

Summary
Glial cell line–derived neurotrophic factor (GDNF) is a distant member of the TGFβ protein family that is essential for neuronal survival and renal morphogenesis. We show that mice who are deficient in the glycosyl-phosphatidyl inositol (GPI) -linked protein GFRα1 (GDNFRα) display deficits in the kidneys, the enteric nervous system, and spinal motor and sensory neurons that are strikingly similar to those of the GDNF-and Ret-deficient mice. GFRα1-deficient dopaminergic and nodose sensory ganglia neurons no longer respond to GDNF or to the structurally related protein neurturin (NTN) but can be rescued when exposed to GDNF or neurturin in the presence of soluble GFRα1. In contrast, GFRα1-deficient submandibular parasympathetic neurons retain normal response to these two factors. Taken together with the available genetic and biochemical data, these findings support the idea that GFRα1 and the transmembrane tyrosine kinase Ret are both necessary receptor components for GDNF in the developing kidney and nervous system, and that GDNF and neurturin can mediate some of their activities through a second receptor.
Introduction
Glial cell line–derived neurotrophic factor (GDNF) (Lin et al., 1993), neurturin (NTN) (Kotzbauer et al., 1996), and persephin (PSP) (Milbrandt et al., 1998) constitute a class of secreted proteins that is structurally related to the transforming growth factor protein family. Studies in primary neuronal cultures, as well as in lesioned animal models, have provided evidence that GDNF is a survival factor for embryonic midbrain dopaminergic neurons that degenerate in Parkinson’s disease (Lin et al., 1993; Beck et al., 1995; Tomac et al., 1995), spinal motor neurons that degenerate in amyotrophic lateral sclerosis and spinal muscular atrophies (Henderson et al., 1994; Oppenheim et al., 1995; Yan et al., 1995), locus coeruleus noradrenergic neurons (Arenas et al., 1995), and subpopulations of peripheral sensory, sympathetic, and parasympathetic neurons (Buj-Bello et al., 1995; Trupp et al., 1995). Likewise, NTN was shown to be effective in attenuating the death of cultured embryonic dopaminergic, motor (Klein et al., 1997), sympathetic, and sensory neurons (Kotzbauer et al., 1996), while PSP was shown to promote the survival of cultured dopaminergic and motor, but not peripheral, neurons (Milbrandt et al., 1998).
The essential physiological role of the GDNF/NTN/PSP protein family is illustrated by the phenotype of mice in whom the GDNF gene has been disrupted. These mice display deficits in primary sensory, sympathetic, and motor neurons and also fail to develop metanephric kidneys, ureters, and most of the enteric nervous system (Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996). Consequently, although these mice are born, they die shortly after birth, owing to an inability to consume milk and to the lack of renal function.
Despite the physiological and clinical significance of the GDNF protein family, the mechanism by which these growth factors transduce signals is not fully understood. Biochemical and cell culture studies have suggested that GDNF and NTN bind one of several glycosyl-phosphatidyl inositol (GPI) -linked proteins (designated GDNF family receptors GFRα1–4) and that they also require the presence of the transmembrane tyrosine kinase Ret for signal transduction and neuronal survival (Jing et al., 1996; Treaner et al., 1996; Baloh et al., 1997; Buj-Bello et al., 1997; Klein et al., 1997; Sanicola et al., 1997; Masure et al., 1998; Naveilhan et al., 1998; Thompson et al., 1998; Worby et al., 1998) (data not shown). These studies further revealed that GDNF and NTN facilitate the formation of a physical complex between GFRα and Ret and lead to activation of the Ret tyrosine kinase (Jing et al., 1996; Treanor et al., 1996; Klein et al., 1997). Taken together, the findings supported the proposal that cellular responses to the GDNF protein family are mediated through multicomponent receptor complexes composed of a shared signaling subunit, the orphan tyrosine kinase, Ret, and one of several ~55 kDa ligand binding subunit proteins, which do not have an intracellular domain, and which bind the extracellular membrane leaflet via the GPI lipid modification.
Despite the available information, several major issues concerning the validity of the multicomponent receptor model remained. Most importantly, to date there is no evidence that the GPI-linked proteins are, in fact, functional and necessary receptor subunits for the GDNF protein family in vivo. In addition, the ligand specificity of the endogenous GPI-linked proteins remained unclear—this since, in a cell free system, recombinant GDNFRα/GFRα1 and NTNRα/GFRα2 displayed a high degree of binding specificity for GDNF and NTN, respectively, when tested alone (Klein et al., 1997) but bound GDNF equally well in the presence of Ret (Sanicola et al., 1997). Likewise, GDNF and NTN were shown to be equally effective in activating Ret through GFRα1, and NTN appeared only 30-fold more efficient than GDNF in activating Ret through GFRα2 in a fibroblast cell line (Baloh et al., 1997). Nonetheless, in primary neurons, GDNF displayed a preference for GFRα1, and NTN could promote survival only through GFRα2 (Buj-Bello et al., 1997).
To examine the physiological significance and ligand specificity of the GFRα proteins, we have generated and analyzed mice who are deficient in GFRα1 (GFRα1−/−). We show that GFRα1−/− mice display neuronal and renal deficits that are strikingly similar, but not identical to, those of the GDNF−/− and Ret−/− mice. Moreover, nodose ganglia and midbrain dopaminergic neurons derived from the GFRα1−/− embryos no longer survive in the presence of GDNF and NTN, whereas the response of GFRα1−/− submandibular parasympathetic neurons to these two factors is indistinguishable from that of their wild-type counterpart. The findings verify the physiological importance of the GFRα receptors and validate the multicomponent receptor hypothesis for the GDNF protein family; they further support the idea that, although GDNF and NTN display receptor preferences, they can use multiple GFRα receptors in vivo.
Results
Generation of the GFRα1−/− Mice
DNA fragments containing the first three of nine exons in the GFRα1 gene (Eng et al., 1998) were isolated and used to generate a targeting construct in which part of exon 2, encoding for amino acid 14–66, had been deleted (Figure 1A). This targeting construct was electroporated into embryonic stem (ES) cells (Moore et al., 1996), and clones in which the GFRα1 gene had been disrupted by homologous recombination (Figure 1B) were injected or aggregated into blastocysts to produce GFRα1 mutant mice (Figure 1C).
Figure 1. Disruption of the GFRα1 Gene.
