

Abstract
Many alleles of human disease genes have mutations within splicing consensus sequences that activate cryptic splice sites. In Caenorhabditis elegans, the unc-73(e936) allele has a G-to-U mutation at the first base of the intron downstream of exon 15, which results in an uncoordinated phenotype. This mutation triggers cryptic splicing at the −1 and +23 positions and retains some residual splicing at the mutated wild-type (wt) position. We previously demonstrated that a mutation in sup-39, a U1 snRNA gene, suppresses e936 by increasing splicing at the wt splice site. We report here the results of a suppressor screen in which we identify three proteins that function in cryptic splice site choice. Loss-of-function mutations in the nonessential splicing factor smu-2 suppress e936 uncoordination through changes in splicing. SMU-2 binds SMU-1, and smu-1(RNAi) also leads to suppression of e936. A dominant mutation in the conserved C-terminal domain of the C. elegans homolog of the human tri-snRNP 27K protein, which we have named SNRP-27, suppresses e936 uncoordination through changes in splicing. We propose that SMU-2, SMU-1, and SNRP-27 contribute to the fidelity of splice site choice after the initial identification of 5′ splice sites by U1 snRNP.
PRE-mRNA splicing takes place in a large ribonucleoprotein complex called the spliceosome (Burge et al. 1999). Components of this splicing machinery assemble at conserved signal sequences within the pre-mRNA. The 5′ splice site consensus sequence M−3A−2G−1 | G+1U+2R+3A+4G+5U+6 and the 3′ splice site consensus sequence Y−3A−2G−1 | R+1 (M is either A or C; R is a purine, and Y is a pyrimidine) define the limits of the intron. Base-pairing interactions between the 5′ end of the U1 snRNA and the 5′ splice site consensus sequence occur early in spliceosome assembly. It is the nearly invariable GU dinucleotide at the first two positions of the 5′ end of the intron that defines the beginning of the intron. The 5′ consensus sequence is essential but insufficient for splice site selection, as 5′ splice sites with weaker consensus matches may require additional determinants for proper activation (Sanford et al. 2005).
Mutations that disrupt the 5′ consensus splice signal can lead to genetic disease in humans (Nelson and Green 1990; Cohen et al. 1994). Approximately 15% of point mutations that cause genetic diseases affect pre-mRNA splicing consensus sequences (Krawczak et al. 1992). For some specific disease genes, as many as 50% of the known heritable alleles alter splicing (Teraoka et al. 1999; Ars et al. 2000; Roca et al. 2003; Pagenstecher et al. 2006). Among all the positions of the 5′ splice site consensus sequence, the highest proportion of human disease mutations occur at the +1G position (Buratti et al. 2007). The fidelity of pre-mRNA splice site choice is largely disrupted by this defect, since this mutation causes splicing at this site to be either abolished or outcompeted by the activation of nearby cryptic 5′ splice sites (Nelson and Green 1990; Cohen et al. 1994). Cryptic splice sites are used only when the wild-type splice donor is disrupted by mutation, as they tend to have very weak splice donor consensus sequences outside of a 5′-GU dinucleotide that defines the beginning of the intron (Roca et al. 2003). Suppression of mutations to the 5′ splice site consensus sequence in vivo has been achieved through the expression of U1 snRNAs containing compensatory base substitutions (Zhuang and Weiner 1986); however, suppression of mutations to the +1 position of the intron using reverse genetic approaches has not been successful (Newman et al. 1985; Nelson and Green 1990; Cohen et al. 1994).
We have used a specific allele of the Caenorhabditis elegans unc-73 gene, e936, which contains a G-to-U mutation at the first nucleotide of intron 16 (Steven et al. 1998), as a model for studying cryptic splice site choice (Roller et al. 2000; Zahler et al. 2004). unc-73 encodes a RAC guanine nucleotide exchange factor that is expressed in neurons and is important for axon guidance (Steven et al. 1998). The e936 allele induces the use of three different cryptic 5′ splice sites (Figure 1A). Two of these 5′ splice sites, located at the −1 and +23 positions, define introns beginning with GU. The third 5′ splice site used is at the mutated wild-type (wt) position and is referred to as “wt” since splicing at this site still produces wild-type unc-73 mRNA and protein, even though the intron begins with UU (Roller et al. 2000). Use of either the −1 or the +23 cryptic site causes a shift in the reading frame and loss of gene function. In e936 animals, 90% of the stable messages of unc-73 are out-of-frame, yet the phenotype is not as severe as for other alleles in this gene. This indicates that the 10% of steady-state messages that are in frame have some functional role.
Figure 1.—

(A) Diagram of the unc-73 gene between exons 15 and 16. The positions of the −1 and +23 cryptic 5′ splice sites are indicated by arrows. The intronic e936 (+1G → U) point mutation is highlighted. (B) γ-32P-labeled RT–PCR results across the cryptic splicing region of unc-73(e936) for different strains. Lanes 1, 2, and 3 are loaded with RT–PCR reactions from wild type (N2), unc-73(e936);sup-39(je5), and unc-73(e936) RNA, respectively. The lines carrying the suppressor alleles and e936 follow in lanes 4–10 as indicated. (C) The unc-73 genomic sequence from exon 15 (uppercase letters) and intron 15 (lowercase letters). The locations of the az23 and e936 mutational substitutions are indicated below. The position of the −9 cryptic splice donor activated in e936az23 is indicated by an arrow above.
