

Abstract
Epigenetic phenomena, and in particular heritable epigenetic changes, or transgenerational effects, are the subject of much discussion in the current literature. This article presents a model of transgenerational epigenetic inheritance and explores the effect of epigenetic inheritance on the risk and recurrence risk of a complex disease. The model assumes that epigenetic modifications of the genome are gained and lost at specified rates and that each modification contributes multiplicatively to disease risk. The potentially high rate of loss of epigenetic modifications causes the probability of identity in state in close relatives to be smaller than is implied by their relatedness. As a consequence, the recurrence risk to close relatives is reduced. Although epigenetic modifications may contribute substantially to average risk, they will not contribute much to recurrence risk and heritability unless they persist on average for many generations. If they do persist for long times, they are equivalent to mutations and hence are likely to be in linkage disequilibrium with SNPs surveyed in genomewide association studies. Thus epigenetic modifications are a potential solution to the problem of missing causality of complex diseases but not to the problem of missing heritability. The model highlights the need for empirical estimates of the persistence times of heritable epialleles.
THE modern definition of epigenetics is the study of heritable changes in gene expression that are not caused by changes in DNA sequence (Richards 2006; Bird 2007; Bossdorf et al. 2008). Epigenetic effects include methylation of the cytosine residue in DNA and the modification of chromatin proteins that package DNA (Youngson and Whitelaw 2008). Although this definition of epigenetics includes inheritance during both mitosis and meiosis, I am concerned in this article only with epigenetic changes that are transmitted to offspring, what has been called “transgenerational epigenetic inheritance” (Morgan and Whitelaw 2008; Youngson and Whitelaw 2008). The modern definition of epigenetics arose from the original definition of Waddington (1957; Holliday and Pugh 1975).
The possibility of nongenetic inherited effects on phenotype has excited great interest among both evolutionary biologists and human geneticists because it provides an additional mechanism of inherited variability and one that is not detectable in genomic surveys of sequence variation. Inherited epigenetic changes have been proposed as an explanation for the “missing heritability,” meaning inherited causes of risk of complex genetic diseases that have not yet been identified in genomewide association studies (GWAS) (Maher 2008; McCarthy and Hirschhorn 2008). Inherited epigenetic changes that contribute to disease risk would not be detectable in GWAS but may contribute to average risk and to similarities among relatives.
In this article, I present a simple model of the inheritance of epigenetic changes. The goal is to quantify the potential contribution they can make to average risk and recurrence risk. The model is developed in a standard population genetics framework and can be regarded as a generalization of previous multilocus models of complex diseases, particularly that of Risch (1990).
I assume that epigenetic effects are caused by the presence or the absence of epigenetic modifications of specific chromosomal locations. Bird (2007), Haig (2007), Richards (2008), and others have emphasized that, although epigenetic changes differ in many ways from mutations, their transmission to offspring is the same as the transmission of mutations, except for the possibility that they might be spontaneously lost. If the gain and loss of epigenetic changes are controlled by a locus elsewhere in the genome, as modeled by Bjornsson et al. (2004), then the resulting phenotypic effects are attributable to variation at that locus (Richards 2006; Johannes et al. 2008). The epigenetic changes are simply the mechanism by which that locus affects phenotype. If, however, the appearance of an epigenetic change at a location in the genome is not attributable to any particular locus or loci, then the phenotypic effects of the presence or the absence of an epigenetic change are attributable to the genomic location itself. That is the case I am concerned with here.
I begin by introducing the basic model of a randomly mating population and extend standard genetic theory to the case of epigenetic inheritance. Then I consider nonequilibrium populations in which environmental changes cause an increase in the rate of gain of epigenetic changes.