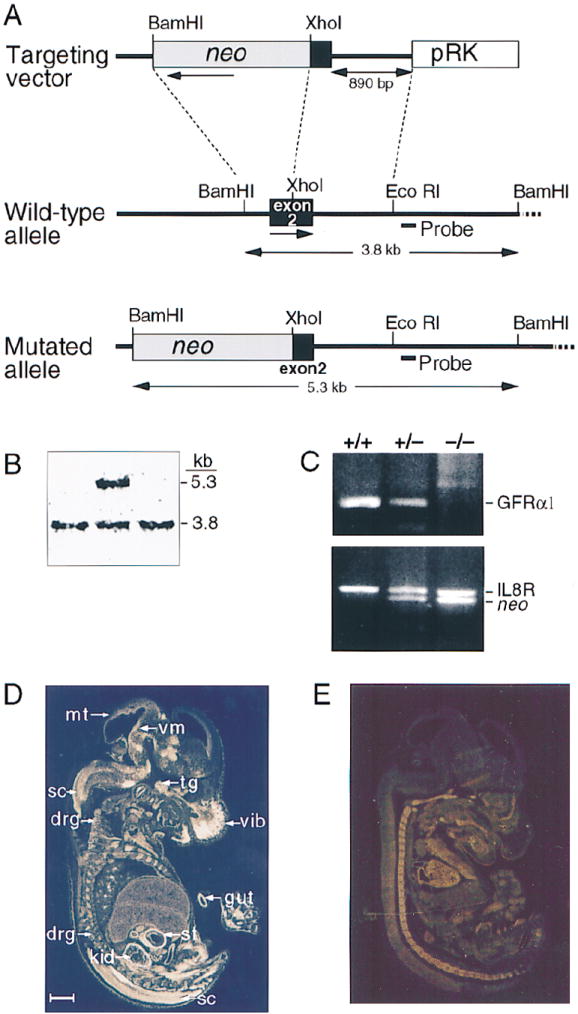
(A) Targeting vector, wild-type GFRα1 allele and the disrupted allele. Amino acids 14–66 are missing from the disrupted gene. The location of the probe used in Southern blot is indicated (Probe). The directions of gene transcription are marked by horizontal arrows.
(B) Detection of homologous recombination event in an ES clone by a Southern blot.
(C) Genotype analysis of wild-type (+/+), heterozygous mutant (+/−), and homozygous mutant (−/−) animals by polymerase chain reaction. The band in the upper panel (GFRα1) is specific for the wild-type GFRα1 gene. The upper band in the lower panel (IL8R) represents a control fragment from the IL8 receptor gene. The lower band in this panel (neo) is specific for the neo gene.
(D and E) In situ hybridization of wild-type (D) and GFRα1−/− (E) E15 mouse embryos with exon 2 GFRα1 probe. Abbreviations: drg, dorsal root ganglia; gut, gut; kid, kidney; sc, spinal cord; st, stomach; tg, trigeminal gangilon; vib, vibrissa; and vm, ventral midbrain. Scale bar, 1 mm.
Whereas mRNA for GFRα1 was found in the kidney, gut, and nervous system of wild-type and GFRα1 heterozygous embryos by in situ hybridization, no GFRα1 transcripts encoding amino acids 14–66 were detected in null, mutant littermates (Figures 1D and 1E). Heterozygous mice were viable, normal in size, fertile, and did not display any gross morphological or behavioral abnormalities. In contrast, GFRα1−/− mice died 1–1.5 days after birth, even though they were initially able to suckle and had normal limb and body movements.
Neuronal Deficits in the GFRα1 Null Mice
Since GDNF, NTN, and PSP are potent survival factors for embryonic midbrain dopaminergic (Lin et al., 1993; Beck et al., 1995; Tomac et al., 1995; Milbrandt et al., 1998), noradrenergic (Arenas et al., 1995), motor (Henderson et al., 1994; Oppenheim et al., 1995; Yan et al., 1995), sensory, and sympathetic (Buj-Bello et al., 1995; Trupp et al., 1995; Kotzbauer et al., 1996) neurons in culture and/or in animal models in vivo, we first examined whether the GFRα1−/− mutant mice displayed any neuronal deficits. As was observed in the GDNF−/− mice (Moore et al., 1996; Sánchez et al., 1996), the GFRα1−/− embryos exhibited small losses of lumbar spinal (24%) and trigeminal nucleus (22%) motor neurons, but not of facial motor neurons (Table 1). In addition, like their GDNF−/− counterparts, the GFRα1−/− embryos had a normal complement of tyrosine hydroxylase–positive dopaminergic neurons in the substantia nigra (Table 1; Figures 2A and 2B), did not display a significant reduction in the density of dopaminergic projections in the striatum (Table 1; Figures 2C and 2D), and possessed a normal number of noradrenergic neurons in the locus coeruleus (Table 1).
Table 1.
Neuronal Counts
| CNS catecholaminergic neurons | +/+ | −/− | GRFα−/− % Deficit |
| Dopaminergic (SN) | 4822 ± 336 (3) | 5060 ± 405 (3) | n.s. (n.s.) |
| Noradrenergic (LC) | 1912 ± 82 (3) | 2198 ± 130 (3) | n.s. (n.s.) |
| Sensory ganglia | |||
| Trigeminal | 42700 ± 996 (3) | 39886 ± 2048 (3) | n.s. (n.s.) |
| Vestibular | 3804 ± 339 (3) | 4060 ± 134 (5) | n.s. (n.s.) |
| Petrosal-Nodose | 7884 ± 403 (3) | 6714 ± 127 (5) | 15** (40%) |
| L5 dorsal root | 7428 ± 493 (3) | 7486 ± 925 (3) | n.s. (23%) |
| Sympathetic ganglia | |||
| Superior cervical | 21790 ± 616 (3) | 21364 ± 938 (3) | n.s. (35%) [100%] |
| Motor nuclei | |||
| Facial (VII) | 4586 ± 251 (4) | 4236 ± 148 (6) | n.s. (n.s.) |
| Trigeminal (V) | 1047 ± 27 (4) | 816 ± 20 (6) | 22*** (19%) |
| Spinal lumbar (L1–L6) | 3272 ± 92 (3) | 2503 ± 320 (4) | 24* (22%) |
Cell counts are expressed as the mean number of neurons ± SEM. The number of F2 129 × CD-1 animals that were analyzed is shown in parentheses. The percent values depicted in parentheses represent the observed deficits in the GDNF−/− embryos. The percent values depicted in brackets represent the published deficits in the Ret−/− embryos.
One-tailed Student’s t test: n.s., not significant,
p < 0.05,
p < 0.01,
p < 0.001.
Figure 2. Neuronal Populations in P0 wild-type (+/+) and GFRα1−/− (−/−) Mice.
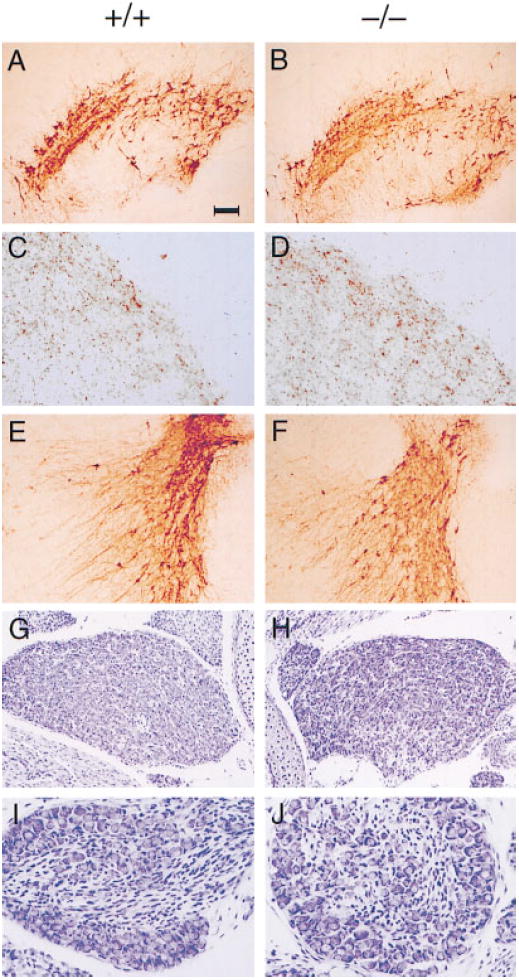
Tyrosine hydroxylase staining of substantia nigra (A and B), striatum (C and D), and locus coeruleus (E and F). Tyrosine hydroxylase is the rate-limiting enzyme in dopamine and noradrenaline synthesis. Cresyl violet staining of superior cervical ganglia (G and H) and petrosal nodose ganglia (I and J) neurons from 129 × CD-1, F2 mice. No deficits or abnormalities in neuronal number, morphology, or innervation pattern are detected in the GFRα1−/− mice. Scale bar: ~100 μm in (A), (B), (E), and (F), 30 μm in (C), (D), (I), and (J), and 50 μm in (G) and (H).