In a previous genetic screen for extragenic suppressors of e936 movement defects, Way and colleagues identified sup-39 (Run et al. 1996). It was subsequently shown that mutations in sup-39 alter cryptic splice site choice of e936 (Roller et al. 2000). sup-39 encodes a U1 snRNA gene with a compensatory mutation at the position that normally base pairs with the +1G. This allows sup-39 to base pair with an intron with a +1U (Zahler et al. 2004). This dominant suppressor increases usage of the mutated splice site and improves the fraction of in-frame messages from e936 from 10 to 33%, with a dramatic improvement in coordination. A similar mutant U1 snRNA suppressor with a different compensatory substitution, sup-6(st19), was found to suppress the intronic +1G to A transition of unc-13(e309) to allow for splicing at the mutated wild-type site, even though the intron begins with AU instead of GU (Zahler et al. 2004).
We are interested in identifying additional factors that play a role in cryptic 5′ splice site choice. To do this, we took advantage of unc-73(e936), in which modest increases in the use of the wt splice site lead to dramatic increases in coordination, as a sensitive screen for changes in cryptic splice site choice. In this article we report that the proteins SMU-1 and SMU-2, which are nonessential factors previously shown to have a role in alternative splicing (Spartz et al. 2004), have a role in selection of cryptic 5′ splice sites. We also report the identification of a new dominant suppressor of cryptic splicing, snrp-27, which encodes a C. elegans homolog of the human tri-snRNP 27K protein.
MATERIALS AND METHODS
Growth and maintenance of worms:
Worms were grown at 20° on (NGM) hardened agar in petri dishes (9 and 5 cm) with a limited supply of Escherichia coli for food (Lewis and Fleming 1995). The CB936 [unc-73(e936)], JW101 [unc-73(e936); sup-39(je5)], Hawaiian CB4856, and Bristol N2 strains were provided by the Caenorhabditis Genetics Center (CGC) at the University of Minnesota.
N-Ethyl-N-nitrosurea mutagenesis:
The protocol previously developed for identifying e936 suppressors with EMS mutagenesis (Run et al. 1996) was adapted for mutagenesis with N-ethyl-N-nitrosourea (ENU). Synchronized L4 worms were washed with M9 solution and placed into a 15-ml conical tube. Worms were concentrated by centrifugation at 3000 × g and resuspended in 2 ml of M9. A 50-mm ENU stock was diluted to 0.33 mm in M9 solution and 20 μl of this were added to the vial with the worm suspension. The vial was rotated for 4 hr at room temperature. After rotation, worms were collected by centrifugation and the ENU supernatant was removed. The worm pellet was washed two times with 4 ml of M9, rotating between washes. Then worms were plated onto NGM plates with E. coli as a food source and allowed to recover overnight.
Mutant screen:
The screen for suppressed animals was performed essentially as described by Run et al. (1996). The day following the mutagenesis with ENU described above, four healthy mutagenized worms (P0) were placed on the edge of an E. coli-seeded 10-cm NGM agar plate, and 250 such plates were made. Plates were incubated for 1 week at 20°, allowing sufficient time for the F2 generation to reach adulthood. Putative suppressed animals were identified by their ability to migrate to the far side of the plate containing food. The screening process also involved tapping the head of the migrated worms and looking for coordinated, sinusoidal forward and backward movement. We screened 200,000 mutagenized haploid genomes:
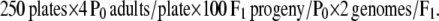 |
Thrashing assay:
To measure how well the suppressed worms move compared to the uncoordinated mutant unc-73(e936) worms, a thrash test was performed (Tsalik and Hobert 2003). Live L4 worms were transferred to a plate filled with liquid M9 solution at 20°. For a period of 60 sec, worms were observed and the number of times that they bent across their body axis in a rhythmic or rapid manner was recorded.
RNA extraction:
RNA extractions were performed as described previously (Roller et al. 2000).
Reverse transcription:
To generate complementary DNAs from extracted RNA, an oligonucleotide primer complementary to a portion of unc-73 exon 17 was used in reverse transcriptase (RT) reactions: 5′-ACT TGT CCA TCA AAA TCT GC-3′. Reverse transcription reactions were performed as previously described (Roller et al. 2000). For the experiment in Figure 4 only, the RT primer used was the same as the exon 16 reverse primer used in the polymerase chain reactions (PCRs) (see below).
Figure 4.—

The az26 allele of snrp-27 is a dominant suppressor of e936 cryptic splicing. (A) Alignment of human tri-snRNP 27K protein and C. elegans SNRP-27. The Met at position 141 that is changed to Thr in az25 and az26 and the corresponding base substitution are indicated. (B) The snrp-27 transgene contents of the four extrachromosomal array lines to be tested. (C) Thrash test and backward movement assay results for various lines. For array-containing strains, GFP+ L4 animals were picked and assayed. (D) RT–PCR results and quantitation of the relative level of the bands for the various lines. For the array-containing lines, 200 individual L4 worms were picked and RNA was extracted and tested by RT–PCR.
PCRs and determination of relative cryptic splice site usage:
Primers corresponding to unc-73 exon 15 (5′-AGA AGT TGT ACG GAT AAG AC-3′) and exon 16 (5′-GAA ACT TCA ATG CGT TTA GC-3′) were used in PCR reactions with Taq DNA polymerase and unc-73 cDNAs as previously described (Roller et al. 2000). PCR cycles were as follows: 94° for 5 min and then 30 cycles of 94° for 1 min, 59° for 1 min for annealing, and 72° for 1 min for extension. γ-32P labeling of oligonucleotides for PCR and separation of labeled unc-73 RT–PCR products on 6% polyacrylamide urea gels were performed as previously described (Roller et al. 2000). Gels were dried onto Whatman paper and visualized with an Amersham Biosciences (Piscataway, NJ) Typhoon. Quantitation of comparative splice site usage was done using ImageQuant software (Molecular Dynamics) as previously described (Roller et al. 2000). For each strain, the average quantitation of at least three independent RNA extractions and RT–PCRs was reported.
Dominant/recessive assay:
Genetic crosses were used to determine whether each individual extragenic suppressor is dominant or recessive. Tests were performed as described by Run et al. (1996). Some of the extragenic suppressors showed partial suppression of the e936 uncoordinated phenotype when heterozygous, indicating semidominant suppression.
