

Abstract
trkB encodes a receptor tyrosine kinase activated by three neurotrophins—brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4/5. In vivo, three isoforms of the receptor are generated by differential splicing—gp145trkB or the full-length trkB receptor, and trkB.T1 and trkB.T2, two cytoplasmically truncated receptors that lack kinases, but contain unique C termini. Although the truncated receptors appear to be precisely regulated during nervous system development and regeneration, their role in neurotrophin signaling has not been directly tested. In this paper, we studied the signaling properties and interactions of gp145trkB, trkB.T1, and trkB.T2 by expressing the receptors in a Xenopus oocyte microinjection assay. We found that oocytes expressing gp145trkB, but not trkB.T1 or trkB.T2, were capable of eliciting45Ca efflux responses (a phospholipase C-γ-mediated mechanism) after stimulation by BDNF. When trkB.T1 and trkB.T2 were coexpressed with gp145trkB, they acted as dominant negative receptors, inhibiting the BDNF signal by forming nonfunctional heterodimers with the full-length receptors. An ATP-binding mutant of gp145trkB had similar dominant inhibitory effects. Our data suggest that naturally occurring truncated trkB receptors function as inhibitory modulators of neurotrophin responsiveness. Furthermore, the homodimerization of gp145trkB appears to be an essential step in activation of the BDNF signaling cascade.
Keywords: BDNF, dominant negative, neurotrophin, truncated trkB, tyrosine kinase, Xenopus oocyte
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family of growth factors (Barde et al., 1982; Leibrock et al., 1989). In the developing and adult nervous systems, BDNF functions in the regulation of neuronal survival, outgrowth, and differentiation (Eide et al., 1993). Recently, BDNF has attracted interest from the clinical community because of its therapeutic potential in the treatment of Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (Eide et al., 1993; Koliatsos et al., 1993; Morse et al., 1993; Hyman et al., 1994).
Although the exact mechanism of BDNF receptor activation has not been delineated, a 145 kDa product of the trkB proto-oncogene (gp145trkB) is believed to play a critical role in this process. Non-neuronal cells transfected with trkB cDNA acquire BDNF-binding sites and biological responsiveness to picomolar concentrations of the growth factor (Soppet et al., 1991; Squinto et al., 1991). Also, in vivo expression of gp145trkB appears to correlate well with neuronal populations known to respond to the growth factor (Middlemas et al., 1991; Eide et al., 1993).
In the rat, differential splicing of the trkB mRNA yields at least three receptor isoforms: gp145trkB or the full-length trkB receptor, and trkB.T1 and trkB.T2, two kinase-deficient receptors lacking most of the cytoplasmic domain of the full-length receptor, but containing unique short C terminal sequences (Allendoerfer et al., 1994). Postulated functions for truncated trkB receptors have included the following: roles in the facilitation or inhibition of gp145trkB-dependent signaling, roles in ligand clearance or sequestration, and cell–cell adhesive effects, axonal outgrowth/promotion, and synaptic plasticity (Klein et al., 1990a,b; Beck et al., 1993).
In the current paper, we study the signaling properties and interactions of gp145trkB, trkB.T1, and trkB.T2 by expressing these transcripts in microinjected Xenopusoocytes. We find that only gp145trkB expression (not trkB.T1 or trkB.T2) is sufficient to elicit a45Ca efflux response [indicating activation of phospholipase C-γ (PLC-γ)] after stimulation by BDNF; furthermore, when either trkB.T1 or T2 was coexpressed with gp145trkB, it acted as a dominant inhibitor, blocking BDNF signaling by sequestering wild-type (catalytic) receptors within nonfunctional heterodimers. Our study suggests that trkB.T1 and trkB.T2 may act as inhibitory modulators of neurotrophin responsivenessin vivo. In addition, our data support an essential role for BDNF-induced gp145trkB homodimerization in BDNF signal transduction.
MATERIALS AND METHODS
cDNA constructs. cDNAs encoding the full-length rat trkB receptor, trkB.T1, and trkB.T2 were generously provided by Dr. D. S. Middlemas and T. Hunter (The Salk Institute, La Jolla, CA). An ATP-binding mutant of gp145trkB (ATPmutmyc) was constructed by introducing a lysine → methionine substitution at position 560 in the ATP-binding domain (Transformer Mutagenesis Kit, Clontech, Palo Alto, CA). A 38 nucleotide primer (38-mer) was used to introduce the mutation: 5′-CGTCCTTCAGCG TCATCACGG CCACCAG GA TCTTATCC-3′. A second primer mutated a novel SacI site to AATII in the Bluescript SK plasmid: 5′-TGAAAA GCTGGACGTCCA CCGCGGTG-3′.
Wild-type and mutant gp145trkB receptors were tagged at their C termini by PCR. Wild-type receptors were tagged with an epitope from influenza virus hemagglutinin (IVH) (Niman et al., 1983). The mutant receptor was tagged with an epitope from the human c-myc proto-oncogene (myc) (Evan et al., 1985). Primers used included a 20-mer upstream to a unique BglII restriction site (5′-CTGCTTGGTA GGAGA GAACC-3′), as well as epitope primers that included the C terminus of the trkB receptor, mutation of the existing stop codon, epitope tag, a new termination codon, and a novelXbaI site. For IVH: 5′-CCGTCGACTTACCGTGAAGGTCCTC CTAGCGATGCGTAGTCAGGGACAT CGTATGGGT AACTACTTCCCCTCCGAA GAAGACGGAGTGTTGCTCC-3′. For myc: 5′-CCGTCGACTTACAGGTCCTC CTCGGAG-ATCAGCTTCTGCTCGCCTCCGC CTAGGA TGTCCAGGTAGA-CG-3′. PCR products were filled in with Klenow, blunt-cloned intoHincII-digested Bluescript SK, digested withBglII–XbaI, and then unidirectionally cloned into wild-type, truncated, or mutant trkB cDNAs.