MODEL
The model assumes that disease risk is affected by n diallelic genetic loci and by ν sites at which epigenetic changes may be present. For simplicity, I refer to an epigenetic change that affects phenotype as an epigenetic mark, which is a shorthand for the more accurate “metastable epiallele” (Morgan and Whitelaw 2008). Throughout, Greek letters are used for parameters of the epigenetic part of the model. To illustrate the approach taken in the simplest mathematical form, I assume multiplicative interactions across loci and epigenetic sites, thus generalizing Risch's (1990) multiplicative model. Other more complicated models can be formulated using the same methods. The multiplicative model is simple to analyze because the average risk and recurrence risks are computed by calculating contributions from each genetic locus and epigenetic site separately and then multiplying. Also it has not been rejected for any of the SNPs identified in GWAS so far. The multiplicative model can be represented by a product,
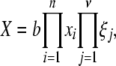 |
(1) |
where X is the disease risk, b is the background risk, xi is the contribution to the risk of locus i, and ξi is the contribution to the risk of epigenetic site j. The average risk, K, is the average of X taken over all genotypes and epigenetic configurations. This model does not allow for epistatic interactions among loci, but the same formalism can be used to analyze other assumptions about disease causation. Nor does this model allow for an interaction between genetic loci and epigenetic modifications, something that has been shown to occur in some species.
The contributions of each locus and epigenetic site to risk are assumed to be independent, implying that K is the product of the average contributions,
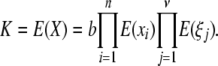 |
(2) |
The recurrence risk ratio for a relative of relationship R is
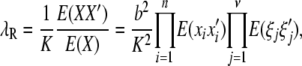 |
(3) |
where the prime indicates the risk in a relative with relationship R.
Following the notation in a previous article (Slatkin 2008), the causative allele at each locus is denoted by + and the other allele is denoted by a –. The frequency of + at locus i is pi. Each + at locus i increases the disease risk by a factor (1 + ri), which implies that interactions within loci are also multiplicative. The quantity 1 + ri is the odds ratio for each + at locus i. The genotypic risk ratios are 1 + ri for +/– heterozygotes and (1 + ri)2 for +/+ homozygotes.
The average contribution of locus i to disease risk is
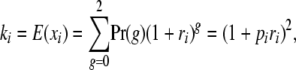 |
(4) |
where g is the number of + alleles (0, 1, and 2) and the second equality follows when genotypes are in Hardy–Weinberg equilibrium.
The contribution of locus i to recurrence risk in relatives with relationship R is
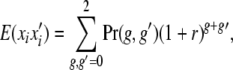 |
(5) |
where the joint probability of g and g′ depends on relatedness.
The epigenetic contribution to risk of site j depends on the presence or the absence of an epigenetic mark. Let 1 denote the presence of a mark and 0 the absence. The difference between an epigenetic mark and an allele is that the mark may be lost or gained in a few generations. The general case can be modeled using a two-state Markov chain. Let αj be the loss rate, defined to be the probability that a chromosome that has a mark at site j gives rise to a chromosome in the next generation that lacks the mark at that site, and let βj be the gain rate, the probability that a chromosome that lacks a mark at site j gives rise to a chromosome that has a mark at that site in the next generation. Both the loss and the gain rates may be time dependent or depend on environmental conditions, but in this section they are assumed to be independent of time. If αj and βj are small, the epigenetic part of the model becomes equivalent to the genetic part. It is easy to allow for more than two epigenetic states by increasing the size of the Markov chain.
Considering the state of an epigenetic site as a two-state Markov chain, the transition matrix in a single generation is
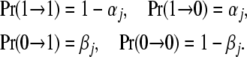 |
(6) |
The standard theory of Markov chains tells us that the equilibrium frequency of a mark at site j is  and that the transition probabilities after m generations are
and that the transition probabilities after m generations are
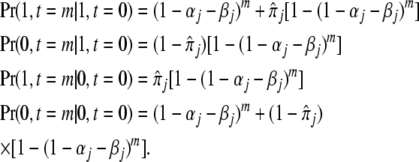 |
(7) |
Because the Markov chain is reversible, these formulas give the probabilities that an epigenetic site in a relative separated by m meioses from a proband is in state 1 or 0 (i.e., has or does not have the epigenetic mark), given the state of the proband. Note that m does not correspond directly to the degree of relationship. For example, a parent–offspring pair and a pair of full siblings are both pairs of first-degree relatives, but m = 1 for parents and offspring and m = 2 for full siblings.