Surprisingly, however, a comparison of GDNF−/− and GFRα1−/− embryos showed differences in other neuronal populations. For instance, whereas the GDNF−/− embryos possessed a 23% deficit in the L5 dorsal root ganglia sensory neurons, the GFRα1−/− embryos had a normal complement of this neuronal population. Similarly, although the GDNF−/− mice displayed a decrease of 40% in the number of petrosal-nodose sensory ganglia neurons, the size of this ganglion was reduced by only 15% in the GFRα1−/− mice. Finally, even though the GDNF−/− and the Ret−/− embryos suffered 35% and 100% losses, respectively, in the sympathetic superior cervical ganglion neurons (Durbec et al., 1996; Moore et al., 1996), GFRα1−/− mice did not display any significant loss of these neurons (Table 1; Figures 2E–2H; data not shown).
The fact that GDNF−/− and the GFRα1−/− mice display mild but identical deficits in the number of lumbar spinal and trigeminal motor neurons is consistent with the notion that GFRα1−/− serves as an essential receptor for GDNF in these neuronal populations. However, the findings that neither the absence of GDNF (Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996), nor the absence of its putative receptor subunit (Table 1; Figure 2), led to a profound degree of neuronal cell losses outside the enteric nervous system suggests that although GDNF can promote the survival of multiple neuronal populations in vitro, it is not a predominant survival factor for central or peripheral neurons in vivo. Since GDNF−/− embryos suffer a more severe loss of petrosal-nodose, dorsal root, and superior cervical ganglia neurons, as compared with the GFRα1−/− mice, some of the survival effects of GDNF on these neuronal populations may be mediated by a second receptor.
The finding that the GFRα1 (no deficit) and GDNF (35% deficit) null mice each had a less severe neuronal loss of superior cervical ganglia neurons than their Ret null counterpart (100% deficit) (Table 1) is in agreement with the hypothesis that Ret is a shared signaling component for the GDNF protein family that can act in conjunction with multiple GFR subunits. Although only a limited survey of neuronal deficiencies has been published for the Ret−/− embryos (Schuchardt et al., 1994; Durbec et al., 1996), these findings suggest that the neuronal deficits in the Ret−/− mice will be at least as severe as those found in their GFRα1 and GDNF counterparts.
Enteric Nervous System Deficits
As GDNF (Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996) and Ret (Schuchardt et al., 1994; Durbec et al., 1996) were shown to be essential for the development of the enteric nervous system, we next determined whether ablation of GFRα1 would lead to deficits in this tissue. In E17 wild-type and GFRα1+/− mice, the neural crest–derived enteric neurons belonging to the myenteric (Auerbach) and submucosal (Meissner) plexi were readily visible along the length of the gastrointestinal tract (Figure 3; data not shown). In contrast, these neurons were completely absent in the intestines and colons of age-matched GFRα1−/− littermates (Figure 3). In addition, the GFRα1−/− animals displayed only a small number of neurons in the stomach (part of the foregut) (data not shown). The absence of myenteric and submucosal neurons in the intestines of the GFRα1−/− mice, and the presence of some neurons in the stomachs of these animals, concurs with previous observations in the GDNF−/− (Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996) and Ret−/− (Schuchardt et al., 1994; Durbec et al., 1996) embryos.
Figure 3. Enteric Nervous System in Wild-Type (+/+) and GFRα1−/− (−/−) Mice.
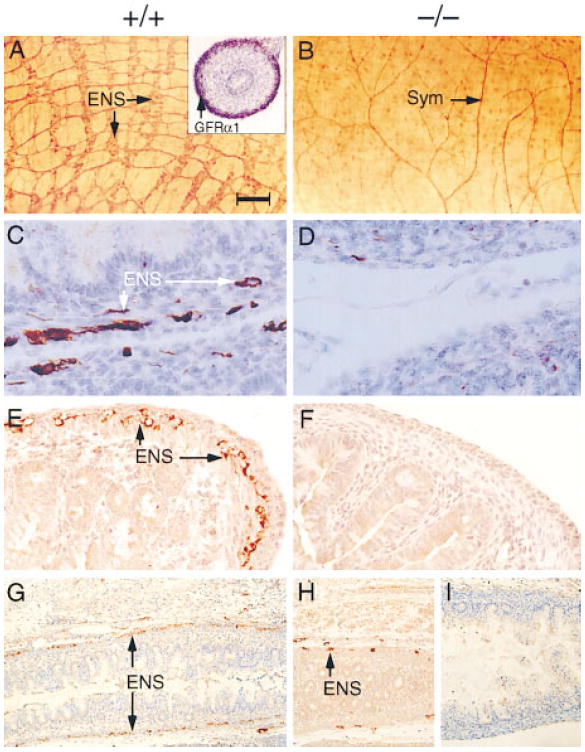
(A and B) Whole mounts of small intestine from E18 mice stained with the general neuronal antibody PGP 9.5. Inset in (A) depicts the expression of GFRα1 mRNA in E18 wild-type mouse gut as detected by in situ hybridization.
(C–I) Section through the small intestine (C and D), colon (E and F), and rectum (G–I) of E17 embryos stained with neurofilament (C and D) or peripherin (E–I). Enteric neurons (ENS) were not found in the intestine and are very rarely found in the stomach and colon of the GFRα1−/− 129 × CD-1, F2 mice. No ENS neurons were detected in the colon of the GDNF−/− mice.
(I) Sym represents afferent fibers probably derived from sympathetic innervation to the gut. Scale bar: ~100 μm in (A) and (B), 30 μm in (C) and (D), 50 μm in (E) and (F), and 300 μm in (G) through (J).
Taken together, these findings strengthen the notion that GFRα1, Ret, and GDNF act in the same signaling pathway and support the idea that a pool of neural crest cells that is derived from the postotic hindbrain and is dependent on the GDNF signal gives rise to most of the enteric nervous system, whereas a distinct lineage that is derived from trunk neural crest and is not completely dependent on the GDNF signal contributes to the enteric nervous system in the foregut (Durbec et al., 1996).
Surprisingly, in the GFRα1−/− (Figure 3H) but not in GDNF−/− (Figure 3I) embryos, a small number of enteric neuron cell bodies were detected in the descending colon, sigmoid colon, and rectum. Thus, it appears as if a subpopulation of enteric neurons that reside in these derivatives of the hindgut may respond to GDNF in part, through a second receptor.