For expression in COS cells, XhoI–XbaI fragments containing tagged constructs were inserted under the cytomegalovirus promoter in pcDNAneo (Invitrogen, San Diego, CA). For expression inXenopus oocytes, constructs were inserted into theEcoRI site of Bluescript SK or theNcoI–HindIII site of pSP64T, a vector that includes 5′ and 3′ Xenopus β-globin-untranslated sequences (Krieg and Melton, 1984). The 5′-untranslated region and anNcoI site (700 bp) of trkB were removed by PCR. The upstream primer was 5′-GAACATACC ATGGCCATGTCGCCCTGGCCGAGGT GGCATGGACCCGCGATGGCGCGGCTCTGG-3′; the downstream primer was 5′-CTTCAGAAACGCCTTGTAAGCC-3′. PCR products were filled in with Klenow, blunt-cloned into HincII-digested Bluescript SK, digested with NcoI–HindIII, and then unidirectionally cloned into pSP64T. The NcoI site was removed with Mung bean nuclease. Cloning was completed by unidirectionally cloning a 2.2 kbHindII–Xba fragments of the tagged mutant or wild-type receptors into the pSP64T construct.
Antibodies. Mouse monoclonal epitope antibodies (Abs) were obtained from Boehringer Mannheim (12CA5-IVH, Indianapolis, IN) or Oncogene Science (9E10-myc). Anti-phosphotyrosine (APT) Abs were purchased from Upstate Biotechnology (4G10, Lake Placid, NY) or were received as a gift from Dr. J. Escobedo (FB2B5, University of California, San Francisco, CA).
Growth factor, COS transfections, and extracts. Purified recombinant human BDNF was received as a gift from Dr. A. Rosenthal (Genentech). In some experiments, recombinant human BDNF was used in the form of baculoviral or CHO supernatants in serum-free media (gifts of Drs. W. Mobley, University of California, San Francisco and Dr. P. Olson, Chiron Corporation, Emeryville, CA). The biological activity and dose–response curves for BDNF were verified by using neurite outgrowth assays of dorsal root ganglion explants or trkB-expressing PC12 NNR5 cells (data not shown).
COS7 cells were transfected with DEAE dextran (400 mg/ml) and chloroquine phosphate (1 mm), then incubated in DMEM (+10% fetal bovine serum) at 5% CO2 for 72 hr. Plates were stimulated with BDNF (20 pm to 20 nm) 5 min before lysis. Cells were lysed in 1% Triton X-100, 20 mm Tris-Cl, pH 8.0, 137 mm NaCl, 2 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 20 μm leupeptin, and 1 mm sodium orthovanadate. Lysates were cleared by centrifugation, immunoprecipitated with Abs for 2 hr at 4°C, and then collected with protein A–Sepharose (Pharmacia, Uppsala, Sweden). For myc immunoprecipitations, protein G-Plus Sepharose (Oncogene Science) or anti-mouse IgG (Sigma, St. Louis, MO) were used. After precipitation and washing, Sepharose beads were boiled in Laemmli’s sample buffer for 5 min (Laemmli, 1970). Proteins were separated on SDS-PAGE gels, then transferred to nitrocellulose. Blots were probed overnight in Tris-buffered saline containing 0.1% Tween-20 and then visualized using alkaline phosphatase-conjugated anti-mouse or anti-rabbit secondary antibody and colorimetric agents (Promega, Madison, WI).
In vitro transcription and expression in Xenopusoocytes.In vitro transcripts containing a 5′-GpppG cap (Pharmacia) were prepared using a kit from Stratagene (La Jolla, CA). Templates were linearized in pBSSK or pSP64T and transcribed under the T7 or SP6 RNA polymerase promoters. Transcripts were analyzed on formaldehyde gels and quantitated by spectrophotometry.
Mature oocytes (Dumont stage V–VI) were defolliculated manually or by 0.5–2 hr treatment with collagenase (Sigma type I, 1 mg/ml). Oocytes were maintained in modified Barth’s saline solution (MBSH) containing Na-HEPES (15 mm, pH 7.6), 1 mg/ml bovine serum albumin, 100 μg/ml penicillin G, and 100 mg/ml streptomycin. Oocytes were injected with 50 nl of cRNA solution or sterile water for controls.
Calcium efflux experiments. Efflux experiments were conducted according to the method of Ueno et al. (1991). Calcium mobilization was quantitated by measuring 45Ca efflux from injected oocytes. Two days after injection, oocytes were incubated in Ca45 (100 mCi/ml) for 3 hr in calcium-free MBSH at 19°C. Groups of eight oocytes were washed and then transferred to 24-well plastic cell dishes at 0.5 ml of calcium-free MBSH per well. Media were collected and replaced every 10 min, and radioactivity was counted in a liquid scintillation counter. After calcium efflux had stabilized (3 values < 1000 counts/min), growth factor was added (BDNF, 5–400 ng/ml).
35S immunoprecipitations. Injected oocytes were incubated overnight at 19°C and then metabolically labeled for 24 hr with [35S]methionine (12 μCi/oocyte). Oocytes were stimulated with BDNF for 3 hr at 4°C before being lysed in ice-cold RIPA buffer (1% Triton X-100, 50 mmNaCl, 50 mm NaF, 30 mmsodium pyrophosphate, 5 mm EDTA, 10 mm Tris-Cl, pH 7.4, 2 mmphenylmethylsulfonyl fluoride, 25 mm leupeptin, 10 mm pepstatin A, and 0.2 U/ml aprotinin). Lysates were cleared by centrifugation at 13,000 × g for 10 min at 4°C, then incubated with APT mAb 4G10 for 3 hr at 4°C. Immune complexes were collected with protein A–Sepharose, rinsed three times in RIPA buffer, separated on SDS-PAGE, and then analyzed using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA) and autoradiography.
Immunoblots of trkB receptors expressed in oocytes. Oocytes were lysed in ice-cold RIPA buffer 36 hr after injection, cleared by centrifugation, partially purified with wheat germ agglutinin–Sepharose (Pharmacia), separated by 7% SDS-PAGE, and then blotted with anti-IVH or anti-myc Abs. After transfer to nitrocellulose, blots were blocked, incubated in anti-mouse IgG conjugated to horseradish peroxidase (Zymed, San Francisco, CA), and then visualized using enhanced chemiluminescence (ECL; Amersham, Arlington Heights, IL).