Mapping suppressors:
To map unc-73(e936) suppressors to a chromosome, the snip-SNP approach was taken (Wicks et al. 2001). Each suppressor was mated with unc-73(+); sup-39(+) males of the Hawaiian strain CB4856, for which thousands of single-nucleotide polymorphisms (SNPs) relative to the N2 strain are known. The F1 progeny were then segregated and allowed to self-fertilize. Uncoordinated F2 animals were picked onto individual plates and the F3 animals were scored. The Unc F2 animals that had 100% uncoordinated progeny must be homozygous unc-73(e936)/unc-73(e936);sup(CB+)/sup(CB+). DNA was extracted from these worms and PCRs were done across known SNP regions and digested with DraI restriction enzyme, to indicate whether the locus is derived from CB4856 or N2. The location of the suppressor gene in every case is a location that always corresponds to the CB4856 genome.
Bacterial mediated RNAi:
The RNAi bacterial feeding method was used to introduce dsRNA into the worms (Timmons et al. 2001). The following RNAi feeding vectors from the Ahringer RNAi screening set were used: smu-1 I-6J07 and empty vector L4440 (Kamath and Ahringer 2003; Kamath et al. 2003). E. coli bacterial strain HT115 (DE3) transformed with these plasmids was grown in 10 ml of liquid culture in LB with 50 μg/ml Ampicillin. NGM plates containing IPTG and 50 μg/ml carbenicillin were seeded with one drop of overnight culture. After overnight room temperature incubation, unc-73(e936) worms at the L4 stage were transferred to the seeded plates. Worms were allowed to grow at 15° for 72 hr or 22° for 36–40 hr. Adult worms were moved to a new, freshly seeded plate. Adults continued to be moved each day to freshly seeded plates, and unhatched embryos were counted to account for embryonic lethality. The progeny were scored for uncoordination in a thrash assay.
Construction of snrp-27 transgenes:
The snrp-27 genomic region was amplified from the genome of N2 and az26 worms with PCR primers 5′-GTA CCG GTT CGC GAG CAA TGG GAC G-3′ and 5′-GTA TGC TAG CTT CCA ATG AGT TTG TCA TCC-3′, which contained sites for the restriction enzymes AgeI and NheI, respectively. PCR products were ligated into the TOPO TA vector pCR2.1 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Clones containing inserts were sequenced to confirm a 100% match to the worm genome, with the exception of the point mutation in the az26-derived clones. Confirmed inserts were removed from the vector by digestion with AgeI and NheI and cloned into these same sites found in the polylinker pPD103.05 (a gift from the Andrew Fire laboratory). This Fire vector contains the constitutive let-858 promoter followed by a polylinker containing the AgeI and NheI sites followed by the rest of the let-858 gene. These plasmids were mixed with a sur-5∷GFP coexpression marker plasmid (pTG96_2) (Gu et al. 1998) and total N2 DNA was digested with PvuII restriction enzyme as carrier (final concentrations of injection mix are 30 ng/μl Plet-858∷snrp-27 plasmid, 20 ng/μl pTG96_2 plasmid, and 80 ng/μl PvuII-digested N2 total DNA). DNA was injected into the gonads of young adult N2 worms and GFP-expressing lines carrying extrachromosomal arrays were established (Jin 1999). To establish strains homozygous for unc-73(e936) and containing these extrachromosomal arrays, N2 males were crossed to the array-containing hermaphrodites. GFP-expressing males from the first generation were then crossed to unc-73(e936) hermaphrodites. F2 animals from this cross that expressed GFP and had somewhat uncoordinated movement were picked and confirmed to be homozygous for e936.
RESULTS
A screen for suppressors of unc-73(e936) uncoordination yields genes that alter cryptic splicing:
Having previously established that suppression of unc-73(e936) uncoordination provides a very sensitive assay for changes in cryptic splice site selection (Roller et al. 2000), we decided to repeat the previous screen for unc-73(e936) suppressors (Run et al. 1996) to potentially identify additional classes of cryptic splicing factors. The only difference to our approach this time was that we used ENU as the mutagen instead of ethyl methanesulfonate (EMS).
We screened the F2 progeny of 200,000 ENU mutagenized CB936 [unc-73(e936)] genomes for animals that showed suppression of the uncoordination defect. When the suppressed worms were isolated and tapped on the head, they were able to move backward, unlike the CB936 parent strain, but sometimes their heads would flop from side to side, unable to make complete sinusoidal movements. The suppressed worms were able to respond to mechanical stimulus, but appeared slightly lethargic relative to wild-type (N2) worms. Overall, their movement was much more responsive and energetic than that of unc-73(e936) animals, but not as responsive as that of the sup-39 U1 snRNA suppressors of e936 that were previously identified.
We isolated seven independent suppressed animals from the screen, allowed them to propagate and grow, and collected their RNA for RT–PCR analysis to determine whether the suppressors were acting at the level of splicing. All but two of the isolated suppressor strains showed detectable changes in the ratio of cryptic splice site usage compared to that of unc-73(e936) (Figure 1B). The two suppressors that did not show any detectable changes in splicing ratios, az22 and az28, were not studied further. These may represent suppressors of the loss of unc-73 RAC-GEF function, but their suppression does not appear to be at the level of splicing (Figure 1B, lanes 4 and 10).