Renal Deficits
Studies in embryonic kidney cultures have demonstrated that PSP and GDNF can promote the outgrowth and branching of the ureteric bud from the nephric ducts (Milbrandt et al., 1998). In addition, GDNF (Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996) and Ret (Schuchardt et al., 1994) have been shown to be essential for the development of ureters and kidneys by gene targeting. We therefore further examined the GFRα1−/− mice for renal abnormalities.
In agreement with the hypothesis that GFRα1 is an essential receptor component for GDNF in the developing kidney, most of the GFRα1−/− animals (13 of 17) had complete bilateral renal and uretal deficits. In the remaining GFRα1−/− embryos (4 of 17), one rudimentary kidney was detected. Other organs that are derived from the embryonic urogenital intermediate mesoderm, including the pro- and mesonephros, the adrenal glands, and the gonads, as well as the remaining abdominal viscera and thoracic tissues, appeared normal (Figures 4A and 4B; data not shown). Anatomical examination of the GFRα1−/− mice at E12.5, when the metanephric kidney forms, demonstrated the presence of a mesonephric duct and undifferentiated kidney mesoderm but not of a morphologically defined ureter, ureteric bud, or nephrons (Figures 4C and 4D). Moreover, Pax2, a homeo-domain transcription factor that is initially expressed in the early ureteric epithelium and is then induced in the nephrogenic region of the metanephrogenic blastema (Dressler and Douglass, 1992; Rothenpieler and Dressler, 1993), was absent from the kidneys of the E12.5 GFRα1−/− embryos (Figures 4E and 4F). In contrast, the Wilm’s tumor suppressor gene and putative transcriptional repressor WT1, which is initially expressed in the uninduced kidney mesenchyme (Kreidberg et al., 1993), was found in the kidney region of both the wild-type and GFRα1−/− E12.5 embryos (Figures 4G and 4H).
Figure 4. Kidneys in Wild-Type and GFRα1−/− Mice.
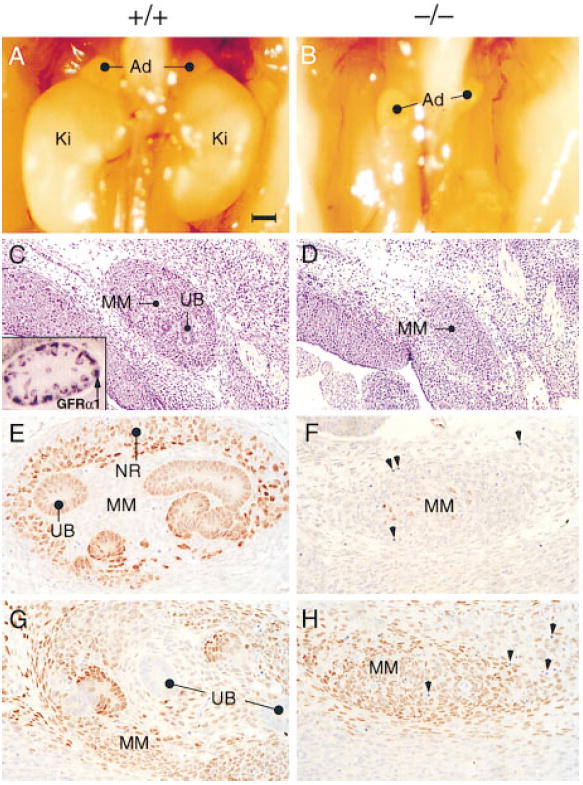
(A and B) Photographs of the abdomen in E17 wild type (A) and GDNF−/− (B) 129 × CD-1, F2 mice. Note the position of the kidneys (Ki) subadjacent to the adrenals (Ad) in (A) and their absence in the mutant (B).
(C–H) Sagittal sections through the kidney region of E12.5 wild-type (+/+) and GFRα1−/− (−/−) embryos stained with hematoxylin and eosin (C and D), Pax2 antibodies (E and F), or WT1 antibodies (G and H). Inset in (C) represents in situ hybridization of GFRα1 cDNA probe to developing nephrons and ureteric bud in wild-type kidney. Abbreviations: UB, uretric bud; MM, metanephric (condensing) mesenchyme; and NR, nephrogenic region (the region that undergoes mesenchymal-to-epithelial conversion and differentiated nephrons). Scale bar: ~500 μm in (A) and (B), 100 μm in (C) and (D), and 20 μm in (E) through (H).
The mammalian kidney develops by reciprocal inductive interactions between the ureteric bud, which is an evagination of the mesonephros/Wolffian duct, and the metanephrogenic blastema, a caudal intermediate mesodermal tissue. The metanephrogenic blastema (which makes GDNF) is thought to induce the ureteric bud (which makes GFRα1 and Ret) to form collecting ducts/ureter. The differentiated ureteric bud, in turn, induces the metanephrogenic blastema to form nephrons (Saxen, 1987). The absence of Pax2-positive ureteric bud cells in GFRα1−/− mice suggests that the ureteric bud either did not branch from the nephric duct or degenerated shortly after its formation. In the absence of differentiated ureters, the renal parenchyma most likely does not express Pax2 and will not undergo a mesenchymal-to-epithelial conversion. Consequently, no differentiated nephrons will be formed. The fact that WT1 is expressed in the metanephrogenic blastema of the GFRα1−/− mice is consistent with the notion that this tissue is dedicated to becoming kidney independent of the ureteric bud and of the GDNF signal.
The Response of GFRα1 Null Neurons to GDNF
To further elucidate whether the neuronal and renal deficits that were observed in the GFRα1−/− mice were indeed caused by a failure in the reception of the GDNF signal, we examined the response of cells derived from the GFRα1-deficient mice to this factor. Embryonic, nodose sensory ganglia neurons were dissected from wild-type, GFRα1+/−, and GFRα1−/− embryos, and their survival in the presence of GDNF and other neurotrophic factors was examined. The majority of wild-type and GFRα1−/− nodose neurons survived in the presence of brain-derived neurotrophic factor, which mediates its signal through the tyrosine kinase receptor TrkB (Klein et al., 1991; Soppet et al., 1991) (Figure 5A). Likewise, GDNF prevented the death of nodose neurons that were derived from wild-type or GFRα1+/− neurons (Figure 5A; data not shown). However, consistent with the idea that GFRα1 is an essential GDNF receptor component, GDNF failed to rescue the majority of GFRα1-deficient nodose neurons, even when applied at high concentrations (50 ng/ml) (Figure 5A). Likewise, primary embryonic dopaminergic neurons that were derived from wild-type embryos survived in the presence of GDNF, while similar neurons that originated from GFRα1−/− littermates no longer responded to this factor at any of the concentrations tested (Figure 5B). Neuronal survival in the presence of GDNF was, however, restored following the addition of exogenous, soluble, recombinant GFRα1 to the GFRα1-deficient neurons (Figure 5B), supporting the idea that these neurons failed to survive in the presence of GDNF solely owing to the absence of GFRα1, and that they did not degenerate at earlier embryonic stages.
Figure 5. Survival of Primary Embryonic Neurons from Wild-Type and GFRα1−/− Mice in the Presence of GDNF.
