

Abstract
Aggressive behavior is observed across animal taxa and is likely to be evolutionarily conserved. Although potentially advantageous, aggression can have social and health consequences in humans, and is a component of a number of psychiatric disorders. As a complex genetic trait, it is modulated by numerous quantitative trait loci (QTL) with allelic effects that can vary in direction and magnitude and that are sensitive to environmental perturbations. Assays to quantify aggressive behavior in Drosophila melanogaster have been developed, making this an ideal model system in which to dissect the genomic architecture underlying manifestation of and variation in aggressive behavior. Here, we map QTL affecting variation in aggression between two wild-type Drosophila strains. We identified a minimum of five QTL in a genomewide scan: two on chromosome 2 and three on chromosome 3. At least three and possibly all five of these QTL interact epistatically. We used deficiency complementation mapping to subdivide two linked, epistatically interacting QTL of large effect on chromosome 3 into at least six QTL, and complementation tests to mutations identified four candidate quantitative trait genes. Extensive epistasis poses a serious challenge for understanding the genetic basis of complex traits.
AGGRESSIVE behavior is observed throughout the animal kingdom and can be essential for survival. Aggression is used to gain access to territory, food, or mates; in defense against predators; to protect offspring; and to gain social status in a dominance hierarchy. On the other hand, aggression can be costly, since violent interactions with others can cause injury, and aggressive displays are energetically expensive and interfere with other essential behaviors, such as feeding and mating. The persistence of high levels of genetic variation for aggression within populations (Dow and Von Schilcher 1975; Hoffmann 1988, 1989; Hoffmann and Cacoyianni 1989) suggests that a balance of evolutionary forces acts to maintain the variation. Theoretical modeling has suggested that frequency-dependent selection could be generating genetic variation in aggression (Maynard Smith and Harper 1988). The observation that many behavioral phenotypes vary widely in a population is often attributed to life history trade-offs (Cabral et al. 2008; Maney 2008); recent work also suggests that animal “personalities” within a population might be adaptive (Wolf et al. 2007). Another potential explanation is that the pleiotropic nature of many genes affecting aggressive behavior subjects them to opposing or multidirectional selective pressures, maintaining variation.
In humans, aggression is often associated with impulsive behavior, and can reach pathological levels. It is a component of a number of behavioral or psychiatric disorders, such as alcoholism, borderline personality disorder, conduct disorder, and Alzheimer's disease. It can also constitute a behavioral pathology in and of itself (e.g., intermittent explosive disorder). Therefore, an understanding of the genetic basis of variation in aggressive behavior is important from the dual perspectives of evolutionary genetics and human health.
Considerable work on the neurobiology and genetics of aggressive behavior in mammals and invertebrates has revealed key molecules required to mediate and modulate aggression, in particular biogenic amines. Normal levels of serotonin (5-hydroxytryptophan, 5-HT) inhibit aggressive behavior in vertebrates, and decreased levels of 5-HT and 5-hydroxyindoleacetic acid (5-HIAA), a metabolite of 5-HT, are associated with increased levels of impulsivity and aggression (Manuck et al. 1999; Nelson and Chiavegatto 2001). In invertebrates, increased 5-HT results in increased aggressive behavior (Edwards and Kravitz 1997; Huber et al. 1997; Baier et al. 2002; Dierick and Greenspan 2007). Monoamine oxidase A (MAOA), which catabolizes 5-HT as well as dopamine and norepinephrine, has also been associated with aggressive behavior in humans; males lacking a functional copy of MAOA (which is X-linked) are highly aggressive (Brunner et al. 1993), and expression levels of brain MAOA are inversely correlated with aggression (Alia-Klein et al. 2008). The noradrenergic (De Almeida et al. 2005) and dopaminergic (Haller et al. 1998) systems also modulate aggressive behavior. The Drosophila homolog of noradrenaline, octopamine, also appears to modulate aggression, since males lacking octopamine display hardly any aggressive behaviors (Baier et al. 2002; Hoyer et al. 2008). Polymorphisms in catecholamine-O-methyltransferase, which degrades dopamine and norepinephrine, are also associated with variation in aggression in schizophrenic men (Han et al. 2004).
The neurotransmitters nitric oxide (NO) and γ-aminobutyric acid (GABA) (Miczek et al. 2003) also modulate aggressive behavior. In humans, haplotypes in the gene encoding nitric oxide synthase (NOS) are associated with increased aggression and/or suicidal behavior (Rujescu et al. 2008). In mice, knocking out expression of neuronal NOS is associated with increased aggression (Nelson et al. 1995), although this may be attributable to the effect of the knockout on 5-HT (Chiavegatto et al. 2001). Neuropeptide Y and its invertebrate homolog, Neuropeptide F, affect aggression in mammals (Karl et al. 2004; Gammie et al. 2007; Karl and Herzog 2007) and Drosophila (Dierick and Greenspan 2007). In Drosophila, correct expression of the male-specific transcript of fruitless, a gene in the sex-determination pathway, is required for executing male aggressive behaviors (Lee and Hall 2000; Vrontou et al. 2006; Certel et al. 2007; Chan and Kravitz 2007).
As a quantitative trait, we expect natural variation in levels of aggression to be influenced by segregating alleles at multiple interacting loci with varying effects that depend on the social and physical environment. Recent studies documenting correlated responses of the transcriptome to artificial selection for divergent levels of aggressive behavior from wild-derived (Edwards et al. 2006) and laboratory (Dierick and Greenspan 2006) strains of Drosophila hint that the genetic basis of natural variation in this behavior may be very complex. However, these studies are unable to discriminate which of the correlated transcriptional responses are caused by polymorphic alleles at loci directly affecting aggressive behavior and which are transregulated by the causal polymorphisms, or hitchhike along with the selected causal polymorphisms. Therefore, we need to use QTL mapping to identify causal loci affecting variation in aggressive behavior between wild-type lines.