One of the suppressors appeared to be intragenic to unc-73. Suppressor az23 did not segregate uncoordinated animals in the F2 generation after crossing to N2 males, and it displayed an extra band in the gel showing the various unc-73(e936) spliced transcripts (Figure 1B, lane 9). This extra band was cloned and sequenced to identify the new splice junction. This new splice site was found nine nucleotides upstream of the original splice donor, generating a message that is in frame. The genomic sequence of unc-73 for this potential intragenic suppressor was obtained and we determined that az23 was a second site A-to-T mutation in exon 15, eight nucleotides upstream of the original splice site (and e936 mutation). This created a new 5′ GU dinucleotide that acted as a splice donor from the −9 position and was used in 31% of the steady-state unc-73 transcripts (Figure 1C). Use of this new splice donor removes only three amino acids from the in-frame message and led to phenotypic suppression of the e936 mutant animals. Run et al. (1996) reported isolating eight intragenic suppressors of e936, but as that report came out before the molecular identification of unc-73 and the e936 allele (Steven et al. 1998), those suppressors were not characterized as leading to a change in splicing, although that is a likely assumption.
Extragenic suppressors of unc-73(e936) alter cryptic splice site usage:
We further investigated how well the four extragenic splicing suppressors identified from our suppressor screen suppress the e936 mutation. We assayed the improved movement of the suppressed animals, using a thrashing assay (Tsalik and Hobert 2003). These four extragenic suppressors showed significant improvement in coordinated movement in comparison to the unc-73(e936) mutant (Figure 2A). There was a range of improvement, with az27 being the weakest of the suppressors. None appeared to move as well as wild type or the previously described sup-39(je5) suppressor.
Figure 2.—

Quantitation of unc-73(e936) movement defect suppression and cryptic splice site usage in the presence of the isolated extragenic suppressor mutations. (A) Results from 10 sets of worm thrashing experiments performed on the indicated strains. (B) Representation of splice site usage as determined by γ-32P-labeled RT–PCR and quantitated from a minimum of three different sets of reactions for each. Representative PhosphorImager images of the RT–PCR reactions for the different strains are shown directly below the column of quantitative data for each strain.
We quantitated the relative levels of steady-state messages that were spliced at the −1, mutated wt, and +23 splice sites for each of these extragenic suppressors (Figure 2B). ImageQuant software (Molecular Dynamics) was used to determine the radioactivity in each band from the PhosphorImager image, and the fraction of RT–PCR product representing each mRNA from each RNA sample was calculated as described previously (Roller et al. 2000). Averages of a minimum of three different RT–PCR reactions for each are presented, and a representative RT–PCR reaction for each of the suppressors is shown at the bottom of Figure 2B. We previously demonstrated that the relative stabilities of the three isoforms are not regulated by nonsense-mediated decay, so these ratios appear to correspond to the relative usage of the different splice sites by the splicing machinery (Roller et al. 2000). Consistent with the data from the thrashing assay, these four suppressors showed increases in levels of messages that were spliced at the wt position relative to the unc-73(e936) mutant strain. All four suppressors showed approximately a twofold increase in the steady-state level of messages that were spliced at the wt position. The −1 and wt positions appear to be present in a 1:1 ratio, indicating that, like sup-39, these suppressors work to increase usage of the overlapping −1/wt splice donors relative to the +23 site, while the choice between −1 and wt is likely made at a later step in spliceosome assembly (Roller et al. 2000). None of these four suppressor alleles had as strong a suppressor activity at the level of splicing as the U1 snRNA suppressor sup-39(je5). However, je5 also led to 50% embryonic lethality, and we observed no embryonic lethality with any of these new extragenic suppressors (data not shown).
A series of genetic crosses was carried out on each of the four new alleles to determine whether they are dominant or recessive suppressors of the e936 mutation (Run et al. 1996). While doing these crosses, we observed that for three of the suppressors, az24, az25, and az26, there was an easy-to-score intermediate suppressed phenotype for animals that were heterozygous for the suppressor and homozygous for unc-73(e936); the heterozygous suppressor animals move better than the e936 animals, but not as well as the animals that are homozygous for the suppressor. We refer to the heterozygous suppressor animals as having an “unc-ish” phenotype. The 1(wt):2(unc-ish):1(unc) ratio observed from scoring progeny of e936;az24/+, e936;az25/+, and e936;az26/+ animals is consistent with these suppressors being semidominant. In contrast, the e936 suppressor az27 behaved as would be expected for a true recessive allele.
Mapping the isolated extragenic suppressors of unc-73(e936):
We used snip-SNP mapping between N2 and CB4856 (Wicks et al. 2001) to narrow down the location of the extragenic suppressor mutations on the chromosomes of C. elegans. Suppressors az25 and az26 both mapped to chromosome I and were linked to unc-73. The SNP mapping placed both az25 and az26 between base positions 8,416,835 and 9,910,535 on chromosome I. az24 mapped between base positions 7,763,699 and 9,149,301 on chromosome III. az27 mapped between base positions 3,117,765 and 4,305,268 on chromosome II. We were immediately able to rule out the possibility that any of these four suppressors represent 1 of the 12 C. elegans U1 snRNA genes on the basis of these genomic positions. To narrow down the possible identity of potential suppressor genes, we compared the SNP mapping results of the extragenic suppressors to a table of chromosomal locations of C. elegans homologs of mammalian splicing factors, spliceosome-associated proteins, and spliceosomal core components identified from proteomic experiments (mammalian spliceosomal protein table courtesy of Melissa Jurica). Using this approach combined with sequencing of candidate genes, we found that the az27 suppressor strain had a premature termination codon in smu-2, the worm homolog of the human RED protein. For both az25 and az26 we found an identical missense mutation in the gene R05D11.7, the worm homolog of the human tri-snRNP 27K protein. We were unable to identify a potential gene for az24 with this approach. We next set out to confirm whether these mutations in smu-2 and R05D11.7 are indeed suppressors of e936.