The model assumes that presence of an epigenetic mark at site j increases disease risk by a factor 1 + ρj. If the epigenetic sites are at equilibrium under the current rates of gain and loss of marks, the expected contribution of site j to average risk is
 |
(8) |
where γ = 0, 1, 2 is the number of marks at site j (cf. Equation 4). Similarly
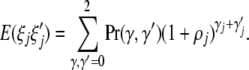 |
(9) |
As shown in the appendix, the joint probability of the epigenetic states in relatives is calculated in the same way as the corresponding probabilities for genetic loci, 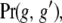 but with the additional complication caused by the possibility that marks may be gained or lost during the meioses that separate close relatives. Although the resulting expressions (Equations A2) are not especially simple in form, they are easily evaluated.
but with the additional complication caused by the possibility that marks may be gained or lost during the meioses that separate close relatives. Although the resulting expressions (Equations A2) are not especially simple in form, they are easily evaluated.
Can epigenetic sites account for missing causality and heritability?
In discussions of missing heritability, there is a tendency to assume genetic and other factors that contribute most to individual risk also contribute most to recurrence risk. But in reality, factors that increase recurrence risk substantially do not necessarily have much effect on average risk and vice versa. The solution to the problem of missing heritability is not necessarily the same as the solution to the problem of missing causality, as has been pointed out by Hemminki et al. (2008).
To illustrate the difference between causality and heritability, consider the comment of McCarthy and Hirschhorn (2008, p. R153) in their discussion of the potential importance of low-frequency alleles: “For instance, the locus-specific sibling relative risk attributable to a variant with control MAF [minor allele frequency] of 1% and a per-allele odds ratio of 3 exceeds that of the strongest common T2D susceptibility variant currently known (TCF7L2) and around 30 such variants distributed across the genome could explain all the residual missing inherited risk for this disease.” In the notation used here, McCarthy and Hirschhorn assume p = 0.01 and r = 2. Thirty such loci would increase the risk ratio for full siblings, λS, by 3.1, which is roughly λS for T2D, thus confirming their statement. The effect on average risk of these 30 loci is relatively minor, however. They would elevate the average risk only by a factor of (1 + 0.01 × 2)60 = 3.28 over the background risk. The reason is that an average individual will carry only one + allele that elevates risk by a factor of 3 over an individual homozygous for – at all 30 loci. Thus such rare alleles can easily account for missing heritability but not for the relatively high average risk of T2D. In contrast, 30 loci with + in frequency 0.2 and an allele-specific odds ratio of 1.25 (r = 0.25) will together increase average risk by a factor of 18.7 but increase λS by a factor of only 1.31.
These numbers provide a convenient reference point to ask what would have to be assumed about epigenetic sites to account for the same contributions to average risk and recurrence risk. The contributions of each epigenetic site can be calculated from the formulas above and in the appendix. The contribution to the average risk depends on the equilibrium frequency of epigenetic marks, 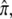 and the effect of each mark on risk, ρ. If there were 30 epigenetic sites with
and the effect of each mark on risk, ρ. If there were 30 epigenetic sites with 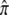 = 0.01 at each and if ρ = 2 for each mark, together they would increase average risk by the same factor as above, 3.28. The contribution to λS depends on the turnover rate of marks, α + β. With
= 0.01 at each and if ρ = 2 for each mark, together they would increase average risk by the same factor as above, 3.28. The contribution to λS depends on the turnover rate of marks, α + β. With 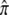 = 0.01, α = 99β. If α = 0.495 and β = 0.005, λS = 1.32 for these 30 sites together, not enough to account for much of the concordance of full siblings. If, instead, α = 0.0495 and β = 0.0005, λS = 2.75. Thus, only if the per generation rate of loss, α, is small can epigenetic marks account for a substantial part of the inherited risk. If α = 0.0495, an epigenetic mark would have to persist for a average of slightly more than 20 generations.
= 0.01, α = 99β. If α = 0.495 and β = 0.005, λS = 1.32 for these 30 sites together, not enough to account for much of the concordance of full siblings. If, instead, α = 0.0495 and β = 0.0005, λS = 2.75. Thus, only if the per generation rate of loss, α, is small can epigenetic marks account for a substantial part of the inherited risk. If α = 0.0495, an epigenetic mark would have to persist for a average of slightly more than 20 generations.
If marks are more common at each site, they can contribute substantially more to average risk. If π = 0.2 and ρ = 0.25, then the contribution to average risk is 18.7 for 30 such sites. However, such sites contribute little to recurrence risk. For example, if α = 0.2 and β = 0.05, then together they increase λS by only 1.16.