RESULTS
BDNF increases 45Ca efflux in oocytes injected with trkB cRNA; no increase is seen with trkB.T1 or trkB.T2
The Xenopus 45Ca efflux assay has been used as a paradigm of receptor activation and signal transduction (Ueno et al., 1991). Receptors capable of hydrolyzing phosphatidylinositol are able to mobilize intracellular calcium stores and increase 45Ca release in response to stimulation by ligand in this assay (Williams et al., 1988; Johnson et al., 1990). In the present study, cRNA encoding the gp145trkB, trkB.T1, or trkB.T2 was transcribedin vitro, then microinjected into stage V–VIXenopus oocytes (see Materials and Methods). Forty-eight hours later, oocytes were loaded with 45Ca, washed in 0.5 ml of Ca2+-free medium, and then tested for 45Ca efflux using a liquid scintillation counter. After 45Ca efflux levels had stabilized to <1000 cpm, oocytes were stimulated with BDNF. The data are represented graphically in Figure1A. Each point represents the average of 4 trials ± SD.
Fig. 1.

Dominant inhibitory effect of naturally occurring truncated trkB receptors in 45Ca efflux assay. Oocytes were injected with cRNA (2 ng/oocyte) and were then loaded with45Ca as described in Materials and Methods. After45Ca efflux levels stabilized, 250 ng/ml BDNF was added to the medium (indicated by arrow). Mean values from four determinations ± SD are shown. A, Comparison of BDNF-induced 45Ca efflux in oocytes expressing the full-length trkB receptor (closed squares) trkB.T1 (closed circles), trkB.T2 (closed triangles), or water control (open triangles). B, Dominant inhibitory effect of trkB.T1. Oocytes were injected with trkB cRNA (2 ng/oocyte) + varying quantities of trkB.T1 (0, 2, 8, or 18 ng/oocyte).C, Dominant inhibitory effect of trkB.T2. Oocytes were injected with 2 ng/oocyte trkB cRNA + varying quantities of trkB.T2 (0, 2, 8, or 18 ng/oocyte). D, Dominant inhibitory effect of trkB.T1 and trkB.T2 correlates with loss of trkB homodimers. A binomial model of dimer association (see Results) was used to predict the number of trkB homodimers (open squares). Peak45Ca efflux values for trkB.T1-injected (closed circles) and trkB.T2-injected (closed triangles) oocytes are plotted as a function of truncated:full-length trkB cRNA.
When full-length trkB receptors were expressed in oocytes, a 15-fold increase in 45Ca efflux was seen after stimulation by BDNF (Fig. 1A, closed squares). No change from baseline was seen in pools of oocytes expressing trkB.T1 (closed circles), trkB.T2 (closed triangles), or sterile water (Control; open triangles). Comparable expression levels of gp145trkB, trkB.T1, and trkB.T2 were verified by antigen blotting (data not shown).
Our finding that full-length (but not truncated) trkB receptors were capable of eliciting 45Ca efflux responses was consistent with the hypothesis that these receptors are sufficient to activate PLC-γ-dependent BDNF signaling pathways. The activation of PLC-γ by BDNF stimulation has been shown previously in transfected cells lines and in primary neuronal cultures (Widmer et al., 1993;Middlemas et al., 1994).
Inhibition of BDNF-induced 45Ca efflux by coexpression of trkB.T1 or trkB.T2
To test whether trkB.T1 or trkB.T2 would facilitate, inhibit, or have no effect on gp145trkB-dependent signaling, we coexpressed these receptors at various concentrations inXenopus oocytes. As shown in Figure 1, B andC, both trkB.T1 and trkB.T2 inhibited gp145trkB-dependent BDNF signaling in a dose-dependent manner. At 250 ng/ml BDNF, a greater than ninefold excess of truncated receptor was necessary to abolish the45Ca efflux response. These data supported a role for naturally occurring truncated trkB receptors as inhibitory modulators of gp145trkB-dependent signaling.
Inhibition of BDNF signaling by truncated trkB receptors parallels a predicted decline in the number of gp145trkBhomodimers
Tyrosine kinase-mediated growth factors such as epidermal growth factor, basic fibroblast growth factor, or platelet-derived growth factor (PDGF) are believed to activate their receptors according to a process of allosteric dimerization (White, 1991; Ueno et al., 1993). Binding of a dimeric growth factor induces receptors to pair, causing a conformational change that in turn triggers the intermolecular phosphorylation of intracellular tyrosines and subsequent activation of downstream proteins. Less is known about the activation of neurotrophin signaling pathways, but it is generally believed that the neurotrophins transduce their signals by a similar process.
If BDNF activates its receptors by allosteric dimerization, then a binomial model of dimer association should be able to predict the number of receptor homodimers or heterodimers formed in oocytes expressing different trkB receptor isoforms (Ueno et al., 1991). If the ratio of truncated to full-length receptors is x, then the probability of full-length receptors forming homodimers would be 1/(1 + x)2; for truncated homodimersx2/(1 + x)2; and for full-length–truncated heterodimers 2x/(1 + x)2(Ueno et al., 1991). The number of full-length homodimers should be 1/(1 + x) times the number of homodimers found in oocytes expressing full-length receptors only. This model is displayed graphically in Figure 1D (open squares, dotted lines). At a truncated to full-length trkB receptor ratio of 1:1, oocytes should have 1/2 the number of full-length homodimers found in oocytes expressing full-length receptors only; at a ratio of 4:1, the number of full-length homodimers should drop to 1/5; and at 9:1, the number of full-length homodimers should drop to 1/10.
When percent maximum 45Ca efflux levels for co-injected oocytes (trkB.T1 + full-length, closedcircles; trkB.T2 + full-length, closedtriangles) were compared with the predicted number of gp145trkB homodimers (Fig. 1D,open squares), a close approximation of graphs was seen. These data suggested both that homodimerization of gp145trkB molecules was essential for BDNF-induced PLC-γ activation and that truncated trkB receptors inhibited signaling by reducing the number full-length receptors available to form homodimers. No difference in the stoichiometry of trkB.T1 or trkB.T2 was seen.