Here, we map QTL affecting the difference in aggressive behavior between two laboratory stocks: Oregon-R (Ore), a standard laboratory wild-type strain (Lindsley and Zimm 1992), and 2b, a Russian strain selected for low male mating ability (Kaidanov 1990). Recombinant inbred lines (RILs) derived from these strains have been used previously to map QTL affecting a wide variety of quantitative traits: life span (Nuzhdin et al. 1997; Leips and Mackay 2000; Pasyukova et al. 2000; Vieira et al. 2000; Leips and Mackay 2002; Reiwitch and Nuzhdin 2002; Lai et al. 2007); starvation (Vieira et al. 2000; Harbison et al. 2004); heat and cold stress resistance (Morgan and Mackay 2006); reproductive success (Fry et al. 1998; Wayne et al. 2001); numbers of sensory bristles (Gurganus et al. 1998); sex comb teeth (Nuzhdin and Reiwitch 2000) and ovarioles (Wayne et al. 2001); flight velocity and metabolic traits (Montooth et al. 2003); courtship song (Gleason et al. 2002); and olfactory (Fanara et al. 2002), male mating (Moehring and Mackay 2004), and locomotor (Jordan et al. 2006) behavior. We used a population of introgression lines derived from the recombinant inbred lines to map QTL affecting aggressive behavior. We inferred that there are a minimum of five QTL affecting aggressive behavior in these strains and that at least three of these QTL interact epistatically. Two of the three epistatic QTL were localized to a small region on the left arm of the third chromosome. We used complementation tests to deficiencies and their co-isogenic wild-type strains to map the epistatic QTL in this region with higher resolution. Surprisingly, the region fractionated into at least six QTL and complementation tests to mutations implicated four novel candidate genes affecting variation in aggression between Ore and 2b. The expression and function of these genes informs our understanding of the genetic influences on aggression and lays the foundation for future research.
MATERIALS AND METHODS
Drosophila stocks:
We derived 20 introgression lines from the population of 98 Ore × 2b RILs (Nuzhdin et al. 1997). We screened the RIL genotypes, one chromosome at a time, for partially overlapping segments of the 2b genome in an otherwise Ore background, and chose 20 segmental introgressions that together constitute the entire 2b genome. We then substituted each of the chromosomes containing a 2b introgressed fragment into the Ore background, by standard crosses using balancer chromosomes that had themselves previously been substituted into Ore. The cytological limits of the introgressed portion of the 2b genome, as assessed by insertion sites of roo transposable element markers, are given in supporting information, Table S1.
We obtained DrosDel (Ryder et al. 2007) and Exelixis (Parks et al. 2004) deficiency (Df) stocks with defined molecular breakpoints that had been generated in a w1118 isogenic background and stocks with mutations in positional candidate genes from the Bloomington Drosophila Stock Center, Bloomington, Indiana.
Assay to quantify aggressive behavior:
Eight male flies of the same genotype, aged 3–7 days, were transferred without anesthesia to an empty vial and deprived of food for 90 minutes. They were then exposed to a droplet of standard food and allowed to acclimate for 2 minutes, after which they were observed for 2 minutes. The number of aggressive encounters observed during this period was recorded as the aggression score for the replicate. The average of these scores is the mean aggression score (MAS). All assays were performed between 8 and 11 am, at 25° and 75% humidity.
QTL mapping using introgression lines:
We scored 20 replicate samples of each introgression line for aggressive behavior. We partitioned variance in aggressive behavior between (L) and within (E) introgression lines using the one-way random effects ANOVA model Y = μ + L + E. We estimated the broad sense heritability (H2) of aggressive behavior as H2 = σL2/(σL2 + σE2), where σL2 and σE2 are the among- and within-line variance components, respectively. We then used t-tests to assess whether the mean level of aggression in each introgression line was significantly different from Ore and/or 2b. Introgression lines with significantly different MAS scores from Ore contain a 2b QTL allele affecting aggressive behavior.
Deficiency complementation mapping:
The DrosDel and Exelixis deficiency stocks have isogenic genomic background control lines (w1118). We crossed Ore and 3-88R to each deficiency stock and to the control, and assessed aggressive behavior for 10 replicates of each of four genotypes: Df/Ore, Df/3-88R, w1118/Ore, and w1118/3-88R. The statistical model used to analyze the behavioral data is a two-way ANOVA: Y = μ + L + G + L × G + E, where μ is the overall mean, L is the main effect of line (Ore or 3-88R), G is the main effect of genotype (Df or w1118), and E is the environmental variance. A significant L × G term indicates quantitative failure to complement. As for all complementation tests, failure to complement could arise from different effects of Ore and 2b alleles at one locus in the region uncovered by the deficiency, or nonallelic interactions. Note that failure to complement due to epistasis is minimized in these tests, because the Df strains have no additional mutations and a co-isogenic control strain rather than a balancer chromosome is used as the wild-type allele. Also, the introgression design limits epistasis to other QTL in the introgressed region.
Mutant complementation tests:
The test for quantitative failure to complement using mutants is similar to that for deficiencies. However, since no genomic control line is available, we instead assessed aggressive behavior for 10 replicates of each of the following genotypes: mutant/Ore, mutant/3-88R, Bal/Ore, and Bal/3-88R, where Bal is the balancer chromosome over which the mutation is maintained. The same ANOVA model was used, and the same criteria were applied to assess quantitative complementation. We excluded cases of failure to complement where the difference between Bal/Ore and Bal/3-88R means was not significantly different from that between mutant/Ore and mutant/3-88R means. Mutants meeting these criteria were retested using 20 replicates per genotype. Results from both blocks of testing were pooled and tested together using the model
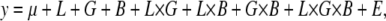 |
where L is the line term, G is the genotype term, and B is the block term.
All statistical analyses were conducted using Statistical Analysis Software Version 8.2 or JMP Version 7.0 (SAS; Cary, NC).