Loss-of-function mutations in smu-2 and smu-1 are extragenic suppressors of the e936 mutation:
We sequenced the smu-2 gene from the az27 animals and found a lesion in the third exon, a T-to-G transversion that results in a nonsense mutation at codon 111 (there are 547 amino acids in the protein). This mutation is predicted to be a null mutation and fits with the genetic evidence that strain az27 is a recessive suppressor of unc-73(e936). smu-1 and smu-2 were isolated in a screen for suppressors of the synthetic lethality of mec-8 and unc-52 mutations (Lundquist and Herman 1994). unc-52 encodes a homolog of the mammalian perlecan extracellular matrix protein and mec-8 encodes an RNA recognition motif-containing (RRM) protein whose loss leads to a variety of mechanosensory and chemosensory defects (Lundquist et al. 1996). unc-52 is an essential gene, but a subset of alleles containing mutations in a region of three consecutive alternative exons has a less severe late larval-onset paralysis phenotype. These viable unc-52 mutations combined with loss of mec-8 lead to synthetic lethality through changes in the ability to skip the mutant exon by alternative splicing. smu-1 and smu-2 mutants were identified as suppressors of this mec-8;unc-52 synthetic lethality. They cause changes in unc-52 splicing and can suppress some of the alleles found in the alternatively spliced region by changing the relative use of the exons (Lundquist and Herman 1994; Spartz et al. 2004). The smu-2 gene encodes a ubiquitously expressed nuclear protein with 40% homology to human RED protein, a component of purified spliceosomes (Jurica and Moore 2003; Spartz et al. 2004; Deckert et al. 2006). SMU-2 interacts with SMU-1, a protein 62% identical to human spliceosome-associated protein fSAP57 (Jurica and Moore 2003; Deckert et al. 2006). SMU-2 has been shown to be required for the stability of the SMU-1 protein (Spartz et al. 2004).
We confirmed that az27 has a mutation in smu-2 by testing a known recessive allele of smu-2 for the ability to suppress unc-73(e936). A double-mutant strain was made, containing both unc-73(e936) and smu-2(mn416), which carries a recessive mutation in the first intron of the gene at the 3′ splice site (Spartz et al. 2004). We compared spliced e936 transcripts from mn416 suppressed animals to that of az27 and found that the ratios of splice site usage are the same between the two (Figure 3A). Consistent with the RT–PCR results, we measured the same amount of coordinated movement between unc-73(e936);smu-2(mn416) double-mutant animals and e936;az27 animals. These results confirm that mutant alleles of the smu-2 gene are suppressors of e936 uncoordination and that they function at the level of splicing (Figure 3B).
Figure 3.—

A comparison of az27 and a known allele of smu-2, smu-2(mn416) in suppression of unc-73(e936). (A) Measurements of unc-73(e936) splice site usage in the presence of smu-2(mn416) and az27 as determined by γ-32P-labeled RT–PCR. (B) Results from 11 sets of worm thrashing experiments each performed on L4 larvae from wild-type (N2), unc-73(e936), unc-73(e936);smu-2(mn416), and unc-73(e936);smu-2(az27) strains.
Previously, SMU-2 was shown to bind to and stabilize another protein called SMU-1, and loss of either protein had a similar ability to suppress unc-52;mec-8 synthetic lethality (Spartz et al. 2004). Since the SMU-2 and SMU-1 proteins interact, we asked whether smu-1(RNAi) could also suppress the unc-73(e936) defect. The smu-1 gene was knocked down in unc-73(e936) animals by RNAi feeding (Timmons et al. 2001; Kamath et al. 2003). After RNAi, the animals were measured for coordinated movement by a thrashing assay. The unc-73(e936); smu-1(RNAi) animals suppressed the e936 phenotype and moved in a more coordinated manner [77 ± 43 thrashes/min for unc-73(e936);smu-1(RNAi) animals vs. 2 thrashes/min for unc-73(e936) animals]. We also fed bacteria harboring the empty L4440 RNAi plasmid vector to unc-73(e936) as a negative feeding control, and these showed no change in uncoordinated movement relative to untreated unc-73(e936). The γ-32P-labeled RT–PCR products of e936 showed a slight increase in the usage of the −1 and mutated wild-type wt splice sites in smu-1(RNAi) animals, compared to unc-73(e936) (data not shown). However, this change in splice site usage was fairly subtle and the standard deviation was quite high, perhaps consistent with the high standard deviation found in the thrash assay for the RNAi suppressed worms. Still, the clear ability to find wild-type moving e936 worms with RNAi of smu-1 is consistent with a loss of SMU-1/SMU-2 leading to suppression of e936 through alteration of cryptic splicing.
A dominant suppressor of e936 is the C.elegans homolog of human tri-snRNP 27K:
The semidominant suppressor strains carrying az25 and az26 were found to have identical lesions in the C. elegans gene R05D11.7, leading to a missense mutation in the protein-coding region. R05D11.7 is the C. elegans homolog of the human tri-snRNP 27K (SNRNP27), a phosphoprotein component of the U4/U6.U5 spliceosomal tri-snRNP complex (Fetzer et al. 1997). We have named this C. elegans gene snrp-27. Figure 4A shows an alignment between the human and worm proteins; these two proteins share 54% amino acid identity. The position of the Met to Thr amino acid substitution found in az25 and az26 at codon 141 is indicated. This amino acid substitution is contained within a 17-amino-acid stretch of 100% sequence identity between worms and humans for this protein; there is no known function for this C-terminal domain.
Confirming the molecular identity of snrp-27(az26) as a suppressor of unc-73(e936) presents two challenges. One challenge is due to the semidominant nature of the mutation in az26. A transgene experiment to confirm that this allele is an e936 suppressor requires expressing the dominant mutant version of the gene in an e936 background. The second challenge is that snrp-27 is the sixth gene in an eight-gene operon, so it is not possible to clone it in its native promoter context, as the entire operon is too big to fit on a plasmid. We reasoned that a core component of the spliceosome is likely to be ubiquitously expressed, so we made two constructs in which we placed the wild-type or az26 forms of snrp-27 under the control of the promoter for the ubiquitously expressed gene let-858 (Kelly et al. 1997). Extrachromosomal arrays carrying either snrp-27(+) or snrp-27(az26) under the control of the let-858 promoter were successfully obtained, including three independent lines for snrp-27(az26) (Figure 4B). Genetic crosses were done so that these extrachromosomal arrays were moved into an unc-73(e936) homozygous background, and in this context they were tested for phenotypic and molecular suppression of e936.
