immunohistochemistry, and Drs. M. Barbacid and I. Silo-Santiago for providing the TrkC kinase-deficient mutant strain and for sharing data before publication. This work has been supported by research grants from the United States Public Health Service (NIH grant MH48200) and the Howard Hughes Medical Institute. E.J.H. is a recipient of Postdoctoral Fellowship for Physicians and a Research Associate of the Howard Hughes Medical Institute; I.F. was the recipient of a long-term fellowship from the Human Frontier Science Program Organization; and L.F.R. is an Investigator of the Howard Hughes Medical Institute.
References
- Arumae U, Pirvola U, Palgi J, Kiema T-R, Palm K, Moshnyakov M, Ylikoski J, Saarma M. Neurotrophins and their receptors in rat trigeminal system during maxillary nerve growth. J Cell Biol. 1993;122:1053–1065. doi: 10.1083/jcb.122.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamji SX, Madjan M, Poznink CD, Belliveau DJ, Alyz R, Kohn J, Causing CG, Miller FD. The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol. 1998;140:911–927. doi: 10.1083/jcb.140.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbacid M. The trk family of neurotrophin receptors. J Neurobiol. 1994;11:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- Barker PA, Lomen-Hoerth C, Gensch EM, Meakin SO, Glass DJ, Shooter EM. Tissue-specific alternative splicing generates two isoforms of the TrkA receptor. J Biol Chem. 1993;268:15150–15157. [PubMed] [Google Scholar]
- Buchman VL, Davies AM. Different neurotrophins are expressed and act in a developmental sequence to promote the survival of embryonic sensory neurons. Development. 1993;118:989–1001. doi: 10.1242/dev.118.3.989. [DOI] [PubMed] [Google Scholar]
- Clary DO, Weskamp G, Austin LR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey F, Davies AM. TrkB signalling inhibits p75-mediated apoptosis induced by nerve growth factor in embryonic proprioceptive neurons. Curr Biol. 1998;8:915–918. doi: 10.1016/s0960-9822(07)00371-5. [DOI] [PubMed] [Google Scholar]
- Davies AM, Lee KF, Jaenisch R. p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron. 1993;11:565–574. doi: 10.1016/0896-6273(93)90069-4. [DOI] [PubMed] [Google Scholar]
- Davies AM, Minichiello L, Klein R. Developmental changes in NT-3 signaling via TrkA and TrkB in embryonic neurons. EMBO J. 1995;14:4482–4489. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AM. Neurotrophin switching: where does it stand? Curr Opinion Neurobiol. 1997;18:111–118. doi: 10.1016/s0959-4388(97)80128-6. [DOI] [PubMed] [Google Scholar]
- ElShamy WM, Ernfors P. Requirement of neurotrophin-3 for the survival of proliferating trigeminal cells. Development. 1996;122:2405–2414. doi: 10.1242/dev.122.8.2405. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Merlio JP, Persson H. Cells expressing mRNA for neurotrophins and their receptors during embryonic rat development. Eur J Neurosci. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- Fariñas I, Jones KR, Backus C, Wang XW, Reichardt LF. Targeted mutation of the neurotrophin-3 gene results in severe sensory and sympathetic deficits. Nature. 1994;369:658–661. doi: 10.1038/369658a0. [DOI] [PubMed] [Google Scholar]
- Fariñas I, Wilkinson GA, Backus C, Reichardt LF, Patapoutian A. Characterization of neurotrophin and trk receptor functions in developing sensory ganglia: Direct NT-3 activation of trkB neurons in vivo. Neuron. 1998;21:325–334. doi: 10.1016/s0896-6273(00)80542-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel RH, Schnürch H, Stubbusch J, Barde YA. Identification of genes differentially expressed by nerve growth factor- and neurotrophin-3-dependent sensory neurons. Proc Natl Acad Sci USA. 1997;94:12670–12675. doi: 10.1073/pnas.94.23.12670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip NY, Stitt TN, Tapely P, Klein R, Glass DJ, Fandl J, Greene LA, Barbacid M, Yancopoulos GD. Similarities and differences in the way neurotrphins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron. 1993;10:137–149. doi: 10.1016/0896-6273(93)90306-c. [DOI] [PubMed] [Google Scholar]
- Klein R, Martin-Zanca D, Barbacid M, Parada LF. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development. 1990;109:845–850. doi: 10.1242/dev.109.4.845. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Barde YA. Physiology of the neurotrophins. Ann Rev Neurol. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Martin-Zanca D, Barbacid M, Parada LF. Expression of the trk proto-oncogene is restricted to the sensory cranial and spinal ganglia of neural crest origin in mouse development. Genes Dev. 1990;4:683–94. doi: 10.1101/gad.4.5.683. [DOI] [PubMed] [Google Scholar]