RESULTS AND DISCUSSION
QTL mapping using introgression lines:
A mapping population of RILs derived from Ore and 2b has been used to map QTL affecting a large number of complex traits. We assessed whether these strains also differed in aggressive behavior and found that the MAS of Ore males (20.82 ± 2.25) was greater than twice that of 2b males (8.55 ± 1.17) (t = 2.09, P < 0.0001). We hypothesized that we could improve the power to map QTL while reducing the number of lines to be scored if we constructed a population of segmental introgression lines, each of which contains a fragment of the 2b genome in an otherwise Ore background, but which together comprise the entire 2b genome (Figure 1, Table S1).
Figure 1.—

QTL mapping using introgression lines. Gray bars represent the region of the genome derived from the 2b parental line for each introgression line on (A) the X chromosome, (B) chromosome 2, and (C) chromosome 3. The cytological locations of roo transposable element markers are depicted below each set of lines. Gray bars denote introgressions with a mean aggression score (MAS) that is not significantly different from Ore but is different from 2b; black bars denote introgressions with MAS that are significantly different from Ore but not significantly different from 2b, and gray/black bars denote introgressions with MAS that are intermediate between Ore and 2b, and significantly different from both parental lines. The green bar denotes an introgression with a MAS that is significantly greater than either parent line. The QTL locations are inferred using this physical map in conjunction with the MAS of each introgression line and statistical comparisons of the MAS between the introgression line and the parental lines. Orange bars represent QTL with epistatic effects; red bars indicate QTL with positive additive effects; and blue bars represent QTL with negative additive effects. Three possible models explain the QTL on chromosome 2, shown here in different rows (see text for explanation). Significance levels were determined using Student's t-tests. *P < 0.05, **P < 0.01, ***P < 0.001.
There was significant variation in mean aggression scores among the 20 introgression lines (F19, 359 = 10.72, P < 0.0001; Figure 1). The estimate of broad sense heritability of aggression levels in the introgression lines is H2 = 0.33. All but two lines (2-16 and 3-34R) exhibited significantly (P < 0.05) higher levels of aggression than 2b. Five lines differed from Ore: 2-16, 3-17, 3-31R, and 3-34R were less aggressive and line 3-88R was more aggressive.
We used these data to infer the locations of QTL affecting the difference in aggressive behavior between Ore and 2b. None of the X chromosome introgressions were significantly different from Ore, and all were different from 2b. The most parsimonious inference is that there are no X-linked QTL affecting the difference in aggressive behavior between these strains (Figure 1).
The MAS of chromosome 2 introgression line 2-16 is significantly different from Ore, but not 2b, and therefore contains at least one QTL affecting aggressive behavior. However, the introgressions in lines 2-18 and 2-81R together include the entire 2b fragment in 2-16, which is not consistent with a single QTL model. There are three possible two-QTL models that explain this observation (Figure 1). (1) There is a QTL with a negative effect on aggressive behavior between 29F;30D and another with an equal positive effect in the region of 27B. Line 2-16 contains only the negative effect QTL, while lines 2-78R and 2-18 contain both QTL. (2) There is a QTL with a negative effect on aggressive behavior between 34EF;38E and another with an equal positive effect between 39A;50B. Line 2-16 contains only the negative effect QTL, while line 2-81R contains both QTL. (3) Line 2-16 contains two QTL between 29F;30D and 34EF;38E that interact epistatically to reduce the MAS; neither QTL on its own has a detectable effect on aggressive behavior. These models are indistinguishable on the basis of the available data.
There are at least three QTL on chromosome 3. Lines 3-34R, 3-31R, 3-17, and 3-27 do not have statistically different MAS from each other (data not shown); 3-34R, 3-31R, and 3-17 have significantly lower MAS than Ore, and 3-31R, 3-17, and 3-27 have significantly higher MAS than 2b. Taken together, these results are consistent with a QTL affecting reduced aggressive behavior in the 2b region common to all these introgressions, from 97D;100F. Surprisingly, the MAS of line 3-88R, which includes only the 65D;69D region of the 2b genome, is significantly different from both Ore and 2b, and is nearly twice as aggressive as Ore. Therefore, the effect of one or more QTL in this interval must be suppressed by another QTL elsewhere on the 2b chromosome 3. Further, this introgression is completely overlapped by the introgressions in 3-15 and 3-5, indicating that there must be a least two epistatic QTL in the 65D;69D region, one between 65D;67D and another in the region of 69D (the region of nonoverlap of 3-15 and 3-5). We must further postulate that neither epistatic QTL has an effect on its own, but that the effect of both are suppressed by the QTL at 97D;100F, since line 3-34R contains all three QTL, but is not significantly different from lines 3-17 and 3-27, which only contain the 97D;100F QTL (Figure 1).
Therefore, of the five QTL affecting aggressive behavior, at least three and possibly all five interact epistatically. We previously inferred that epistatic interactions were prevalent among loci affecting natural variation in aggressive behavior, since the estimate of additive genetic variance from response to artificial selection (Edwards et al. 2006) was much less than the estimate of the total genetic variance among inbred lines derived from the same base population as the selection lines (A. C. Edwards, J. F. Ayroles, E. A. Stone, M. A. Carbone, R. F. Lyman and T. F. C. Mackay, unpublished data). In the light of extensive epistasis shaping the genetic architecture of aggressive behavior, we cannot be certain that there are no QTL for aggressive behavior on the X chromosome, only that we cannot detect them with the available genotypes. Furthermore, this experimental design enables us to detect QTL affecting aggressive behavior and infer the presence of interactions among them, but we do not have enough information to estimate all additive and epistatic effects.