At present, too little is known about the frequency or gain and loss rates of inherited epigenetic marks to estimate any parameters of this model and hence to resolve the question of whether inherited epigenetic changes can account for either missing causality or missing heritability. These numerical results indicate, however, that unless epigenetic marks persist for many generations, they are unlikely to contribute much to missing heritability because identity by descent does not imply identity in state. They may well contribute to missing causality despite their having a weak effect on heritability.
If epigenetic marks do persist for very long times, they are equivalent to mutations and hence have the same opportunity to be in significant linkage disequilibrium with linked marker SNPs as do other mutations. In that case, they would be detected in GWAS to the same extent as other mutations.
Environmental effects:
There is some evidence that in mammals parental environment is associated with epigenetic inheritance. In mice, nutritional supplements provided to a mother induce heritable epigenetic changes (Cropley et al. 2006). In humans, several epidemiological studies have indicated that malnutrition in mothers affects their offspring's health (Lumey et al. 2007), suggesting that there are transgenerational epigenetic effects. Recently Heijmans et al. (2008) demonstrated that less DNA methylation of the imprinted IGF2 gene was detected in adults who were prenatally exposed to famine. If environmentally induced heritable epigenetic changes are common and influence disease risk, they would result in transient changes in both average risk and recurrence risk. The model developed here can be used to quantify the potential effect.
To illustrate, consider an epigenetic site at which no mark is present because the rate of gain, β, in the above notation, is 0. At t = 0, the environment changes in such a way that β becomes positive. Epigenetic marks begin to accumulate at the site but they are lost with probability α. Initially, the frequency of marks at that site, π, is 0. If the environmental change is permanent, marks at that site will ultimately reach an equilibrium frequency, 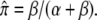 Before the equilibrium frequency is reached, π will increase from 0 according to the following equation:
Before the equilibrium frequency is reached, π will increase from 0 according to the following equation:
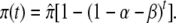 |
(10) |
The population average follows the same course as the probability of the gain of a mark in a single lineage (cf. Equation 7). Equation 10 describes an exponential curve as shown in Figure 1. The time necessary to reach 90% of the equilibrium frequency is
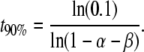 |
(11) |
The approach to equilibrium can take a relatively small number of generations. For example, t90% = 3.3 generations if α = 0.005 and β = 0.495 (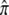 = 0.01) and t90% = 8 generations if α = 0.05 and β = 0.2 (
= 0.01) and t90% = 8 generations if α = 0.05 and β = 0.2 (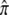 = 0.2). However, if a human generation is ∼25 years, the transient period can easily be ≥75 years.
= 0.2). However, if a human generation is ∼25 years, the transient period can easily be ≥75 years.
Figure 1.—

Illustration of the transient frequency of an epigenetic mark. The curve is a graph of Equation 10, with α = 0.05, β = 0.02.
Before an equilibrium is reached, π(t) will be smaller than its equilibrium value, possibly much smaller. As epigenetic marks at this site become more frequent, they will affect both average risk and recurrence risk in the same way as derived in the previous section. The contribution to the average risk of epigenetic marks at the site in generation t is [1 + π(t)ρ]2. Computing the recurrence risk to relatives is simple if the relatives are members of the same generation (i.e., full or half siblings, first cousins, etc.). In that case Equations A1 and A2 in the appendix can be used with π replaced by π(t). The computation is more complicated for pairs of relatives who are not in the same generation (i.e., parent–offspring, grandparent–offspring, aunt/uncle–niece/nephew, etc.). In that case, formulas similar to Equations A1 and A2 must be derived for each relative pair separately because it is necessary to allow for changes in π(t) between generations. To simplify the results, I restrict the analysis of the case of relatives in the same generation because in practice most interest is in full and half siblings.
If the effect of each epigenetic mark on risk, ρ, does not change with time, then during the transient period before the frequency of marks reaches an equilibrium, the effect on average risk and the recurrence risk to full siblings both change. One example is given in Figure 2. After t90% generations (eight in Figure 2), the contributions of this site to average risk and recurrence risk are nearly at their equilibrium values. The graph shows what was found in other cases as well, that λS responds more quickly to environmental changes than does K. The reason is that, under this model, an epigenetic change that appears in any generation can be transmitted to offspring in the next generation and hence increases recurrence risk, while the average risk increases somewhat more slowly. If such environmental perturbations were frequent, epigenetic changes might increase recurrence risk more than average risk, but that would require that the frequency of perturbations just matches the response time of epigenetic changes at each site.