A trkB receptor ATP-binding mutant functions like trkB.T1 and trkB.T2 to inhibit BDNF signaling
To test whether the dominant inhibitory effect of trkB.T1 and trkB.T2 on BDNF signaling was attributable to their lack of kinase activity, we constructed a mutant of gp145trkBthat contained a lysine → methionine substitution at the ATP-binding site (Lys560). A receptor containing this point mutation would be identical to a full-length receptor in every respect, except that it would be unable to bind ATP and therefore be devoid of kinase activity. As shown in Figure2A (closed circles), when expressed alone in oocytes, the ATP-binding mutant was incapable of inducing a 45Ca efflux response after stimulation by BDNF (Fig. 2A, closed circles). However, when coexpressed with the wild-type gp145trkBreceptor, the mutant caused a dose-dependent inhibition of BDNF-induced45Ca efflux (Fig. 2B). At a BDNF concentration of 250 ng/ml, a ninefold excess of mutant receptor virtually abolished the 45Ca response (closed triangles). The inhibitory effect of the ATP-binding mutant could not be distinguished from that of trkB.T1 or trkB.T2 (Fig.1B,C).
Fig. 2.
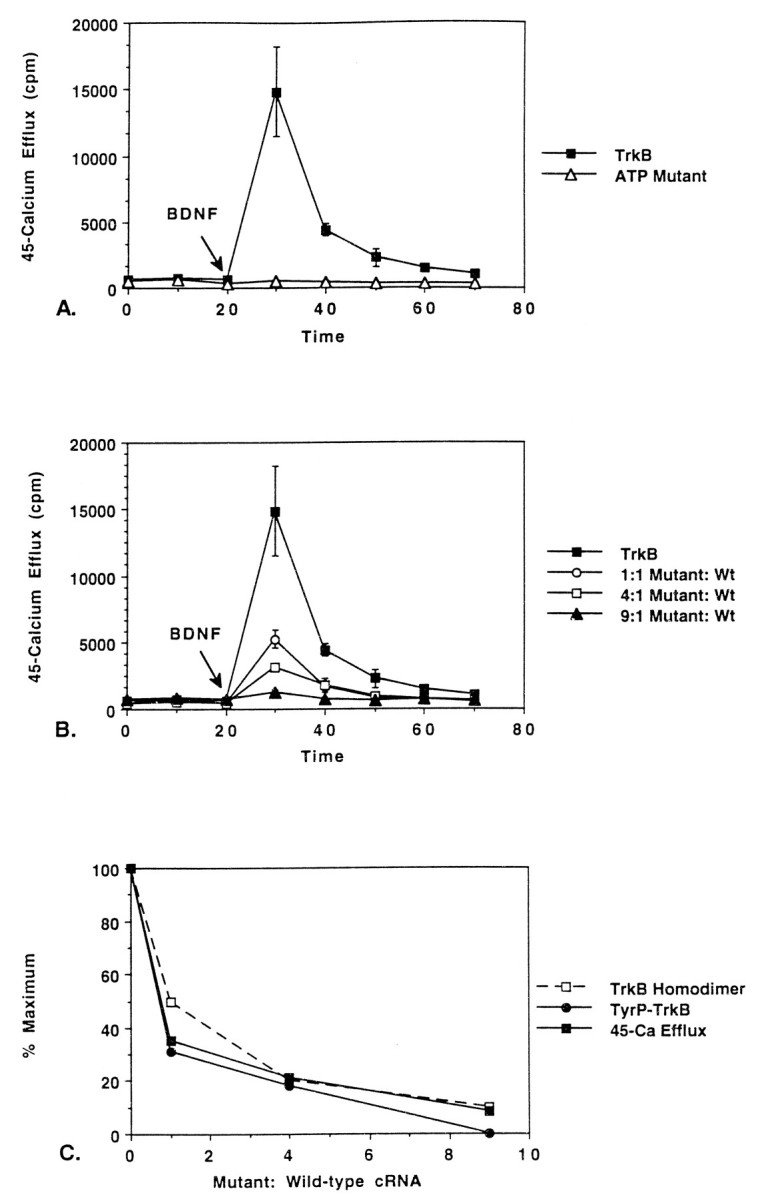
ATP-binding mutant of trkB inhibits BDNF signaling at the level of receptor tyrosine phosphorylation. A, ATP-binding site mutation abolishes BDNF-induced45Ca efflux response. Oocytes were injected with cRNA (2 ng/oocyte) encoding the full-length trkB receptor (closed squares) or its ATP-binding mutant. After45Ca efflux had stabilized, BDNF was added to the medium (250 ng/ml, arrow). B, Dominant inhibitory effect of the ATP-binding mutant. Oocytes were injected with 2 ng/oocyte trkB cRNA + varying quantities of ATP-binding mutant (0, 2, 8, and 18 ng/oocyte). C, Decrease in maximal45Ca efflux responses and trkB tyrosine phosphorylation correlate with a loss of trkB homodimers. A binomial model (see Results) was used to predict the number of trkB homodimers (open squares). Percent maximum trkB tyrosine phosphorylation was determined by labeling trkB ± ATP-binding mutant-expressing oocytes with 35S, stimulating with BDNF, immunoprecipitating lysates with APT antibody, separating on SDS-PAGE gels, and then quantitating bands by PhosphorImager analysis (see Materials and Methods). Data are displayed graphically as percent maximum tyrosine phosphorylation (closed circles). Finally, peak 45Ca efflux responses for ATP-binding mutant-expressing oocytes are plotted as a function of the ratio of truncated:full-length trkB cRNA (closed squares).
Epitope tags distinguish ATP-binding mutant and wild-type trkB receptors
To study mutant–wild-type interactions in microinjected oocytes and transfected cell lines, short epitope tags were attached to the C termini of the trkB ATP-binding mutant and wild-type receptors using PCR (see Materials and Methods). Wild-type trkB receptors were tagged with an epitope from influenza virus hemagglutinin (trkBIVH); ATP-binding site mutants were tagged with an epitope from the c-myc proto-oncogene (ATPmutmyc). Receptors could be distinguished on Western blots of transient COS cell transfections (Fig. 4A,B) or cRNA-injected oocytes (Fig. 5).
Fig. 4.