We performed the thrash test assay on e936 worms expressing the various snrp-27 extrachromosomal arrays (Figure 4C). For the thrash assay, we picked L4 worms that express the GFP coexpression marker contained in the arrays, without regard to their movement capability on the plate, and counted thrashes per minute. For the snrp-27(az26)-expressing arrays we found an improvement in the thrashes per minute relative to the snrp-27(+)-expressing array, which showed no significant difference from the e936 strain lacking the array. Consistent with the high standard deviations in thrashes per minute for the snrp-27(az26)-expressing worms, some animals moved as well as wild type (up to 126 thrashes/min) and others behaved more like e936 (<10 thrashes/min), suggesting a broad range of movement capabilities for the strains carrying the snrp-27(az26) arrays. We decided on another movement assay to measure whether these arrays could rescue the e936 movement defect. When wild-type animals are tapped on the head, 100% of them complete two full sinusoidal cycles of backward movement, while only 8% of e936 animals tested were capable of this movement. One hundred percent of suppressed unc-73(e936) snrp-27(az26) animals were capable of this movement. For e936 worms expressing the snrp-27(+) extrachromosomal array, only 5% were capable of this backward movement, while between 65 and 75% of e936 worms carrying snrp-27(az26) extrachromosomal arrays were capable of sinusoidal backward movement. This is consistent with expression of the mutant form of snrp-27 serving as a dominant suppressor of the e936 uncoordination phenotype.
We next decided to test whether the snrp-27(az26) transgene suppression of e936 occurs at the level of splicing. Because only ∼50% of the progeny of the snrp-27 array-carrying worms inherit the extrachromosomal array, and because only about two-thirds of snrp-27(az26) array-carrying worms show improved movement, we needed to isolate RNA from array-carrying worms showing improved movement to have a reasonable chance of observing changes in the ratio of unc-73(e936) cryptic splicing products. For each of the e936; Ex snrp-27(az26) lines, 200 L4 worms expressing GFP and capable of backward movement were picked and RNA was extracted. RNA was also extracted from 200 e936; Ex snrp-27(+) worms, although none showed wild-type movement. RT–PCR assays on unc-73 were performed as in previous experiments (Figure 4D) along with e936 and e936 az26 controls. The quantitation of the relative splice site usage indicates that the snrp-27(az26) arrays improve splicing at the −1/wt sites relative to e936, but the snrp-27(+) array does not. This is consistent with suppression at the level of splice site choice. The dominant suppressor transgene arrays do not change splicing to the same extent as found in the unc-73(e936);snrp-27(az26) suppressor strain; this may be due to the semidominance of az26 and the fact that the strains that express the dominant extrachromosomal arrays for snrp-27(az26) also carry, and presumably still express, two chromosomal copies of snrp-27(+).
DISCUSSION
We have taken advantage of an extremely sensitive assay, in which small changes in cryptic splice site choice lead to dramatic phenotypic changes in movement, to identify extragenic and intragenic suppressors with a role in cryptic splice site determination. We identified extragenic suppressors of e936 that led to ∼2-fold increases in the usage of the mutated wild-type splice site, as verified by RT–PCR analysis. These splicing suppressors led to dramatic increases in coordinated movement, ranging from 8-fold to 20-fold in a thrash assay. The intragenic suppressor that we identified, az23, helped confirm that changes in splicing lead to movement defect suppression.
We identified an apparent null mutation in the smu-2 gene as an extragenic suppressor of e936 that functions at the level of splicing. smu-2(az27) acts as an extragenic suppressor of the e936 splicing defect, as does a previously described allele, smu-2(mn416). SMU-2 binds and stabilizes SMU-1 (Spartz et al. 2004), and we provide evidence that smu-1(RNAi) can also suppress the e936 unc phenotype. Our study uncovers potential new roles for the SMU-1/SMU-2 complex as fidelity factors for splicing. It was shown previously that recessive mutations in smu-2 and smu-1 affect the accumulation of alternatively spliced transcripts of unc-52 (Spartz et al. 2004). In addition to promoting the skipping of unc-52 exon 17, it was also demonstrated that smu-1(mn415) can enhance the phenotype of unc-52(e1421) (Spike et al. 2001). e1421 contains a splice donor mutation, from g | gtaag to g | gtaaa, in the intron downstream of alternative exon 16 (Rogalski et al. 1995). This previously reported result is also consistent with a role for the smu genes in altering usage of weak 5′ splice sites. smu-1 and smu-2 are not essential for splicing or viability, but when the splicing machinery is challenged with suboptimal cryptic splice sites, these proteins appear to have a role in the 5′ splice site selection process. RED protein, the mammalian homolog of SMU-2, was previously reported to have been found in the spliceosomal C-complex by mass spectrometry (Jurica and Moore 2003). It was also reported to be found in all the active B-complexes of the spliceosome, along with fSAP57, the mammalian homolog of SMU-1 (Deckert et al. 2006). These findings suggest that SMU-2 interacts with the spliceosome after the assembly of the tri-snRNP.