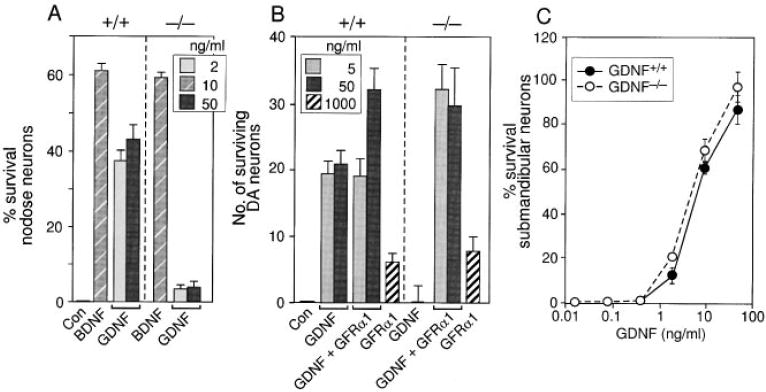
The response of primary embryonic wild-type (+/+) and GFRα1−/− (−/−) nodose (A), dopaminergic (B), and submandibular (C) neurons from 129 × CD-1, F2 mice to GDNF. Neuronal survival is presented as percent or absolute number over control.
Surprisingly, although the response of both nodose and dopaminergic neurons to GDNF was completely dependent on GFRα1, we found that GFRα1−/− parasympathetic, submandibular neurons survived in the presence of GDNF as well as did their wild-type counterparts (Figure 5C). Taken together, these findings support the hypothesis that GFRα1 is an essential GDNF receptor component in many, but not all, populations of neuronal cell types. In addition, it appears as if distinct classes of neurons and nonneuronal cells may be able to respond to GDNF, possibly with a lower sensitivity, via an alternative receptor.
The Response of GFRα1−/− Neurons to NTN
In view of reports that cells expressing GFRα1 and Ret can respond to the GDNF-related protein NTN (Baloh et al., 1997), we have determined whether the survival of GFRα1−/− neurons is still stimulated by this factor.
Examination of wild-type embryonic nodose sensory ganglia neurons revealed that at low concentrations (2 ng/ml), NTN rescued only 25% of the neurons that were rescued by a similar concentration of GDNF. In contrast, at higher concentrations (50 ng/ml), NTN was able to rescue most of the GDNF-responsive neurons (Figure 6A). Nodose ganglia neurons from the GFRα1−/− retained their response to the low concentrations of NTN, but no longer responded to high concentrations of this factor (Figure 6A). Thus, it appears as if NTN promotes survival of nodose neurons through two distinct receptors, a high affinity receptor (possibly GFRα2 [Buj-Bello et al., 1997; Klein et al., 1997]), which might be expressed by a small subpopulation of the embryonic nodose neurons, and GFRα1, which appears to be more abundantly expressed, and which functions as a lower affinity receptor for NTN in this neuronal cell type.
Figure 6. Survival of Primary Embryonic Neurons from Wild-Type and GFRα1−/− Mice in the Presence of NTN.
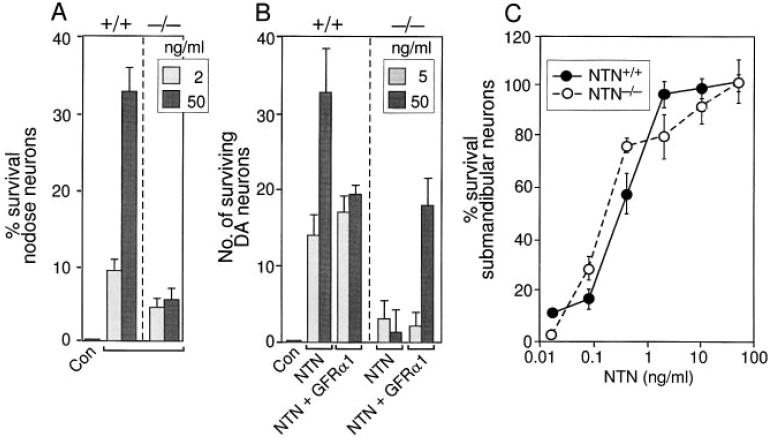
The response of primary embryonic wild-type (+/+) and GFRα1−/− (−/−) nodose (A), dopaminergic (B), and submandibular (C) neurons from 129 × CD-1, F2 mice to NTN. Neuronal survival is presented as percent or absolute number over control.
Analysis of wild-type embryonic midbrain dopaminergic neurons revealed that this neuronal population survives equally well in the presence of NTN and GDNF (Figure 6B; data not shown). Surprisingly, given previous evidence that NTN could mediate its activities through a distinct receptor (Buj-Bello et al., 1997; Klein et al., 1997), neither GDNF (Figure 5B) nor NTN (Figure 6B) were able to support the survival of dopaminergic neurons that were taken from GFRα1−/− embryos. As before, the survival of GFRα1−/− dopaminergic neurons in the presence of NTN was restored following addition of exogenous, soluble recombinant GFRα1 (Figure 6B). Thus, GFRα1−/− functions as a receptor for both NTN and GDNF in developing dopaminergic neurons in vitro.
Among the neuronal populations tested, primary embryonic, parasympathetic submandibular neurons were found to be most sensitive to NTN (Figure 6C). Whereas GFRα1−/− nodose and dopaminergic neurons no longer respond to NTN, the ability of GFRα1−/− parasympathetic submandibular neurons to survive in the presence of this factor, even at very low concentrations (0.08 ng/ml), was indistinguishable from that of wild-type neurons (Figure 6C). Thus, the response of submandibular neurons to NTN does not require GFRα1 and must be mediated by a distinct receptor that could be GFRα2 (Buj-Bello et al., 1997; Klein et al., 1997) or GFRα3 (Masure et al., 1998; Naveilhan et al., 1998; Worby et al., 1998). Interestingly, although GFRα1−/− submandibular neurons survive in the presence of GDNF as well as their wild-type counterparts, GDNF was less effective than NTN in promoting the survival of this neuronal population. Thus, in contrast to NTN, GDNF did not elicit significant survival of parasympathetic submandibular neurons at 0.08 ng/ml or 0.4 ng/ml (Figure 5C) and was effective only at concentrations above 2 ng/ml. The fact that GDNF was less potent than NTN in promoting the survival of submandibular neurons is consistent with the idea that the receptors present on these cells are preferentially activated by NTN.
Taken together, these data favor the hypothesis that GFRα1 is the major high affinity receptor for GDNF on most cell types (Jing et al., 1996; Treanor et al., 1996; Buj-Bello et al., 1997), but that GDNF can also interact with GFRα2 (Baloh et al., 1997; Sanicola et al., 1997) or other GFRα receptors. Conversely, it appears as if NTN has a high affinity receptor (Buj-Bello et al., 1997; Klein et al., 1997) but can mediate signals also through GFRα1 in vitro (Baloh et al., 1997) and possibly in vivo.
Discussion
By creating GFRα1−/− mice, we were able to demonstrate that GFRα1 is essential for GDNF signal transduction in the developing kidney and enteric nervous system, and in subpopulations of motor, sensory, and sympathetic neurons. These findings substantiate the hypothesis that the receptors for the GDNF protein family are composed of two subunits; a GPI-linked ligand-binding protein that belongs to the GFRα family and a signaling component that is represented by the transmembrane tyrosine kinase Ret.