Epitope tags distinguish the ATP-binding mutant from the wild-type trkB receptor. A, COS cells transfected with either vector control (lane 1) or trkBIVH (lane 2). Lysates were immunoprecipitated with IVH Ab (12CA5), separated by 6% SDS-PAGE, then immunoblotted with IVH Ab. B, COS cells transfected with either vector control (lane 1) or ATPmutmyc (lane 2). Lysates were immunoprecipitated with anti-myc Ab (9E10), separated by 6% SDS-PAGE, and then blotted with anti-myc Abs.
Fig. 5.

Expression levels of wild-type and ATP-binding mutant reflect quantities of cRNA injected. A, IVH ECL blot of oocytes injected with trkBIVH ± ATPmutmyc. Oocytes were injected as described in Materials and Methods, lysed 36 hr later with RIPA buffer, separated on 7% SDS-PAGE gels, and then immunoblotted with IVH Ab (12CA5). Oocytes were injected with sterile water (lane 1), trkBIVH (1 ng; lane 2), or ATPMutmyc + trkBIVH at a ratio of 4:1 (4 ng/1 ng/oocyte; lane 3) or 9:1 (9 ng/1 ng/oocyte;lane 4). Lysates collected from 6 oocytes were loaded per lane. B, Myc ECL blot of oocytes co-injected with trkBIVH ± ATPmutmyc. Oocytes were injected and lysed as described above. Lysates from 2 oocytes were loaded per lane. Proteins were separated on 7% SDS-PAGE gels and then immunoblotted with myc Ab (9E10). Oocytes were injected with trkBIVH only (1 ng/oocyte; lane 1) or ATPMutmyc:trkBIVH at a ratio of 9:1 (9 ng/1 ng/oocyte; lane 2) or 4:1 (4 ng/1 ng/oocyte; lane 3).
In oocyte experiments, presence of the epitope tags allowed us to verify that levels of mutant or wild-type receptor expression reflected ratios of cRNA injected (Fig. 5A,B). Slight decreases in trkBIVH expression could be seen at maximal mutant:wild-type ratios (Fig. 5A, lane 4). The slight limitation in translation could be seen at the high ratios of cRNA required to visualize tagged receptors on blots (fivefold higher cRNA concentrations for Western blotting experiments compared with45Ca efflux studies).
Figure 3 shows that presence of an epitope tag did not appear to interfere with the ability of the receptor to become activated by its ligand. Figure 3B is an APT blot of COS cells transfected with vector control or trkBIVH, and then immunoprecipitated with anti-IVH antibody. Lanes 3 and4 show that cells transfected with trkBIVH demonstrated a specific tyrosine phosphorylation product at 145 kDa, the predicted size of the full-length trkB receptor. Some autophosphorylation was seen in the absence of ligand (lane 3); however, stimulation by BDNF significantly increased this level of receptor tyrosine phosphorylation (lane 4). Because the levels and timing of trkB tyrosine phosphorylation correlated well with previous observations of COS-transfected untagged trkA receptors (Jing et al., 1992), no interference with receptor activation was believed to occur secondary to attachment of the tag. Vector controls showed no reaction (lanes 1, 2).
Fig. 3.
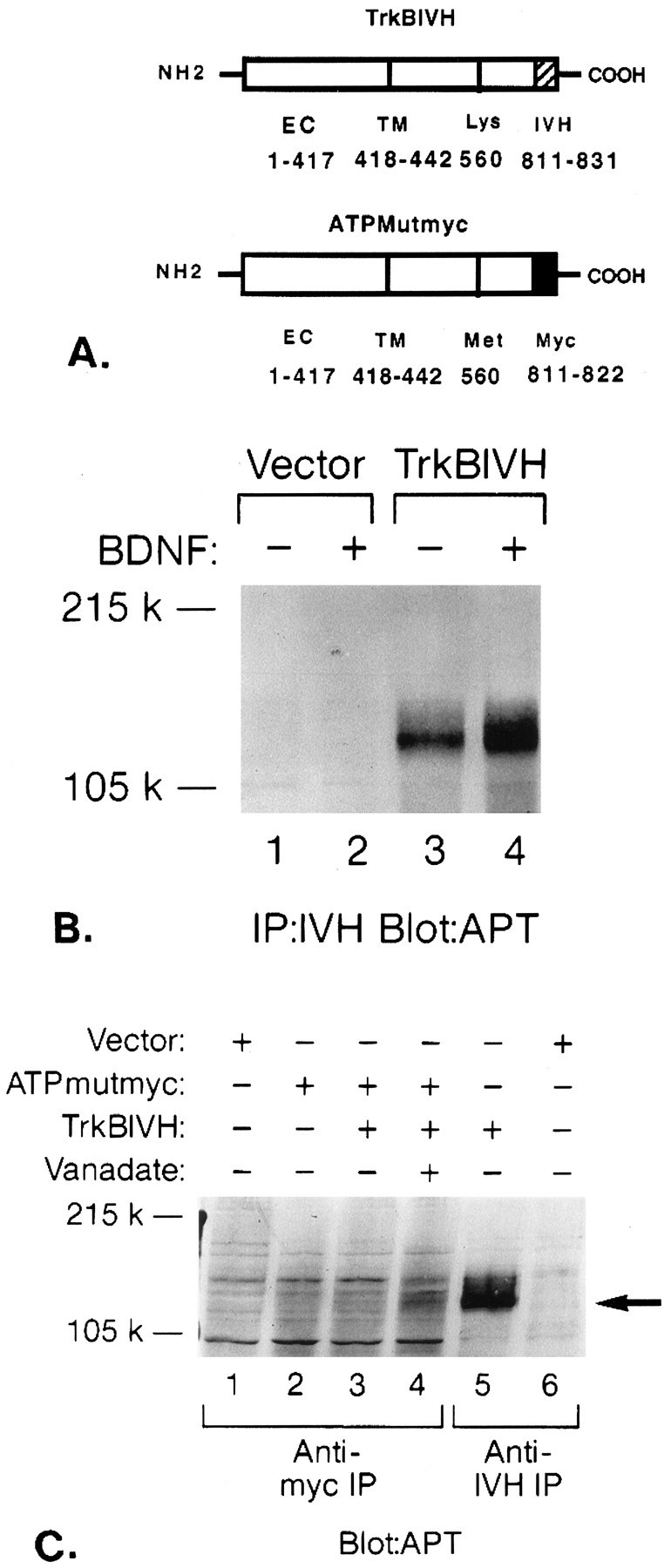
Intermolecular phosphorylation of ATP-binding mutant by wild-type gp145trkB.A, Schematic diagram of epitope-tagged wild-type and mutant trkB receptors. PCR was used to attach an IVH tag to the C terminus of the wild-type trkB receptor (trkBIVH) and myc tag to the C terminus of the ATP-binding mutant (ATPMutmyc) as described in Materials and Methods. Amino acid numbers are listed for the extracellular (EC) or transmembrane domains (TM), ATP-binding site (Lys or Met), and epitope tags (IVH or Myc). B, BDNF induces rapid tyrosine phosphorylation of trkBIVH receptors. COS cells were transfected with vector control (lanes 1, 2) or trkBIVH (lanes 3, 4). Cells in lanes 2 and 4 were stimulated for 5 min with 100 ng/ml BDNF. Lysates were immunoprecipitated with IVH Ab, separated by 6% SDS-PAGE, and then immunoblotted with the APT Ab 4G10. C, Intermolecular tyrosine phosphorylation of ATPMutmyc by trkBIVH. COS cells were transfected with vector alone (lanes 1, 6), ATPMutmyc (lane 2), ATPmutmyc + trkBIVH (lanes 3, 4), or trkBIVH alone (lane 5). Cells in lane 4 were pretreated with 50 mm sodium orthovanadate for 3 hr before lysis. Cells were stimulated with BDNF, then immunoprecipitated with either anti-myc (lanes 1–4) or anti-IVH Abs (lanes 5, 6). Proteins were separated by 6% SDS-PAGE and were then immunoblotted with APT Ab. The location of gp145trkB is indicated by anarrow.