Figure 2.—

Illustration of the time-dependent behavior of the contribution to average risk, Ki, and the contribution to recurrence risk to full siblings, λiS, for the case of ri = 2, αι = 0.05, βi = 0.2.
DISCUSSION AND CONCLUSION
The theory developed in this article is a first step in quantifying the effect of epigenetic change on disease risk and recurrence risk. Epigenetic changes affect phenotype by modifying gene expression. What is not known is whether the modification of the expression of a particular gene that affects disease risk is more likely to be caused by epigenetic change or by mutation. The above results show that if an epigenetic change and a mutation have the same effect on disease risk and are found in the same population frequency, they will contribute equally to average risk but the mutation will contribute more to recurrence risk. The reason is that the higher rate of loss of epigenetic modifications means that identity by descent does not imply identity in state. Consequently, it will be difficult for inherited epigenetic changes to account for the missing heritability of complex diseases unless they are more common than mutations or have more pronounced effects on disease risk.
The model analyzed is a generalization of a standard population genetic model. The difference is that gains and losses of epigenetic changes are more rapid than is usually assumed for mutations. The analysis was restricted to cases in which the rates of gain and loss, α and β, and the effect of epigenetic changes on disease risk, ρ, were assumed constant and independent of genetic loci and other epigenetic sites. Those assumptions can easily be relaxed but a more elaborate analysis in the absence of any data seems unnecessary at present. The goal of this article is to show that epigenetic inheritance can be modeled in a simple framework that is easily extended to more realistic sets of assumptions.
This model calls attention to the need for empirical studies of the frequency and persistence of transgenerational epigenetic modifications. Such studies are needed to assess the potential importance epigenetic factors for complex inherited diseases. Inherited epigenetic changes must persist for tens of generations or more for them to contribute significantly to similarities of close relatives. Until estimates of persistence times of inherited epigenetic changes are available, it will be difficult to draw firm conclusions about their potential role.
Web resources:
A mathematic program implementing the equations in the appendix is available from the Slatkin laboratory web site, ib.berkeley.edu/labs/slatkin.
Acknowledgments
I thank J. Hollick and M. McCarthy for helpful discussions about this topic and the reviewers for helpful comments on earlier versions of this manuscript. This research was supported in part by National Institutes of Health grant R01-GM40282.
APPENDIX
The problem is to find the joint probability of epigenetic states in pairs of relatives with relationship R. The underlying idea traces to Cotterman's unpublished Ph.D. thesis (Cotterman 1974). In a randomly mating population, the joint probability depends on two coefficients: cR, the probability that pairs of relatives share exactly one allele identical by descent (IBD), and uR, the probability that they share both alleles IBD. When used in the context of epigenetic sites, IBD does not imply identity in state because of the possible gain and loss of epigenetic marks.
The joint probabilities are found by using the equilibrium frequencies of marks at pairs of sites when they are not IBD and using Equations 7 to compute the joint probability when sites are IBD. Consider an epigenetic site j that is identical by descent on two chromosomes, one from the proband and one from the relative. Define Q11 to be the probability that both sites have the epigenetic mark, Q10 to be the probability that the proband has the mark and the relative does not, Q01 to be the probability that the proband lacks the mark and relative has it, and Q00 to be the probability that both lack the mark. From (4)
 |
(A1) |
where the j is dropped for notational convenience.