Analyses of the GFRα1−/− mice further revealed that GFRα1 can function as a receptor for other GDNF-like proteins, such as NTN, and that in some cell types, GDNF can elicit a response through an alternative receptor, possibly another member of the GFR family.
Comparing the GFRα1−/−, Ret−/−, and GDNF−/− Mice
The striking similarities in the phenotypes of the GFRα1−/−, Ret−/−, and GDNF−/− mice, which include deficits in the kidneys and enteric neurons, strongly support the proposal that these three molecules are components of the same signaling cascade, and that Ret and GFRα1 serve as coreceptors for GDNF. Mechanistically, the fact that GFRα1−/−, GDNF−/−, and Ret−/− mice all display a similar loss-of-function phenotype indicates that GFRα1 acts as a coactivator, rather than as a ligand-regulated suppresser of Ret, as suggested for other coreceptors (Stone et al., 1996).
Despite the overall similarities in phenotype, some differences between these mice are notable. First, whereas the GFRα1−/− mice have a normal complement of superior cervical ganglion neurons (Table 1), there is a partial (35%) loss of this neuronal population in GDNF−/− embryos (Moore et al., 1996), and a complete loss in the Ret−/− mice (Durbec et al., 1996). Likewise, while the GDNF−/− embryos display deficits of 40% and 23%, respectively, in the number of petrosal-nodose and dorsal root ganglia neurons, the GFRα1−/− mice display only a small deficit (15%) in the number of petrosal-nodose ganglia neurons, and they have a normal complement of dorsal root ganglia neurons.
We cannot exclude the possibility that these differences stem in part from the genetic background of these mice. Nevertheless, the severity of the phenotype of the Ret−/− mice, when compared with that of the GFRα1−/− mice, is consistent with the idea that Ret is an essential, shared signaling component for the GDNF family of receptors (Jing et al., 1996; Treanor et al., 1996; Buj-Bello et al., 1997; Klein et al., 1997). Likewise, the fact that GDNF−/− mice display more significant neuronal deficits, as compared with their GFRα1−/− counterparts, supports the proposal that GDNF can mediate signals through GFRα2 (Baloh et al., 1997; Sanicola et al., 1997) or through another member of the GFRα receptor family. Further evidence of this hypothesis is provided by our findings that some populations of neurons respond to GDNF in the absence of GFRα1 (Figures 5C and 6C).
Surprisingly, although the comparison between the GDNF−/− and GFRα1−/− mice suggests that GDNF rescues petrosal-nodose neurons independent of GFRα1 in vivo (Table 1), GDNF was not able to promote the survival of a significant number of GFRα1−/− petrosal-nodose neurons in vitro (Table 1; Figure 5A). Thus, it is possible that the effects of GDNF on this neuronal population in vivo are indirect. Alternatively, GDNF may influence the development of a subpopulation of these neurons at a stage before they were cultured.
Ligand Specificity of the GFRα Receptors
Studies in a cell-free system indicated that GFRα1 selectively binds to GDNF, whereas GFRα2 selectively binds NTN (Klein et al., 1997). Surprisingly, although Ret by itself does not bind any known member of the GDNF protein family with a high affinity (Jing et al., 1996; Treanor et al., 1996; Klein et al., 1997), it can, when coexpressed with the GFRαs, change their ligand-binding specificity, allowing, for example, the binding of GDNF to GFRα2 (Sanicola et al., 1997). The promiscuousness of the GFRαs, in the presence of Ret, is further illustrated by the findings that cells that express GFRα2 together with Ret can respond to multiple ligands (Baloh et al., 1997; Buj-Bello et al., 1997).
To examine the ligand specificity of the endogenous GFRα1 and the receptor specificity of GDNF and NTN, we have analyzed multiple classes of primary neurons derived from the GFRα1−/− embryos. This analysis revealed that GDNF can, in certain cell types, mediate its response through an alternative receptor, as illustrated by its ability to promote the survival of GFRα1−/− submandibular neurons. Similarly, examination of GFRα1−/− neurons for their response to NTN showed that GFRα1 serves as a receptor for NTN on dopaminergic and nodose sensory ganglia, while submandibular neurons respond to this factor via another receptor, most likely GFRα2. Although these findings are consistent with the proposal that GDNF and NTN can activate multiple receptors, it is important to note that these receptors are activated with a different potency. Thus, GFRα1 appears to be activated preferentially by GDNF, whereas the receptor present on submandibular neurons is preferentially activated by NTN (Figures 5C and 6C).
The GFRα Receptors May Display Distinct Functions In Vivo
As revealed by experiments in cultured cells (Baloh et al., 1997) and neurons (Buj-Bello et al., 1997), and through the analysis of the GFRα1−/− mice, it appears that both NTN and GDNF can interact with multiple receptors (e.g., GFRα1 and GFRα2), and that GFRα1 can mediate the activity of multiple ligands. Surprisingly, however, although such promiscuous receptor–ligand interactions can take place in an experimental setting (Figures 5 and 6) (Baloh et al., 1997), they do not appear to be prevalent in vivo, since most of the major deficits in the GFRα1−/− mice are recapitulated in the GDNF−/− mice. The limited ability of GFRα2 to compensate for GFRα1 in vivo or in vitro is consistent with the distinct tissue distribution of these two receptors. For example, developing dopaminergic neurons, which do not respond to GDNF in vitro, in the absence of GFRα1, express only GFRα1, whereas GFRα2 transcripts are found in high abundance adjacent to, but not on, this neuronal cell type (M. H. and L.-C. W., unpublished data). Likewise, high levels of GFRα1 mRNA, but not of GFRα2 mRNA, are found in nodose sensory ganglia neurons (Buj-Bello et al., 1997) and on spinal motor neurons (Klein et al., 1997; Widenfalk et al., 1997), which show deficits in the GFRα1−/− mice. Finally, GFRα1, but not GFRα2, transcripts are expressed in the embryonic day 14 mouse kidney, an organ that fails to develop in the absence of GFRα1 (Baloh et al., 1997). In contrast, cells such as dorsal root and superior cervical ganglia neurons, which suffer minor or no deficits in GFRα1−/− embryos as compared with the GDNF−/− embryos, appear to express both GFRα1 and GFRα2 during development (Baloh et al., 1997; Nosrat et al., 1997; Widenfalk et al., 1997). Likewise, parasympathetic neurons, which retain responsivenes to GDNF and NTN in the absence of GFRα1, appear to express multiple GFRs (A. F. and A. D., unpublished data in the chick embryo). The one possible exception to this rule that we observed is enteric neurons. Despite the fact that both GFRα1 and GFRα2 are expressed in the developing gut (Jing et al., 1996; Treanor et al., 1996; Baloh et al., 1997; Buj-Bello et al., 1997; Klein et al., 1997), GFRα2 fails to compensate for GFRα1 in this tissue. However, the cell type that expresses GFRα2 in the gut has not been identified, and it is possible that the inability of GFRα2 to substitute for GFRα1 is due to its absence from the enteric neurons. This may be analogous to the situation for dopamine neurons, in which both GFRα1 and GFRα2 are expressed in the vicinity of dopamine neurons, but only GFRα1 is expressed at significant levels on the dopamine neurons themselves (M. H. and L. Wang, unpublished data).