Deficiency complementation mapping:
There are at least two epistatically interacting QTL with a large effect on aggressive behavior in the 65D;69D region. Therefore, we used complementation to deficiencies (Pasyukova et al. 2000) to map these QTL with higher resolution. Deficiency mapping has been used to successfully fine-map QTL affecting variation between Ore and 2b for many quantitative traits (Pasyukova et al. 2000; Fanara et al. 2002; De Luca et al. 2003; Harbison et al. 2004; Moehring and Mackay 2004; Jordan et al. 2006). These analyses typically reveal that single QTL fractionate into multiple linked QTL.
The precise breakpoints of this QTL are somewhere between 65A;65D and 69D;70C; therefore we used deficiencies spanning the 65A;70C cytological interval. We introduced two improvements over previous deficiency complementation tests. First, introgression line 3-88R contains this region of the 2b genome in an otherwise Ore genetic background. Therefore we used 3-88R and Ore as the parent lines for the complementation tests. This limits the potential for noncomplementation due to epistatic interactions to interactions between QTL in this region only. Second, we used deficiency stocks from the DrosDel and Exelixis (Parks et al. 2004; Ryder et al. 2007) collections. These deficiencies have been constructed in isogenic backgrounds and do not contain any additional mutations, which reduces the likelihood of identifying a region that fails to complement due to epistatic interactions between mutations carried on the deficiency chromosome, and not the deficiency itself. Furthermore, the breakpoints of the deficiencies are molecularly defined, enabling more precise mapping.
We crossed Ore and 3-88R to 27 deficiency stocks (Df/Bal, where Bal is a balancer chromosome) with overlapping breakpoints spanning the QTL region (Figure 2), and to a w1118 control line, which had the appropriate isogenic background in which the deficiency stocks were constructed. We scored the aggressive behavior of F1 males of four genotypes: Df/Ore, Df/3-88R, w1118/Ore, and w1118/3-88R-3. We analyzed the data for each deficiency by factorial ANOVA (Table 1), which partitions variation among the genotypes into the cross-classified main effects of line (Ore, 3-88R), genotype (Df, w1118) and the line × genotype interaction. We inferred complementation if the line × genotype interaction term was not significant, as this indicates that the difference in phenotype between Ore and 3-88R was the same in the Df and w1118 chromosome backgrounds. We inferred failure to complement if the line × genotype interaction was significant, which indicates the difference in aggressive behavior between Ore and 3-88R varied between the Df and w1118 chromosome backgrounds. We then delineated the QTL locations by the region of nonoverlap of deficiencies complementing the trait phenotype with those that fail to complement the trait phenotype.
Figure 2.—

Deficiency complementation mapping. The bars depict the approximate cytological locations of deficiencies used for complementation mapping. Red bars indicate deficiencies that fail to complement QTL affecting aggressive behavior; green bars represent deficiencies that complement QTL affecting aggression.
TABLE 1.
Deficiency complementation tests
| Deficiency breakpoints | Mean aggression score (SE)
|
ANOVA P-values
|
||||
|---|---|---|---|---|---|---|
| Deficiency genotype | Df/88 MAS | Df/Ore MAS | Line (L) | Genotype (G) | L × G | |
| w1118; Df(3L)ED4341/TM6C, Sb1 | 63F6;64B11 | 12.1 (1.12) | 12.0 (1.21) | 0.9820 | 0.0082 | 0.9224 |
| w1118; Df(3L)ED4342/TM6C, Sb1 | 64B1;64B13 | 15.6 (1.54) | 16.1 (1.89) | 0.8175 | 0.8947 | 0.9054 |
| w1118; Df(3L)ED210/TM6C, Sb1 | 64B11;64D1 | 22.0 (1.75) | 18.3 (1.86) | 0.2237 | 0.0026 | 0.1850 |
| w1118; Df(3L)Exel6105/TM6B, Tb1 | 64D1;64D6 | 16.9 (0.98) | 19.9 (0.66) | 0.0499 | 0.0103 | 0.4879 |
| w1118; Df(3L)Exel6106/TM6B, Tb1 | 64D6;64E2 | 22.8 (1.04) | 22.5 (1.83) | 0.6360 | <0.0001 | 0.4709 |
| w1118; Df(3L)Exel6107/TM6B, Tb1 | 64E5;64F5 | 30.3 (2.30) | 21.8 (1.87) | 0.0089 | <0.0001 | 0.0003* |
| w1118; Df(3L)Exel7210/TM6B, Tb1 | 65A1;65A5 | 17.5 (0.98) | 20.2 (2.23) | 0.0997 | 0.0080 | 0.6151 |
| w1118; Df(3L)Exel8101/TM6B, Tb1 | 65A3;65A9 | 21.1 (1.84) | 18.5 (1.46) | 0.6404 | 0.0007 | 0.1048 |
| w1118; Df(3L)ED211/TM6C, Sb1 | 65A9;65B4 | 11.9 (1.57) | 11.3 (0.94) | 0.8709 | 0.0036 | 0.7789 |
| w1118; Df(3L)ED212/TM6C, Sb1 | 65A9;65D5 | 21.4 (2.77) | 18.8 (1.05) | 0.4203 | 0.0043 | 0.3622 |
| w1118; Df(3L)Exel6110/TM6B, Tb1 | 65E4;65E8 | 16.5 (1.15) | 17.0 (1.11) | 0.4007 | 0.2697 | 0.6828 |
| w1118; Df(3L)Exel6111/TM6B, Tb1 | 65E7;65F4 | 24.3 (1.24) | 16.9 (1.02) | 0.0114 | <0.0001 | 0.0002* |
| w1118; Df(3L)Exel8104/TM6B, Tb1 | 65F7;66A4 | 19.3 (1.63) | 15.7 (1.16) | 0.4032 | 0.1167 | 0.0519 |
| w1118; Df(3L)ED4408/TM6C, Sb1 | 66A22;66C5 | 12.8 (1.32) | 10.2 (1.44) | 0.3737 | 0.0032 | 0.3146 |
| w1118; Df(3R)Exel7317/TM6B, Tb1 | 66C5;66D3 | 17.8 (0.88) | 16.2 (1.12) | 0.9453 | 0.1820 | 0.1838 |