- Moshnyakov M, Arumäe U, Saarma M. mRNAs for onr, two or three members of trk receptor family are expressed in single rat trigeminal ganglion neurons. Mol Brain Res. 1996;43:141–148. doi: 10.1016/s0169-328x(96)00168-4. [DOI] [PubMed] [Google Scholar]
- Mu X, Silos-Santiago I, Carroll SL, Snider WD. Neurotrophin receptor genes are expressed in distinct patterns in developing dorsal root ganglia. J Neurosci. 1993;13:4029–4041. doi: 10.1523/JNEUROSCI.13-09-04029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkina N, Adu J, Fischer A, Pinon LGP, Buchman VL, Davies AM. Expression and function of TrkB variants in developing sensory neurons. EMBO J. 1996;15:6385–6393. [PMC free article] [PubMed] [Google Scholar]
- Ninkina N, Grashchuck M, Buchman VL, Davies AM. TrkB variants with deletions in the leucine-rich motifs of the extracellular domain. J Biol Chem. 1997;272:13019–13025. doi: 10.1074/jbc.272.20.13019. [DOI] [PubMed] [Google Scholar]
- Paul G, Davies AM. Trigeminal sensory neurons require extrinsic signals to switch neurotrophin dependence during the early stages of target field innervation. Dev Biol. 1995;171:590–605. doi: 10.1006/dbio.1995.1307. [DOI] [PubMed] [Google Scholar]
- Piñon LGP, Minichiello L, Klein R, Davies AM. Timing of neuronal death in trkA, trkB and trkC mutant embryos reveals developmental changes in sensory neuron dependence on trk signaling. Development. 1996;122:3255–3261. doi: 10.1242/dev.122.10.3255. [DOI] [PubMed] [Google Scholar]
- Reichardt LF, Fariñas I. Neurotrophic factors and their receptors: roles in neuronal development and function. In: Cowan MW, Jessell TM, Zipursky L, editors. Molecular Approaches to Neural Development. New York: Oxford University Press; 1997. pp. 220–263. [Google Scholar]
- Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Philips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution and expression of extracellular domain immunoadhesions. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibayama E, Koizumi H. Cellular localization of the Trk neurotrophin receptor family in human non-neuronal tissues. Am J Pathol. 1996;148:1807–1818. [PMC free article] [PubMed] [Google Scholar]
- Stucky CL, Koltzenberg M. The low-affinity neurotrophin receptor p75 regulates the function but not the selective survival of specific subpopulations of sensory neurons. J Neurosci. 1997;17:4398–4405. doi: 10.1523/JNEUROSCI.17-11-04398.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Martin-Zanca D, Gilbert DJ, Jenkins NA, Copeland NG, Parada LF. trkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in non-neuronal tissues. Development. 1993;118:463–475. doi: 10.1242/dev.118.2.463. [DOI] [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Donovan MJ, Palko ME, Blair-Flynn J, Hempstead BL, Parada LF. Targeted deletion of all isoforms of the trkC gene suggests the use of alternate receptors by its ligand neurotrophin-3 in neuronal development and implicates trkC in normal cardiogenesis. Proc Natl Acad Sci SA. 1997;94:14776–14781. doi: 10.1073/pnas.94.26.14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiler K. The House Mouse: Atlas of Embryonic Development. New York: Springer-Verlag; 1989. [Google Scholar]
- Tsoulfas P, Soppet D, Escandon E, Tessarollo L, Mendoza-Ramirez JL, Rosenthal A, Nikolics K, Parada LF. The rat trkC locus encodes multiple neurogenic receptors that exhibit differential response to neurotrophin-3 in PC12 cells. Neuron. 1993;10:975–990. doi: 10.1016/0896-6273(93)90212-a. [DOI] [PubMed] [Google Scholar]
- Valenzuela DM, Maisonpierre PC, Glass DJ, Rojas E, Nuñez L, Kong Y, Gies DR, Stitt TN, Ip NY, Yancopoulos GD. Alternative forms of rat trkC with different functional capabilities. Neuron. 1993;10:963–974. doi: 10.1016/0896-6273(93)90211-9. [DOI] [PubMed] [Google Scholar]
- Van der Zee CE, Ross GM, Riopelle RJ, Hagg T. Survival of cholinergic forebrain neurons in developing p75NGFR-deficient mice. Science. 1996;274:1729–1732. doi: 10.1126/science.274.5293.1729. [DOI] [PubMed] [Google Scholar]
- Williams R, Bäckström A, Kullander K, Halböök F, Ebendahl T. Developmentally regulated expression of mRNA for neurotrophin high-affinity (trk) receptors within chick trigeminal sensory neurons. Eur J Neurosci. 1995;7:116–128. doi: 10.1111/j.1460-9568.1995.tb01026.x. [DOI] [PubMed] [Google Scholar]
- Wilkinson GA, Fariñas I, Backus C, Yoshida CK, Reichardt LF. Neurotrophin-3 is a survival factor in vivo for early mouse trigeminal neurons. J Neurosci. 1996;16:7661–7669. doi: 10.1523/JNEUROSCI.16-23-07661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt S, Davies AM. Regulation of expression of mRNAs encoding the nerve growth factor receptors p75 and trkA in developing sensory neurons. Development. 1993;119:635–647. doi: 10.1242/dev.119.3.635. [DOI] [PubMed] [Google Scholar]