In summary, the striking similarities between the GFRα1−/−, GDNF−/−, and Ret−/− mice support the hypothesis that GFRα1 is an important receptor component for GDNF and validate the physiological significance of GFRα and the multicomponent receptor hypothesis (Jing et al., 1996; Treanor et al., 1996; Baloh et al., 1997; Buj-Bello et al., 1997; Klein et al., 1997; Sanicola et al., 1997). Although GDNF and NTN display some preferential interactions with different GFRα receptors in vivo, their distinct functions may be controlled, to a large extent, by their unique tissue distribution.
Experimental Procedures
Production of the GFRα1−/− Mice
A genomic library, derived from the 129 mouse strain, was screened with synthetic oligonucleotides, and four positive phages encoding for the GFRα1 gene were identified. A neomycin gene under the control of the phosphoglycerate kinase I promoter was then fused to an 890 bp Eco-XhoI fragment, representing a portion of the GFRα1 gene N-terminal to amino acid 14, and to a 6.8 kbp Bam HI fragment, representing a portion of the GFRα1 gene, which is C-terminal to amino acid 66, resulting in a deletion of 52 amino acids from the second exon. A thymidine kinase cDNA under the control of the CMV promoter was added to the targeting construct, and the construct was electroporated into R1 ES (Nagy et al., 1993) and ES D3-C12 (Moore et al., 1996) lines. Clones were selected in 400 μg/ml G418 and 2 μM gancyclovir, expanded, and screened by Southern analysis for homologous recombination. The targeted R1 ES was injected into the blastocoel cavity of 3.5-day-old C57BL/6J blastocysts, and six highly chimeric male founders were chosen for further studies. In parallel, clumps of the targeted R1 ES cells were aggregated with diploid embryos from 2.5-day-old C57BL/6J superovulated females (Nagy and Rossant, 1993), and three additional founders were isolated. The resulting embryos were recovered by caesarean section, genotyped, and analyzed.
GFRα1−/− mice were also produced by tetraploid embryo aggregation (Nagy and Rossant, 1993; Nagy et al., 1993). For this, the targeted ES D3-C12 cells were subjected to 2 mg/ml of G418, and clones in which the second GFRα1 allele was mutated by gene conversion were identified. Clumps of the double mutant ES cells were sandwiched between, or aggregated with, tetraploid embryos that were generated from 2.5-day-old CD-1 mice and implanted. E11–E12 and E14–E16 embryos were recovered by caesarean section, genotyped by polymerase chain reaction and by eye color (the GFRα1−/− embryos, which were derived from the 129 mouse, have pigmented eyes, while the wild-type CD-1 embryos have nonpigmented eyes), and serially sectioned for examination. Since the GFRα1−/− offspring of the different founders displayed a similar phenotype (data not shown), extensive analysis was performed only on the R1 ES–derived embryos in both the C57BL/6J and CD-1 background, as specified.
Histological Analysis
For histological analysis, embryos and neonate pups were fixed with 10% neutral buffered formalin, embedded in paraffin, serially sectioned, and stained with hematoxylin and eosin or the indicated antibody for microscopic examination. Antibody staining was performed using antiperipherin (1:300; Chemicon), antineurofilament 150 kDa (1:1000; Chemicon), anti-Pax2 (10 μg/ml; Bablo), and anti–wild type 1 (1:500; Santa Cruz Biotechnology) and the peroxidase Vectastain kit (Vector Labs), as previously described (Moore et al., 1996). Intestines from animals perfused with 4% paraformaldehyde were stained in whole mount with antibodies to the neuronal marker PGP9.5 (1:4000; Biogenesis) as previously described (Moore et al., 1996). For neuronal counts, neonate pups were fixed in Carnoy’s fixative and embedded in paraffin. Serial sections (7 μm thick) through the head and the lumbar back of homozygous mutants and wild-type littermates were obtained and stained with cresyl violet. Peripheral ganglia and motor nuclei neurons were counted every sixth section (Moore et al., 1996). Some series were stained with antibodies to tyrosine hydroxylase (1:200; Pel-Freeze), and tyrosine hydroxylase–positive neurons were counted in the substantia nigra and locus coeruleus (Liching and Anderson, 1995; Moore et al., 1996).
Survival Assays
E12 nodose sensory and E17 submandibular parasympathetic ganglia neurons were isolated from wild-type, GFRα1+/−, or GFRα1−/− embryos and analyzed as described (Davies et al., 1995). The nodose survival assays for each factor were performed on 21 wild-type and 14 GFRα1−/− embryos. The submandibular survival assays were done on 5 wild-type, 9 GFRα1+/−, and 12 GFRα1−/− embryos for each factor. Individual embryos were analyzed. Dopaminergic neurons were isolated from E12 embryos, and 3–5 embryos were analyzed for each condition as described (Poulsen et al., 1994).
Acknowledgments
We thank E. Berry and W. Anstine for preparing the manuscript and figures, Tina Rarick for photography, Thuy-Nhung Nguyen for help with in situ hybridization, Shadid Sultan and Julio Ramirez for help with histology, and Audrey Goddard for sequencing. I. F. is supported by the Human Frontier Science Program. L. F. R. is an investigator of the Howard Hughes Medical Institute. A. M. D. and A. F. are supported by grants from the Wellcome Trust.