| w1118; Df(3L)ED4421/TM6C, Sb1 | 66D14;67B1 | 11.9 (1.89) | 8.8 (1.89) | 0.3173 | 0.0005 | 0.2679 |
| w1118; Df(3L)Exel6114/TM6B, Tb1 | 67B10;67C5 | 18.5 (1.21) | 15.3 (0.87) | 0.4438 | 0.2143 | 0.0450 |
| w1118; Df(3L)Exel9048/TM6B, Tb1 | 67D1;67D2 | 32.2 (1.33) | 18.1 (1.27) | <0.0001 | <0.0001 | <0.0001* |
| w1118; Df(3L)ED4470/TM6C, Sb1 | 68A6;68E1 | 14.0 (1.86) | 11.6 (2.20) | 0.4551 | 0.0590 | 0.3935 |
| w1118; Df(3L)ED4483/TM6C, Sb1 | 69A4;69D3 | 17.7 (1.17) | 17.0 (2.03) | 0.8489 | 0.2353 | 0.7615 |
| w1118; Df(3L)ED215/TM6C, Sb1 | 69B5;69C4 | 16.9 (1.93) | 10.9 (0.72) | 0.0372 | 0.2946 | 0.0282 |
| w1118; Df(3L)ED4486/TM6C, Sb1 | 69C4;69F6 | 23.9 (2.16) | 14.3 (1.59) | 0.0018 | 0.0212 | 0.0013* |
| w1118; Df(3L)Exel6261/TM6B, Tb1 | 69F6;70A3 | 18.5 (1.44) | 15.8 (1.09) | 0.5931 | 0.1556 | 0.0805 |
| w1118; Df(3L)ED4502/TM6C, Sb1 | 70A3;70C10 | 14.9 (1.08) | 20.3 (1.45) | 0.0424 | 0.1539 | 0.0554 |
| w1118; Df(3L)ED4543/TM6C, Sb1 | 70C6;70F4 | 14.6 (2.34) | 11.9 (1.18) | 0.3929 | 0.1078 | 0.3366 |
| w1118; Df(3L)ED217/TM6C, Sb1 | 70F4;71E1 | 20.0 (1.75) | 17.8 (1.43) | 0.4725 | 0.0267 | 0.4068 |
| w1118; Df(3L)ED218/TM6C, Sb1 | 71B1;71E1 | 17.6 (0.90) | 15.6 (0.91) | 0.4800 | 0.4708 | 0.4072 |
| w1118 (Exel control) | N/A | 14.8 (0.67) | 16.2 (0.96) | N/A | N/A | N/A |
| w1118 (ED control) | N/A | 15.6 (0.80) | 15.7 (1.16) | N/A | N/A | N/A |
Deficiency stocks used in quantitative complementation tests. ED denotes DrosDel deficiencies and Exel indicates Exelixis deficiencies. The isogenic background stocks for both sets of deficiencies are listed as controls. Boldface type indicates deficiencies that failed to complement the QTL for aggressive behavior (P-values of the L × G term from ANOVA < 0.05). *Significant following a Bonferroni correction for multiple tests. Df(3L)ED4502 (results in italics) was marginally significant and was therefore included in complementation tests to mutations.
Note that the interpretation of failure to complement in these tests is complicated by the epistatic gene action of the QTL in the 65A;65D region from 2b, when introgressed in the Ore background. The QTL in the 65D;67D region do not have a main effect on aggressive behavior; neither do the QTL in 69D. The hyperaggressive phenotype only occurs when both QTL alleles from the 2b strain co-occur. Thus, if we observe that a deficiency fails to complement the aggressive phenotype, it must simultaneously both uncover one of the QTL in the introgressed region, and interact epistatically with another.
Six of the deficiency stocks exhibited significant (P < 0.05) quantitative failure to complement (22% of those tested). Four of these tests were significant following a Bonferroni correction for multiple tests (Table 1). Thus, the QTL region was fractionated into multiple smaller QTL. Four of the QTL were in the first of the epistatic QTL inferred from the analyses of the introgression lines (64E5;64F5, 65E8;65F4, 67B10;67C5, and 67D1;67D2), while the other two QTL were in the region of the second epistatic QTL (69B5;69C4 and 69C4;69F6). The effects of all six of these QTL were in the same direction, with the Df/3-88R genotype, which elucidates the effect of the 2b allele in that region, exhibiting higher aggression than the Df/Ore genotype. Each of these significant deficiencies must uncover one of the QTL alleles and interact epistatically with another QTL allele. We cannot map the QTL with which the Dfs interact; all could interact with a single QTL not uncovered by any of the deficiencies, or genes in the regions uncovered by the deficiencies could interact with each other.
An additional QTL in the region of the second epistatic QTL, 70A3;70C10, approached formal significance (P = 0.0554). We included candidate genes from this region in complementation tests to mutants, although the direction of effect of this QTL was opposite to the others, with the Df/Ore allele exhibiting higher levels of aggression.
The 65A;70C cytological interval encompasses nearly 1000 genes mapped to the sequence. Deficiency complementation mapping narrowed the QTL intervals to ∼300 genes: 43 in 64E5;F5, 17 in 65E8;F4, 49 in 67B10;C5, 14 in 67D1;D2, 22 in 69B5;C4, 86 in 69C4;F6, and 70 in 70A3;70C10. Many of the genes in these regions affect metabolism and development.
Mutant complementation tests:
We used quantitative complementation tests to mutations in positional candidate genes in QTL intervals defined by deficiency complementation tests to identify candidate genes corresponding to QTL affecting the difference in aggressive behavior between Ore and 2b. The logic of quantitative complementation tests to mutations is the same as that for deficiency complementation tests. However, the mutations are maintained over balancer chromosomes with visible markers, and they are not all available in a common isogenic background. Therefore, we crossed each mutant stock to both 3-88R and Ore and measured the aggressive behavior of F1 males of the four genotypes: mutant/3-88R, mutant/Ore, Bal/3-88R, and Bal/Ore, where Bal is the balancer chromosome.