In other experiments, trkBIVH cDNA was stably transfected under neomycin selection in PC12 NNR5 cells (PC12 pheochromocytoma cell line lacking trkA and trkB). Nontransfected PC12 NNR5 cells showed no response to BDNF, whereas PC12NNR5 cells expressing trkBIVH responded with extensive neurite outgrowth after stimulation by picomolar concentrations of BDNF (not shown). From these data, we concluded that tagging trkB receptors did not interfere with transduction of the biological effects of the trkB receptors.
In Figure 3C, presence of the Lys → Met560 substitution at the ATP-binding site blocked BDNF-induced tyrosine phosphorylation of trkB (Fig.3C, lane 2). Adequate expression levels of ATPmutmyc were verified by reprobing blots with anti-myc Abs (data not shown). Also, adequate expression levels of ATPMutmyc in COS were verified in myc blots of immunoprecipitations shown in Figure4B.
Evidence for weak intermolecular phosphorylation between mutant and wild-type trkB receptors
Because previous investigators (Jing et al., 1992) had suggested that noncatalytic trkA mutants could inhibit wild-type (catalytic) trkA receptors, but not at the level of receptor tyrosine phosphorylation, additional mutant–wild-type phosphorylation experiments were performed in transiently cotransfected COS cells. Figure 3C shows an APT blot of cotransfected COS cells immunoprecipitated with either anti-myc or anti-IVH Abs. Cells were transfected with vector control (lane 1), ATPMutmyc (lane 2), or both ATPmutmyc and trkBIVH (lanes 3, 4). Under routine culture conditions (Fig. 3C, lane 3), immunoprecipitation by the anti-myc Ab showed no evidence of mutant receptor tyrosine phosphorylation; however, if cells were pretreated with sodium orthovanadate (phosphatase inhibitor) 3 hr before lysis, then a faint tyrosine-phosphorylated product was seen at 145 kDa (Fig.3C, lane 4).
These data were consistent with previous observations by Jing et al. (1992) involving the trkA receptor. However, because we could observe intermolecular phosphorylation of the trkB ATP-binding mutant at only high levels of receptor overexpression (transient COS transfections) pretreated with tyrosine phosphatase inhibitors (sodium orthovanadate), we were uncertain whether mutant receptors were becoming phosphorylated by wild-type receptors within mutant–wild-type heterodimers (intradimeric), or whether phosphorylation of mutant monomers, heterodimers, or homodimers was occurring in trans(interdimeric) by wild-type homodimers. To distinguish between these two possibilities, additional experiments were performed in microinjected oocytes.
ATP-binding mutant inhibits BDNF signaling at the level of receptor tyrosine phosphorylation
Oocytes were injected with varying ratios of ATP-binding mutant and wild-type trkB receptors, labeled with [35S]methionine, stimulated with BDNF, immunoprecipitated with APT Abs, separated on SDS-PAGE gels, and then quantitated using PhosphorImager analysis. Figure 2Ccompares declines in predicted numbers of trkB homodimers (open squares; see description of model earlier in Results) with peak45Ca efflux levels (closed squares) and extent of trkB tyrosine phosphorylation (closed circles). Declines in trkB tyrosine phosphorylation seen at higher levels of mutant receptor expression in fact closely paralleled declines in peak 45Ca efflux, with intensity of BDNF-induced tyrosine phosphorylation best correlating with predicted losses in numbers of wild-type trkB homodimers (rather than losses of wild-type homodimers + wild-type–mutant heterodimers). Based on these findings, we concluded that the phosphorylation of trkB homodimers was a critical event in activation of BDNF-induced PLC-γ-mediated pathways. Furthermore, we concluded that the dominant negative effect exerted by noncatalytic trkB receptors was most likely attributable to inhibition of the BDNF signaling cascade at the level of receptor tyrosine autophosphorylation. The addition of noncatalytic trkB receptors to cells already expressing wild-type (or kinase-containing) gp145trkB reduces the availability of functional wild-type homodimers in the presence of BDNF stimulation.