References
- Arenas E, Trupp M, Akerud P, Ibáñez CF. GDNF prevents degeneration and promotes the phenotype of brain noradrenergic neurons in vivo. Neuron. 1995;15:1465–1473. doi: 10.1016/0896-6273(95)90024-1. [DOI] [PubMed] [Google Scholar]
- Baloh RH, Tansey MG, Golden JP, Creedon DJ, Heuckeroth RO, Keck CL, Zimonjic DB, Popescu NC, Johnson EM, Milbrandt J. Trnr2, a novel receptor that mediates neurturin and GDNF signaling through Ret. Neuron. 1997;18:793–802. doi: 10.1016/s0896-6273(00)80318-9. [DOI] [PubMed] [Google Scholar]
- Beck KD, Valverde J, Alexi T, Poulsen K, Moffat B, Vandlen RA, Rosenthal A, Hefti F. Mesencephalic dopaminergic neurons protected by GDNF from axotomy-induced degeneration in the adult brain. Nature. 1995;373:339–341. doi: 10.1038/373339a0. [DOI] [PubMed] [Google Scholar]
- Buj-Bello A, Buchman VL, Horton A, Rosenthal A, Davies AM. GDNF is an age-specific survival factor for sensory and autonomic neurons. Neuron. 1995;15:821–828. doi: 10.1016/0896-6273(95)90173-6. [DOI] [PubMed] [Google Scholar]
- Buj-Bello A, Adu J, Pinon L, Horton A, Thompson J, Rosenthal A, Chinchetru M, Buchman VL, Davies AM. Neurturin responsiveness requires a GPI-linked receptor and the Ret receptor tyrosine kinase. Nature. 1997;387:721–724. doi: 10.1038/42729. [DOI] [PubMed] [Google Scholar]
- Davies AM, Minichiello L, Klein R. Developmental changes in NT3 signaling via TrkA and TrkB in embryonic neurons. EMBO J. 1995;14:4482–4489. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressler GR, Douglass EC. Pax-2 is a DNA-binding protein expressed in embryonic kidney and Wilms tumor. Proc Natl Acad Sci USA. 1992;89:1179–1183. doi: 10.1073/pnas.89.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbec PL, Larsson-Blomberg LB, Schuchardt A, Costantini F, Pachnis V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development. 1996;122:349–358. doi: 10.1242/dev.122.1.349. [DOI] [PubMed] [Google Scholar]
- Eng C, Myers SM, Kogon MD, Sanicola M, Hession C, Cate RL, Mulligan LM. Genomic structure and chromosomal localization of the human GDNFRα gene. Oncogene. 1998;16:597–601. doi: 10.1038/sj.onc.1201573. [DOI] [PubMed] [Google Scholar]
- Henderson CE, Phillips HS, Pollock HS, Davies AM, Lemeulie C, Armanini MP, Simpson LC, Moffet B, Vandlen RA, Koliatsos VE, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994;266:1062–1064. doi: 10.1126/science.7973664. [DOI] [PubMed] [Google Scholar]
- Jing SQ, Wen DZ, Yu YB, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, et al. GDNF-induced activation of the Ret protein tyrosine kinase is mediated by GDNFRα, a novel receptor for GDNF. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon CC, Jones KR, Reichardt LF, Barbacid M. The TrkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RD, Sherman D, Ho WH, Stone D, Bennett GL, Moffat B, Vandlen R, Simmons L, Gu QM, Hongo JA, et al. A GPI-linked protein that interacts with Ret to form a candidate neurturin receptor. Nature. 1997;387:717–721. doi: 10.1038/42722. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson EM, Milbrandt J. Neurturin, a relative of glial-cell-line–derived neurotrophic factor. Nature. 1996;384:467–470. doi: 10.1038/384467a0. [DOI] [PubMed] [Google Scholar]
- Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell. 1993;74:679–691. doi: 10.1016/0092-8674(93)90515-r. [DOI] [PubMed] [Google Scholar]
- Liching L, Anderson DJ. Postmigratory neural crest cells expressing c-RET display restricted developmental and proliferative capacities. Neuron. 1995;15:527–539. doi: 10.1016/0896-6273(95)90142-6. [DOI] [PubMed] [Google Scholar]
- Lin L-FH, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line–derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Masure S, Cik M, Pangalos MN, Bonaventure P, Verhasselt P, Lesage AS, Leysen JE, Gordon RD. Molecular cloning, expression and tissue distribution of glial-cell-line–derived neurotrophic factor family receptor α3 (GFRα3) Eur J Biochem. 1998;251:622–630. doi: 10.1046/j.1432-1327.1998.2510622.x. [DOI] [PubMed] [Google Scholar]
- Milbrandt J, de Sauvage FJ, Fahrner TJ, Baloh RH, Leitner ML, Tansey MG, Lampe PA, Heuckeroth RO, Kotzbauer PT, Simburger KS, et al. Persephin, a novel neurotrophic factor related to GDNF and neurturin. Neuron. 1998;20:245–253. doi: 10.1016/s0896-6273(00)80453-5. [DOI] [PubMed] [Google Scholar]
- Moore MW, Klein RD, Fariñas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J. Production of completely ES cell–derived fetuses. In: Joyner A, editor. Gene Targeting: a Practical Approach. Oxford: IRL Press/Oxford University; 1993. [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture–derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveilhan P, Baudet C, Mikaels A, Shen L, Westphal H, Ernfors P. Expression and regulation of GFRα3, a glial cell line–derived neurotrophic factor family receptor. Proc Natl Acad Sci USA. 1998;95:1295–1300. doi: 10.1073/pnas.95.3.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosrat CA, Tomac A, Hoffer BJ, Olson L. Cellular and developmental patterns of expression of Ret and glial cell line–derived neurotrophic factor receptor alpha mRNAs. Exp Brain Res. 1997;115:410–422. doi: 10.1007/pl00005711. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Houenou LJ, Johnson JE, Lin F-H, Li L, Lo AC, Newsome AL, Prevette DM, Wang S. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;373:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- Pichel JG, Shen L, Sheng HZ, Granholm A-C, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Poulsen KT, Armanini MP, Klein RD, Hynes MA, Phillips HS, Rosenthal A. TGFβ2 and TGFβ3 are potent survival factors for midbrain dopaminergic neurons. Neuron. 1994;13:1245–1252. doi: 10.1016/0896-6273(94)90062-0. [DOI] [PubMed] [Google Scholar]
- Rothenpieler UW, Dressler GR. Pax-2 is required for mesenchyme-to-epithelium conversion during kidney development. Development. 1993;119:711–720. doi: 10.1242/dev.119.3.711. [DOI] [PubMed] [Google Scholar]
- Sánchez MP, Siols-Santiago I, Frisén J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- Sanicola M, Hession C, Worley D, Carmillo P, Ehrenfels C, Walus L, Robinson S, Jaworski G, Wei H, Tizard R, et al. Glial cell line–derived neurotrophic factor–dependent ret activation can be mediated by two different cell-surface accessory proteins. Proc Natl Acad Sci USA. 1997;94:6238–6243. doi: 10.1073/pnas.94.12.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxen L. Organogenesis of the Kidney. Cambridge: Cambridge University Press; 1987. [Google Scholar]
- Schuchardt A, D’Agati V, Larsson-Blomberg L, Costantini F, Pachnis V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature. 1994;367:380–383. doi: 10.1038/367380a0. [DOI] [PubMed] [Google Scholar]
- Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the TrkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384:129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- Thompson J, Doxakis E, Piñón LGP, Strachan P, Buj-Bello A, Wyatt S, Buchman VL, Davies AM. GFRα-4, a new GDNF family receptor. Mol Cell Neurosci. 1998;11:117–126. doi: 10.1006/mcne.1998.0682. [DOI] [PubMed] [Google Scholar]
- Tomac A, Lindqvist E, Lin LH, Ogren SO, Young D, Hoffer BJ, Olson L. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. 1995;373:335–339. doi: 10.1038/373335a0. [DOI] [PubMed] [Google Scholar]
- Treanor J, Goodman L, de Sauvage F, Stone DM, Poulsen KT, Beck KD, Gray C, Armanini MP, Pollock RA, Hefti F, et al. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- Trupp M, Ryden M, Jornvall H, Funakoshi H, Timmusk T, Areans E, Ibáñez CF. Peripheral expression and biological activities of GDNF, a new neurotrophic factor for avian and mammalian peripheral neurons. J Cell Biol. 1995;130:137–148. doi: 10.1083/jcb.130.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widenfalk J, Nosrat C, Tomac A, Westphal H, Hoffer B, Olson L. Neurturin and glial cell line–derived neurotrophic factor receptor-β (GDNFR-β), novel proteins related to GDNF and GDNFR-a with specific cellular patterns of expression suggesting roles in the developing and adult nervous system and in peripheral organs. J Neurosci. 1997;17:8506–8519. doi: 10.1523/JNEUROSCI.17-21-08506.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Vega QC, Chao HH, Seasholtz AF, Thompson RC, Dixon JE. Identification and characterization of GFRα3, a novel co-receptor belonging to the glial cell line–derived neurotrophic receptor family. J Biol Chem. 1998;273:3502–3508. doi: 10.1074/jbc.273.6.3502. [DOI] [PubMed] [Google Scholar]
- Yan Q, Matheson C, Lopez OT. In vivo, neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:41–44. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]