We initially screened 58 mutants in positional candidate genes from all seven QTL defined by the deficiency complementation tests (Table S2). Where possible, we used P-element insertional mutations that were generated in the same background. We also chose stocks with few additional mutations, and included mutations in computationally predicted genes. The interpretation of the complementation tests to mutations is not as clear-cut as that from the complementation tests to deficiencies, because the genetic background of the mutant and control (balancer) genotypes are not co-isogenic. If the difference between 88R and Ore were due to alleles with strictly additive effects, this would not matter. However, if epistasis is extensive, the failure to complement could be attributable to differences between the mutant and balancer genotypes that lie outside the 88R introgression region, but which interact with QTL in this region. Therefore, we imposed the additional filter that the pattern of failure to complement in these tests must be consistent with an interaction with the mutant chromosome and not the balancer chromosome.
Of the 58 mutants tested, 12 (∼21%) exhibited quantitative failure to complement, inferred by a significant (P < 0.05) line × genotype interaction term. We excluded five of these mutations from further analyses because the difference between Bal/Ore and Bal/3-88R means was not significantly different from that between mutant/Ore and mutant/3-88R means. We retested the remaining seven mutants, using twice the number of replicates as in the initial screen, and confirmed four mutations that exhibited quantitative failure to complement the QTL alleles affecting the difference in aggressive behavior between Ore and 3-88R: CG11006, CG10754, mutagen-sensitive 312, and Ral guanine nucleotide exchange factor 2 (Figure 3). None of these loci have been previously associated with aggressive behavior. The 3-88R/mutant genotype exhibited higher levels of aggression than the Ore/mutant genotype for CG10754, CG11006, and Rgl; this observation was reversed for the mus312 mutant. That flies hemizygous for the 88R-3 allele at all loci are not the more aggressive underscores the complex nature of QTL affecting aggression. The cumulative effects and potential epistatic interactions at these and additional, unidentified, loci must be teased apart in great detail, as they are neither necessarily predictable nor intuitive.
Figure 3.—




Mutant complementation tests. Means and standard errors of aggression scores (y-axis) are depicted for the four genes that exhibit quantitative failure to complement Ore and 3-88R alleles. The blue lines represent the 3-88R line, and the red lines represent the Ore line. Genotype is depicted on the x-axis, such that four genotypes are shown: Ore/Bal, 3-88R/Bal, Ore/mutant, and 3-88R/mutant. (A) CG10754f03161; (B) CG11006EY12079; (C) mus312D1; and (D) RglGT-000359.
We did not identify candidate genes in the 64E5;64F5, 67B10;67C5, 67D1;67D2, and 69B5;69C4 QTL, and it is possible there are additional candidate genes in the QTL in which we did identify a candidate gene. Ideally, all positional candidate genes should be screened, but there were many of the 300 genes for which no mutation was available, and we did not test mutations in candidate genes when the stock contained multiple additional mutations.
Candidate quantitative trait genes for aggressive behavior:
CG11006 has four predicted transcripts and is annotated as being involved in “cellular component organization and biogenesis” (Lyne et al. 2007). CG11006 is highly expressed in the brain, ovary, and male accessory glands (Chintapalli et al. 2007). The CG11006 protein interacts with 10 other gene products, many of which are also computationally predicted (Giot et al. 2003). However, those that have been described function in central nervous system development, bristle morphogenesis, and calcium ion binding. It is orthologous to the human DMD gene, which is implicated in both Becker and Duchenne muscular dystrophy (Wood et al. 1987; Boyce et al. 1991).
CG10754 is adjacent to and partially overlapping with CG11006. CG10754 has one predicted transcript, is annotated to function in RNA splicing, metabolism, and zinc binding (Lyne et al. 2007), and is expressed in the ovary and male accessory glands (Chintapalli et al. 2007). No gene ontology information is available for the two predicted gene products with which CG10754 interacts (Giot et al. 2003). Variation in CG10754 transcript abundance is associated with chill coma recovery time (Ayroles et al. 2009) and sleep behavior (Harbison et al. 2009) in a reference panel of inbred lines derived from a natural population.
mutagen-sensitive 312 (mus312) has three transcripts, and functions in DNA damage recognition and repair, protein binding, and metabolism (Lyne et al. 2007). mus312 is expressed in the ovary and testes, as well as other tissues (Chintapalli et al. 2007). The MUS312 protein interacts with CG5410 and CG8942 gene products, which are involved in mitochondrial transport and Wnt receptor signaling, respectively (Giot et al. 2003).
Ral guanine nucleotide exchange factor 2 (Rgl) encodes four transcripts and is involved in G-protein coupled receptor and tyrosine kinase signaling (Lyne et al. 2007). It is highly expressed in the brain, crop, ovary, and male accessory glands (Chintapalli et al. 2007). RGL interacts with CG2091, encoded by a computationally predicted gene with no known function (Giot et al. 2003). Rgl is orthologous to the human gene SOS1, which is implicated in the dysmorphic Noonan syndrome (Roberts et al. 2007; Tartaglia et al. 2007).
The four candidate quantitative trait genes affecting the difference in aggressive behavior between Ore and 3-88R have little in common other than that they are all expressed in reproductive tissues. CG10754 and Rgl are potentially involved in neurotransmission through their roles in zinc binding and as a G-protein coupled receptor, respectively.