DISCUSSION
In this paper, we report that the naturally occurring truncated trkB receptors trkB.T1 and trkB.T2 have dominant inhibitory effects on BDNF-dependent activation of a PLC-γ-dependent signaling pathway. When expressed alone, the truncated receptors are incapable of eliciting 45Ca efflux responses in theXenopus oocyte system; coexpression with gp145trkB, however, causes a dose-dependent inhibition of the BDNF signal. These findings support a role for naturally occurring truncated trkB receptors in the inhibition of neurotrophin responsiveness in vivo. In fact, Kaplan and colleagues have noted a decrease in the apparent responsiveness of cortical tissue to BDNF (as measured by gp145trkBtyrosine phosphorylation) at developmental periods in which increases in the ratio of truncated to full-length trkB receptors are seen (Knusel et al., 1994). Interestingly, cortical tissues appeared to lose responsiveness to BDNF when truncated receptors seemed to outnumber full-length receptors at ratios of 4:1. In our45Ca efflux experiments, expression of similar ratios of trkB.T1 or trkB.T2 to gp145trkBresulted in a dimunition of BDNF responsiveness by 80%.
Until recently, it has been difficult to determine to what extent full-length trk receptors and their truncated isoforms occur within the same neuronal populations. Coexpression has been difficult to determine because of the small size of sequences distinguishing the isoforms. Nevertheless, recent evidence has suggested that selected populations of neurons (e.g., hippocampal pyramidal neurons, dentate granule cells, and neocortical neurons) do coexpress full-length and truncated trkB receptors in vivo under certain circumstances (Kokaia et al., 1993; Rudge et al., 1994; Lindfors et al., 1995). Our data would support a dominant negative function for the truncated isoforms, but we do not exclude the possibility that these receptors could have distinct inhibitory functions in vivo in the modulation of PLC-γ-dependent pathways.
Our finding that levels of observed trkB tyrosine phosphorylation and BDNF-induced 45Ca efflux closely parallel predicted numbers of trkB homodimers supports an essential role for trkB homodimerization in the activation of the BDNF signaling cascade. Noncatalytic trkB receptors can act as dominant negative inhibitors by forming nonfunctional heterodimers with full-length trkB receptors.
In general, the model of BDNF receptor activation presented in this study is in agreement with the model proposed by Barbacid and colleagues for trkA signaling and nerve growth factor (NGF) (Jing et al., 1992). Using stably transfected NIH-3T3 cells, that group had found that the coexpression of noncatalytic and full-length trkA receptors resulted in fewer numbers of NGF-transformed foci. Based on this dominant inhibitory effect, and their observation that gp140trkA molecules were capable of undergoing intermolecular phosphorylation in cotransfected COS, they concluded that trkA homodimerization was responsible for NGF receptor activation.
Our model differs from the one proposed by Jing et al. (1992) in that we believe that noncatalytic trks inhibit wild-type receptor activation at the level of receptor autophosphorylation. The Xenopusoocyte system allowed us to vary ratios of noncatalytic trkB receptors in quantitative assays of trkB tyrosine phosphorylation. Because extent of ligand-induced trkB tyrosine phosphorylation and PLC-γ pathway activation (as measured by 45Ca efflux responses) best correlated with wild-type homodimer number, we believe this species is the critical one for BDNF-induced signaling. In the work byJing et al. (1992), a failure to see declines in wild-type trkA tyrosine phosphorylation (in transient COS transfections) may additionally have been attributable to levels of noncatalytic receptor overexpression at particular ligand concentrations. In experiments involving a kinase-deficient PDGF receptor, Ueno et al. (1991) have reported previously that 10- to 30-fold ratios of truncated:full-length PDGF receptors were needed to block signaling at ligand concentrations of 0.03 nm, compared with 70- to 90-fold ratios at ligand concentrations of 1 nm.
Finally, our finding that a trkB ATP-binding site mutant functions as a dominant inhibitor of BDNF signaling has implications for futurein vivo studies of neurotrophin function. A noncatalytic mutant of the basic fibroblast growth factor receptor has been used to demonstrate a role for this factor in mesodermal induction and skin differentiation (Amaya et al., 1993; Werner et al., 1993).
Footnotes
This research was supported by National Institute on Aging Grant K11AG00568 (F.F.E.), a seed Grant from the Brain Research Foundation, and National Institutes of Mental Health Grant 48200. L.F. Reichardt is an investigator of The Howard Hughes Medical Institute. We thank Drs. R. Kypta, G. Lopez, A. MacNicol, U. Muller, and A. Muslin for many helpful discussions and comments. We also thank Drs. J. Escobedo, T. Hunter, D. Middlemas, W. Mobley, P. Olson, and A. Rosenthal for their generous gifts of constructs and reagents.
Correspondence should be addressed to Dr. Fernette F. Eide, Department of Neurology, MC 2030, University of Chicago, 5841 South Maryland Avenue, Chicago, IL 60637.