Adding over all the possible configurations,
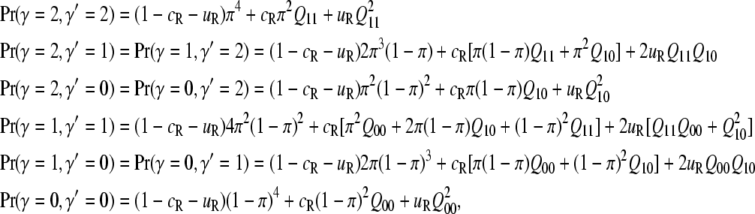 |
(A2) |
where the fact that Q10 = Q01 has been used to simplify the expressions. Although these equations can be expressed in a variety of different algebraic forms, none is particularly simple. If α and β go to zero, Q11 = π, Q00 = 1 – π, Q10 = 0, and (A2) reduces to the corresponding equations for genetic relatedness, which are needed in Equation 4 (with γ replaced by g and π replaced by p). If α and β approach 1, it is straightforward to show that (A2) becomes independent of cR and uR, in which case relatedness does not matter and the recurrence risk, KR, is the average risk, K.
References
- Bird, A., 2007. Perceptions of epigenetics. Nature 447 396–398. [DOI] [PubMed] [Google Scholar]
- Bjornsson, H. T., M. D. Fallin and A. P. Feinberg, 2004. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 20 350–358. [DOI] [PubMed] [Google Scholar]
- Bossdorf, O., C. L. Richards and M. Pigliucci, 2008. Epigenetics for ecologists. Ecol. Lett. 11 106–115. [DOI] [PubMed] [Google Scholar]
- Cotterman, C. W., 1974. A calculus for statistico-genetics (reprint of 1940 Ph.D. Thesis, Ohio State University), pp. 157–272 in Genetics and Social Structure: Mathematical Structuralism in Population Genetics and Social Theory, edited by P. Ballonoff. Dowden, Hutchinson & Ross, Stroudsburg, PA.
- Cropley, J. E., C. M. Suter, K. B. Beckman and D. I. K. Martin, 2006. Germ-line epigenetic modification of the murine A(vy) allele by nutritional supplementation. Proc. Natl. Acad. Sci. USA 103 17308–17312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haig, D., 2007. Weismann rules! OK? Epigenetics and the Lamarckian temptation. Biol. Philos. 22 415–428. [Google Scholar]
- Heijmans, B. T., E. W. Tobi, A. D. Stein, H. Putter, G. J. Blauw et al., 2008. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 105 17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemminki, K., A. Försti and J. L. Bermejo, 2008. The “common disease-common variant” hypothesis and familial risks. PLoS ONE 3 e2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday, R., and J. E. Pugh, 1975. DNA modification mechanisms and gene activity during development. Science 187 226–232. [PubMed] [Google Scholar]
- Johannes, F., V. Colot and R. C. Jansen, 2008. Epigenome dynamics: a quantitative genetics perspective. Nat. Rev. Genet. 9 883–890. [DOI] [PubMed] [Google Scholar]
- Lumey, L. H., A. D. Stein, H. S. Kahn, K. M. van der Pal-de Bruin, G. J. Blauw et al., 2007. Cohort profile: the Dutch Hunger Winter families study. Int. J. Epidemiol. 36 1196–1204. [DOI] [PubMed] [Google Scholar]
- Maher, B. S., 2008. The case of the missing heritability. Nature 456 18–21. [DOI] [PubMed] [Google Scholar]
- McCarthy, M. I., and J. N. Hirschhorn, 2008. Genome-wide association studies: potential next steps on a genetic journey. Hum. Mol. Genet. 17 R156–R165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, D. K., and E. Whitelaw, 2008. The case for transgenerational epigenetic inheritance in humans. Mamm. Genome 19 394–397. [DOI] [PubMed] [Google Scholar]
- Richards, E. J., 2006. Inherited epigenetic variation—revisiting soft inheritance. Nat. Rev. Genet. 7 395–401. [DOI] [PubMed] [Google Scholar]
- Richards, E. J., 2008. Population epigenetics. Curr. Opin. Genet. Dev. 18 221–226. [DOI] [PubMed] [Google Scholar]
- Risch, N., 1990. Linkage strategies for genetically complex traits: I. Multilocus models. Am. J. Hum. Genet. 46 222–228. [PMC free article] [PubMed] [Google Scholar]
- Slatkin, M., 2008. Exchangeable models of complex inherited diseases. Genetics 179 2253–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington, C. H., 1957. The Strategy of the Genes. Allen & Unwin, London.
- Youngson, N. A., and E. Whitelaw, 2008. Transgenerational epigenetic effects. Annu. Rev. Genomics Hum. Genet. 9 233–257. [DOI] [PubMed] [Google Scholar]