Complex genetic architecture of aggressive behavior:
We have identified four novel candidate genes affecting natural variation in aggressive behavior in a small genomic region, in two laboratory strains not selected for aggressive behavior. The QTL regions interact epistatically: it remains to be seen how the candidate genes interact, and what the gene(s) are in the 2b 97D;100F QTL region that suppress the effect of the 2b 65D;69D QTL on aggressive behavior. Extensive epistasis poses a serious challenge for understanding the genetic basis of complex traits using linear models of genotype–phenotype associations. The novel candidate genes were not implicated in previous studies that identified large numbers of potential candidate genes affecting aggressive behavior from whole genome expression profiling of genetically divergent strains recently derived from nature (Edwards et al. 2006, 2008). This is not surprising, since different loci can affect variation in complex traits in different strains. Further, the four loci described in this report may not affect aggression through differences in gene expression; they may differ in expression but these differences were too small to detect in the previous studies; or differences in gene expression at a different developmental stage than young adults (when the transcriptional profiles were obtained) are important. Nevertheless, these results highlight the utility of unbiased genome scans for naturally segregating QTL for identifying novel genes and genetic networks affecting complex behaviors.
Acknowledgments
This work has been supported by National Institutes of Health grants GM-R01 GM076083 (to T.F.C.M.) and F31 MH 74161 (to A.C.E.). This is a publication of the W. M. Keck Center for Behavioral Biology.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.101691/DC1.
References
- Alia-Klein, N., R. Z. Goldstein, A. Kriplani, J. Logan, D. Tomasi et al., 2008. Brain monoamine oxidase A activity predicts trait aggression. J. Neurosci. 28 5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroles, J. F., M. A. Carbone, E. A. Stone, K. W. Jordan, R. F. Lyman et al., 2009. Systems genetics of complex traits in Drosophila melanogaster. Nat. Genet. 41 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier, A., B. Wittek and B. Brembs, 2002. Drosophila as a new model organism for the neurobiology of aggression? J. Exp. Biol. 205 1233–1240. [DOI] [PubMed] [Google Scholar]
- Boyce, F. M., A. H. Beggs, C. Feener and L. M. Kunkel, 1991. Dystrophin is transcribed in brain from a distant upstream promoter. Proc. Natl. Acad. Sci. USA 88 1276–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner, H. G., M. R. Nelen, P. Van Zandvoort, N. G. Abeling, A. H. Van Gennip et al., 1993. X-linked borderline mental retardation with prominent behavioral disturbance: phenotype, genetic localization, and evidence for disturbed monoamine metabolism. Am. J. Hum. Genet. 52 1032–1039. [PMC free article] [PubMed] [Google Scholar]
- Cabral, L. G., B. R. Foley and S. V. Nuzhdin, 2008. Does sex trade with violence among genotypes in Drosophila melanogaster? PLoS ONE 3 e1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Certel, S. J., M. G. Savella, D. C. Schlegel and E. A. Kravitz, 2007. Modulation of Drosophila male behavioral choice. Proc. Natl. Acad. Sci. USA 104 4706–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, Y. B., and E. A. Kravitz, 2007. Specific subgroups of FruM neurons control sexually dimorphic patterns of aggression in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 104 19577–19582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiavegatto, S., V. L. Dawson, L. A. Mamounas, V. E. Koliatsos, T. M. Dawson et al., 2001. Brain serotonin dysfunction accounts for aggression in male mice lacking neuronal nitric oxide synthase. Proc. Natl. Acad. Sci. USA 98 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli, V. R., J. Wang and J. A. Dow, 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 39 715–720. [DOI] [PubMed] [Google Scholar]
- De Almeida, R. M., P. F. Ferrari, S. Parmigiani and K. A. Miczek, 2005. Escalated aggressive behavior: dopamine, serotonin and GABA. Eur. J. Pharmacol. 526 51–64. [DOI] [PubMed] [Google Scholar]
- De Luca, M., N. V. Roshina, G. L. Geiger-Thornsberry, R. F. Lyman, E. G. Pasyukova et al., 2003. Dopa decarboxylase (Ddc) affects variation in Drosophila longevity. Nat. Genet. 34 429–433. [DOI] [PubMed] [Google Scholar]
- Dierick, H. A., and R. J. Greenspan, 2006. Molecular analysis of flies selected for aggressive behavior. Nat. Genet. 38 1023–1031. [DOI] [PubMed] [Google Scholar]
- Dierick, H. A., and R. J. Greenspan, 2007. Serotonin and neuropeptide F have opposite modulatory effects on fly aggression. Nat. Genet. 39 678–682. [DOI] [PubMed] [Google Scholar]
- Dow, M. A., and F. Von Schilcher, 1975. Aggression and mating success in Drosophila melanogaster. Nature 254 511–512. [DOI] [PubMed] [Google Scholar]
- Edwards, A. C., S. M. Rollmann, T. J. Morgan and T. F. C. Mackay, 2006. Quantitative genomics of aggressive behavior in Drosophila melanogaster. PLoS Genet. 2 e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, D. H., and E. A. Kravitz, 1997. Serotonin, social status and aggression. Curr. Opin. Neurobiol. 7 812–819. [DOI] [PubMed] [Google Scholar]
- Fanara, J. J., K. O. Robinson, S. M. Rollmann, R. R. Anholt and T. F. Mackay, 2002. Vanaso is a candidate quantitative trait gene for Drosophila olfactory behavior. Genetics 162 1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry, J. D., S. V. Nuzhdin, E. G. Pasyukova and T. F. Mackay, 1998. QTL mapping of genotype-environment interaction for fitness in Drosophila melanogaster. Genet. Res. 71 133–141. [DOI] [PubMed] [Google Scholar]