REFERENCES
- 1.Allendoerfer KL, Cabelli RJ, Escandon E, Kaplan DR, Nikolics K, Shatz CJ. Regulation of neurotrophin receptors during the maturation of the mammalian visual system. J Neurosci. 1994;14:1795–1811. doi: 10.1523/JNEUROSCI.14-03-01795.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amaya E, Stein PA, Musci TJ, Kirschner MW. FGF signalling in the early specification of mesoderm in Xenopus . Development. 1993;118:477–487. doi: 10.1242/dev.118.2.477. [DOI] [PubMed] [Google Scholar]
- 3.Barde Y-A, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982;1:549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck KD, Lamballe F, Klein R, Barbacid M, Schauwecker PE, McNeill TH, Finch CE, Hefti F, Day JR. Induction of non-catalytic TrkB neurotrophin receptors during axonal sprouting in the adult hippocampus. J Neurosci. 1993;13:4001–4014. doi: 10.1523/JNEUROSCI.13-09-04001.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eide FF, Lowenstein DH, Reichardt LF. Neurotrophins and their receptors: current concepts and implications for neurologic disease. Exp Neurol. 1993;121:200–214. doi: 10.1006/exnr.1993.1087. [DOI] [PubMed] [Google Scholar]
- 6.Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyman C, Juhasz M, Jackson C, Wright P, Ip NY, Lindsay RM. Overlapping and distinct actions of the neurotrophins BDNF, NT-3, and NT-4/5 on cultured dopaminergic and GABAergic neurons of the ventral mesencephalon. J Neurosci. 1994;14:335–347. doi: 10.1523/JNEUROSCI.14-01-00335.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jing S, Tapley P, Barbacid M. Nerve growth factor mediates signal transduction through trk homodimer receptors. Neuron. 1992;9:1067–1079. doi: 10.1016/0896-6273(92)90066-m. [DOI] [PubMed] [Google Scholar]
- 9.Johnson DE, Lee PL, Lu J, Williams LT. Diverse forms of a receptor for acidic and basic fibroblast growth factors. Mol Cell Biol. 1990;10:4728–4736. doi: 10.1128/mcb.10.9.4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell. 1990a;61:647–656. doi: 10.1016/0092-8674(90)90476-u. [DOI] [PubMed] [Google Scholar]
- 11.Klein R, Martin-Zanca D, Barbacid M, Parada LF. Expression of the tyrosine kinase receptor gene trkB is confined to the murine embryonic and adult nervous system. Development. 1990b;109:845–850. doi: 10.1242/dev.109.4.845. [DOI] [PubMed] [Google Scholar]
- 12.Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kokaia Z, Gido G, Ringstedt T, Bengzon J, Kokaia M, Sisjo BK, Persson H, Lindvall O. Rapid increase of BDNF mRNA levels in cortical neurons following spreading depression: regulation by glutamatergic mechanisms independent of seizure activity. Mol Brain Res. 1993;19:277–286. doi: 10.1016/0169-328x(93)90126-a. [DOI] [PubMed] [Google Scholar]
- 14.Koliatsos VE, Clatterbuck RE, Winslow JW, Cayonette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo . Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- 15.Krieg PA, Melton DA. Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res. 1984;12:7057–7070. doi: 10.1093/nar/12.18.7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 17.Leibrock J, Lottspeich F, Hohn A, Hofer M, Gengerer B, Masiakowski P, Thoenen H, Barde Y. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- 18.Lindfors N, Brodin E, Metsis M. Spatiotemporal selective effects on brain-derived neurotrophic factor and trkB messenger RNA in rat hippocampus by electroconvulsive shock. Neuroscience. 1995;65:661–670. doi: 10.1016/0306-4522(94)00550-o. [DOI] [PubMed] [Google Scholar]
- 19.Middlemas DS, Lindberg RA, Hunter T. TrkB, a neural receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Middlemas DS, Meisenhelder J, Hunter T. Identification of TrkB autophosphorylation sites and evidence that phospholipase C-gamma 1 is a substrate of the TrkB receptor. J Biol Chem. 1994;269:5458–5466. [PubMed] [Google Scholar]
- 21.Morse JK, Wiegand SJ, Anderson K, You Y, Cai N, Carnahan J, Miller J, DiStefano PS, Altar CA, Lindsay RM, Alderson RF. Brain-derived neurotrophic factor (BDNF) prevents the degeneration of medial septal cholinergic neurons following fimbria transection. J Neurosci. 1993;13:4146–4156. doi: 10.1523/JNEUROSCI.13-10-04146.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niman HL, Houghten RA, Walker LE, Reisfeld RA, Wilson IA, Hogle JM, Lerner RA. Generation of protein-reactive antibodies by short peptides is an event of high frequency: implication for the structural basis of immune recognition. Proc Natl Acad Sci USA. 1983;80:4949–4953. doi: 10.1073/pnas.80.16.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudge JS, Li Y, Pasnikowski M, Mattsson K, Pan L, Yancopoulos GD, Wiegand SJ, Lindsay RM, Ip NY. Neurotrophic factor receptors and their signal transduction capabilities in astrocytes. Eur J Neurosci. 1994;6:693–705. doi: 10.1111/j.1460-9568.1994.tb00981.x. [DOI] [PubMed] [Google Scholar]
- 24.Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton R, Kaplan DR, Hunter T, Nikolics K, Parada LF. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trk B tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- 25.Squinto SP, Stitt TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, Glass DJ, Masiakowski P, Furth ME, Valenzuela DM, DiStefano PS, Yancopoulos GD. TrkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell. 1991;65:885–893. doi: 10.1016/0092-8674(91)90395-F. [DOI] [PubMed] [Google Scholar]
- 26.Ueno H, Colbert H, Escobedo JA, Williams LT. Inhibition of PDGF beta receptor signal transduction by coexpression of a truncated receptor. Science. 1991;252:844–848. doi: 10.1126/science.1851331. [DOI] [PubMed] [Google Scholar]
- 27.Ueno H, Escobedo JA, Williams LT. Dominant negative mutants of platelet-derived growth factor (PDGF) receptors: inhibition of receptor function by ligand-dependent formation of heterodimers between PDGF alpha- and beta-receptors. J Biol Chem. 1993;30:22814–22819. [PubMed] [Google Scholar]
- 28.Werner S, Weinberg W, Liao X, Peter KG, Blessing M, Yuspa SH, Weiner RC, Williams LT. Targeted expression of a dominant-negative FGF receptor mutant in the epidermis of transgenic mice reveals a role of FGF in keratinocyte organization and differentiation. EMBO J. 1993;12:2635–2643. doi: 10.1002/j.1460-2075.1993.tb05924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White MF. Structure and function of tyrosine kinase receptors. J Bioenerg Biomembr. 1991;23:63–82. doi: 10.1007/BF00768839. [DOI] [PubMed] [Google Scholar]
- 30.Widmer HR, Kaplan DR, Rabin SJ, Beck KD, Hefti F, Knusel B. Rapid phosphorylation of phospholipase C-gamma 1 by brain-derived neurotrophic factor and neurotrophin-3 in cultures of embryonic rat cortical neurons. J Neurochem. 1993;10:2111–2123. doi: 10.1111/j.1471-4159.1993.tb03496.x. [DOI] [PubMed] [Google Scholar]
- 31.Williams JA, McChesney DJ, Calayag MC, Lingappa VR, Logsdon CD. Expression of receptors for cholesystokinin and other Ca2+-mobilizing hormones in Xenopus oocytes. Proc Natl Acad Sci USA. 1988;85:4939–4943. doi: 10.1073/pnas.85.13.4939. [DOI] [PMC free article] [PubMed] [Google Scholar]