We identified a dominant mutation in the highly conserved C-terminal region of snrp-27, the C. elegans homolog of the human tri-snRNP 27K protein. The identical alleles az25 and az26 were isolated independently as suppressors of e936, suggesting that this is a very specific gain-of-function substitution in the protein. Tri-snRNP 27K was identified as a component of the U4/U6.U5 tri-snRNP that was capable of undergoing multiple phosphorylations (Fetzer et al. 1997). The N-terminal half of the protein is a serine- and arginine-rich domain typical of SR proteins and the U1-70K protein, but unlike those proteins snrp-27 contains no RNA recognition motif or any other recognizable domains. It has been shown that stable binding of the U1 snRNP to the 5′ splice site (formation of the spliceosomal E-complex) is mediated by interactions between the SR domains of ASF/SF2, an SR protein, and U1-70K, a protein component of U1 snRNP (Kohtz et al. 1994). Additional studies indicated the requirement for SR proteins in the subsequent recruitment of the U4/U6.U5 tri-snRNP to the assembling spliceosome (Roscigno and Garcia-Blanco 1995). Since phosphorylated SR domains serve as protein–protein intermolecular bridges, it has been proposed that tri-snRNP 27K may be the component of the tri-snRNP that mediates tri-snRNP recruitment to the pre-mRNA through bridging with SR proteins and U1 snRNP (Fetzer et al. 1997). We speculate that this potential interaction may be directly related to a role for SNRP-27 in cryptic splice site choice through mediating the assembling tri-snRNP's interactions with different potential 5′ splice donors.
When the best 5′ splice site is eliminated by mutation, the splicing machinery is left with multiple weaker cryptic 5′ splice sites from which it may choose. The pressure to make a choice is likely the result of SR proteins binding to the exon and recruiting U1 snRNP, which then must choose its best base-pairing partner. Perhaps multiple U1 snRNAs land on the pre-mRNA at these multiple weak sites, and a decision must be made at a subsequent step of splicing to choose which will become a site of catalysis. In alternative splicing of the SV40 T antigen pre-mRNA, there are two alternative 5′ splice sites, and U1 can be directed by a subset of SR proteins to simultaneously assemble at both of them. When this happens, the site closest to the 3′ splice site is chosen (Zahler and Roth 1995). It is very interesting that the mammalian homologs of SMU-1, SMU-2, and SNRP-27 are all factors found to be associated with later steps of spliceosome assembly, as opposed to being splicing factors associated with the initial recognition step of 5′ splice sites (Jurica and Moore 2003). Perhaps the role of SMU-1/SMU-2 is to ensure that when multiple U1 snRNAs bind a message, the one most proximal to the 3′ splice site or that has the best base-pairing interaction with the U1 snRNA is preferred. In the absence of either of these SMU factors, there will be less preference for recruitment of the tri-snRNP components to one specific U1 snRNA. In the case of e936, the loss of SMU proteins might lead to less use of the stronger, more proximal +23 splice site and more use of the other cryptic sites. For the dominant missense suppressor in the SNRP-27 protein, the ability to choose which of the multiple 5′ splice sites to interact with may be altered. Further understanding of the binding partners for SNRP-27, especially in the C-terminal region that contains the dominant mutation, will be helpful in understanding how the spliceosome distinguishes among different weak cryptic splice sites during its assembly transitions.
Acknowledgments
We thank Manny Ares and Melissa Jurica for comments and advice on this manuscript, Robert Herman and Jocelyn Shaw for the gift of smu strains, Brian Ackley for assistance with the ENU mutagenesis, Sergio Barberan-Soler for advice on the analysis of alternative splicing, and Sam Gu for help obtaining plasmids. Some nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health (NIH) National Center for Research Resources. This research is supported by NIH grant R01-GM061646 to A.M.Z. M.D. was supported by the University of California Santa Cruz (UCSC) Center for Biomolecular Science and Engineering Graduate Student Diversity Fellowship in Genomic Sciences and the UCSC Initiative for Maximizing Student Diversity (R25-GM058903). P.E.M. was supported by a fellowship from the UCSC Minority Access to Research Careers program (T34-GM007910).
References
- Ars, E., E. Serra, J. Garcia, H. Kruyer, A. Gaona et al., 2000. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Hum. Mol. Genet. 9 237–247. [DOI] [PubMed] [Google Scholar]
- Buratti, E., M. Chivers, J. Kralovicova, M. Romano, M. Baralle et al., 2007. Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 35 4250–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge, C. B., T. Tuschl and P. A. Sharp, 1999. Splicing of precursors to mRNAs by the spliceosome, pp. 525–560 in The RNA World, Ed. 2, edited by R. F. Gesteland, T. R. Cech and J. F. Atkins. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Cohen, J. B., J. E. Snow, S. D. Spencer and A. D. Levinson, 1994. Suppression of mammalian 5′ splice-site defects by U1 small nuclear RNAs from a distance. Proc. Natl. Acad. Sci. USA 91 10470–10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckert, J., K. Hartmuth, D. Boehringer, N. Behzadnia, C. L. Will et al., 2006. Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol. Cell. Biol. 26 5528–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetzer, S., J. Lauber, C. L. Will and R. Luhrmann, 1997. The [U4/U6.U5] tri-snRNP-specific 27K protein is a novel SR protein that can be phosphorylated by the snRNP-associated protein kinase. RNA 3 344–355. [PMC free article] [PubMed] [Google Scholar]
- Gu, T., S. Orita and M. Han, 1998. Caenorhabditis elegans SUR-5, a novel but conserved protein, negatively regulates LET-60 Ras activity during vulval induction. Mol. Cell. Biol. 18 4556–4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Y., 1999. Transformation, pp. 69–96 in C. elegans: A Practical Approach, edited by I. A. Hope. Oxford University Press, Oxford.
- Jurica, M. S., and M. J. Moore, 2003. Pre-mRNA splicing: awash in a sea of proteins. Mol. Cell 12 5–14. [DOI] [PubMed] [Google Scholar]
- Kamath, R. S., and J. Ahringer, 2003. Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30 313–321. [DOI] [PubMed] [Google Scholar]
- Kamath, R. S., A. G. Fraser, Y. Dong, G. Poulin, R. Durbin et al., 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421 231–237. [DOI] [PubMed] [Google Scholar]
- Kelly, W. G., S. Xu, M. K. Montgomery and A. Fire, 1997. Distinct requirements for somatic and germline expression of a generally expressed Caernorhabditis elegans gene. Genetics 146 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohtz, J. D., S. F. Jamison, C. L. Will, P. Zuo, R. Luhrmann et al., 1994. Protein-protein interactions and 5′ splice-site recognition in mammalian mRNA precursors. Nature 368 119–124. [DOI] [PubMed] [Google Scholar]
- Krawczak, M., J. Reiss and D. N. Cooper, 1992. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum. Genet. 90 41–54. [DOI] [PubMed] [Google Scholar]
- Lewis, J. A., and J. T. Fleming, 1995. Basic culture methods. Methods Cell Biol. 48 3–29. [PubMed] [Google Scholar]
- Lundquist, E. A., and R. K. Herman, 1994. The mec-8 gene of Caenorhabditis elegans affects muscle and sensory neuron function and interacts with three other genes: unc-52, smu-1, and smu-2. Genetics 138 83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundquist, E. A., R. K. Herman, T. M. Rogalski, G. P. Mullen, D. G. Moerman et al., 1996. The mec-8 gene of C. elegans encodes a protein with two RNA recognition motifs and regulates alternative splicing of unc-52 transcripts. Development 122 1601–1610. [DOI] [PubMed] [Google Scholar]
- Nelson, K. K., and M. R. Green, 1990. Mechanism for cryptic splice site activation during pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 87 6253–6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, A. J., R. J. Lin, S. C. Cheng and J. Abelson, 1985. Molecular consequences of specific intron mutations on yeast mRNA splicing in vivo and in vitro. Cell 42 335–344. [DOI] [PubMed] [Google Scholar]
- Pagenstecher, C., M. Wehner, W. Friedl, N. Rahner, S. Aretz et al., 2006. Aberrant splicing in MLH1 and MSH2 due to exonic and intronic variants. Hum. Genet. 119 9–22. [DOI] [PubMed] [Google Scholar]
- Roca, X., R. Sachidanandam and A. R. Krainer, 2003. Intrinsic differences between authentic and cryptic 5′ splice sites. Nucleic Acids Res. 31 6321–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski, T. M., E. J. Gilchrist, G. P. Mullen and D. G. Moerman, 1995. Mutations in the unc-52 gene responsible for body wall muscle defects in adult Caenorhabditis elegans are located in alternatively spliced exons. Genetics 139 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller, A. B., D. C. Hoffman and A. M. Zahler, 2000. The allele-specific suppressor sup-39 alters use of cryptic splice sites in C. elegans. Genetics 154 1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscigno, R. F., and M. A. Garcia-Blanco, 1995. SR proteins escort the U4/U6.U5 tri-snRNP to the spliceosome. RNA 1 692–706. [PMC free article] [PubMed] [Google Scholar]
- Run, J. Q., R. Steven, M. S. Hung, R. van Weeghel, J. G. Culotti et al., 1996. Suppressors of the unc-73 gene of Caenorhabditis elegans. Genetics 143 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford, J. R., J. Ellis and J. F. Caceres, 2005. Multiple roles of arginine/serine-rich splicing factors in RNA processing. Biochem. Soc. Trans. 33 443–446. [DOI] [PubMed] [Google Scholar]
- Spartz, A. K., R. K. Herman and J. E. Shaw, 2004. SMU-2 and SMU-1, Caenorhabditis elegans homologs of mammalian spliceosome-associated proteins RED and fSAP57, work together to affect splice site choice. Mol. Cell. Biol. 24 6811–6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spike, C. A., J. E. Shaw and R. K. Herman, 2001. Analysis of smu-1, a gene that regulates the alternative splicing of unc-52 pre-mRNA in Caenorhabditis elegans. Mol. Cell. Biol. 21 4985–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven, R., T. J. Kubiseski, H. Zheng, S. Kulkarni, J. Mancillas et al., 1998. UNC-73 activates the Rac GTPase and is required for cell and growth cone migrations in C. elegans. Cell 92 785–795. [DOI] [PubMed] [Google Scholar]
- Teraoka, S. N., M. Telatar, S. Becker-Catania, T. Liang, S. Onengut et al., 1999. Splicing defects in the ataxia-telangiectasia gene, ATM: underlying mutations and consequences. Am. J. Hum. Genet. 64 1617–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons, L., D. L. Court and A. Fire, 2001. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263 103–112. [DOI] [PubMed] [Google Scholar]
- Tsalik, E. L., and O. Hobert, 2003. Functional mapping of neurons that control locomotory behavior in Caenorhabditis elegans. J. Neurobiol. 56 178–197. [DOI] [PubMed] [Google Scholar]
- Wicks, S. R., R. T. Yeh, W. R. Gish, R. H. Waterston and R. H. Plasterk, 2001. Rapid gene mapping in Caenorhabditis elegans using a high density polymorphism map. Nat. Genet. 28 160–164. [DOI] [PubMed] [Google Scholar]
- Zahler, A. M., and M. B. Roth, 1995. Distinct functions of SR proteins in recruitment of U1 small nuclear ribonucleoprotein to alternative 5′ splice sites. Proc. Natl. Acad. Sci. USA 92 2642–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler, A. M., J. D. Tuttle and A. D. Chisholm, 2004. Genetic suppression of intronic +1G mutations by compensatory U1 snRNA changes in Caenorhabditis elegans. Genetics 167 1689–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang, Y., and A. M. Weiner, 1986. A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell 46 827–835. [DOI] [PubMed] [Google Scholar]