- Gammie, S. C., A. P. Auger, H. M. Jessen, R. J. Vanzo, T. A. Awad et al., 2007. Altered gene expression in mice selected for high maternal aggression. Genes Brain Behav. 6 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giot, L., J. S. Bader, C. Brouwer, A. Chaudhuri, B. Kuang et al., 2003. A protein interaction map of Drosophila melanogaster. Science 302 1727–1736. [DOI] [PubMed] [Google Scholar]
- Gleason, J. M., S. V. Nuzhdin and M. G. Ritchie, 2002. Quantitative trait loci affecting a courtship signal in Drosophila melanogaster. Heredity 89 1–6. [DOI] [PubMed] [Google Scholar]
- Gurganus, M. C., J. D. Fry, S. V. Nuzhdin, E. G. Pasyukova, R. F. Lyman et al., 1998. Genotype–environment interaction at quantitative trait loci affecting sensory bristle number in Drosophila melanogaster. Genetics 149 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller, J., G. B. Makara and M. R. Kruk, 1998. Catecholaminergic involvement in the control of aggression: hormones, the peripheral sympathetic, and central noradrenergic systems. Neurosci. Biobehav. Rev. 22 85–97. [DOI] [PubMed] [Google Scholar]
- Han, D. H., D. B. Park, C. Na, B. S. Kee and Y. S. Lee, 2004. Association of aggressive behavior in Korean male schizophrenic patients with polymorphisms in the serotonin transporter promoter and catecholamine-O-methyltransferase genes. Psychiatry Res. 129 29–37. [DOI] [PubMed] [Google Scholar]
- Harbison, S. T., A. H. Yamamoto, J. J. Fanara, K. K. Norga and T. F. Mackay, 2004. Quantitative trait loci affecting starvation resistance in Drosophila melanogaster. Genetics 166 1807–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison, S. T., M. A. Carbone, J. F. Ayroles, E. A. Stone, R. F. Lyman et al., 2009. Co-regulated networks that contribute to natural variation in Drosophila sleep. Nat. Genet. 41 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, A. A., 1988. Heritable variation for territorial success in two Drosophila melanogaster populations. Anim. Behav. 36 1180–1189. [Google Scholar]
- Hoffmann, A. A., 1989. Geographic variation in the territorial success of Drosophila melanogaster males. Behav. Genet. 19 241–255. [DOI] [PubMed] [Google Scholar]
- Hoffmann, A. A., and Z. Cacoyianni, 1989. Selection for territoriality in Drosophila melanogaster: correlated responses in mating success and other fitness components. Anim. Behav. 38 23–34. [Google Scholar]
- Hoyer, S. C., A. Eckart, A. Herrel, T. Zars, S. A. Fischer et al., 2008. Octopamine in male aggression of Drosophila. Curr. Biol. 18 159–167. [DOI] [PubMed] [Google Scholar]
- Huber, R., K. Smith, A. Delago, K. Isaksson and E. A. Kravitz, 1997. Serotonin and aggressive motivation in crustaceans: altering the decision to retreat. Proc. Natl. Acad. Sci. USA 94 5939–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, K. W., T. J. Morgan and T. F. Mackay, 2006. Quantitative trait loci for locomotor behavior in Drosophila melanogaster. Genetics 174 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanov, L. Z., 1990. The rules of genetic alteration of Drosophila melanogaster inbred lines determined by selection. Arch. Biol. Nauka 42 131–148. [Google Scholar]
- Karl, T., and H. Herzog, 2007. Behavioral profiling of NPY in aggression and neuropsychiatric diseases. Peptides 28 326–333. [DOI] [PubMed] [Google Scholar]
- Karl, T., S. Lin, C. Schwarzer, A. Sainsbury, M. Couzens et al., 2004. Y1 receptors regulate aggressive behavior by modulating serotonin pathways. Proc. Natl. Acad. Sci. USA 101 12742–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, C. Q., L. D. Parnell, R. F. Lyman, J. M. Ordovas and T. F. Mackay, 2007. Candidate genes affecting Drosophila life span identified by integrating microarray gene expression analysis and QTL mapping. Mech. Ageing Dev. 128 237–249. [DOI] [PubMed] [Google Scholar]
- Lee, G., and J. C. Hall, 2000. A newly uncovered phenotype associated with the fruitless gene of Drosophila melanogaster: aggression-like head interactions between mutant males. Behav. Genet. 30 263–275. [DOI] [PubMed] [Google Scholar]
- Leips, J., and T. F. Mackay, 2000. Quantitative trait loci for life span in Drosophila melanogaster: interactions with genetic background and larval density. Genetics 155 1773–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leips, J., and T. F. Mackay, 2002. The complex genetic architecture of Drosophila life span. Exp. Aging Res. 28 361–390. [DOI] [PubMed] [Google Scholar]
- Lindsley, D. L., and G. G. Zimm, 1992. The Genome of Drosophila melanogaster. Academic Press, San Diego.
- Lyne, R., R. Smith, K. Rutherford, M. Wakeling, A. Varley et al., 2007. FlyMine: an integrated database for Drosophila and Anopheles genomics. Genome Biol. 8 R129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maney, D. L., 2008. Endocrine and genomic architecture of life history trade-offs in an avian model of social behavior. Gen. Comp. Endocrinol. 157 275–282. [DOI] [PubMed] [Google Scholar]
- Manuck, S. B., J. D. Flory, R. E. Ferrell, K. M. Dent, J. J. Mann et al., 1999. Aggression and anger-related traits associated with a polymorphism of the tryptophan hydroxylase gene. Biol. Psych. 45 603–614. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., and D. G. Harper, 1988. The evolution of aggression: can selection generate variability? Phil. Trans. R. Soc. Lond. B Biol. Sci. 319 557–570. [DOI] [PubMed] [Google Scholar]
- Miczek, K. A., E. W. Fish and J. F. De Bold, 2003. Neurosteroids, GABAA receptors, and escalated aggressive behavior. Horm. Behav. 44 242–257. [DOI] [PubMed] [Google Scholar]
- Moehring, A. J., and T. F. Mackay, 2004. The quantitative genetic basis of male mating behavior in Drosophila melanogaster. Genetics 167 1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montooth, K. L., J. H. Marden and A. G. Clark, 2003. Mapping determinants of variation in energy metabolism, respiration and flight in Drosophila. Genetics 165 623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, T. J., and T. F. Mackay, 2006. Quantitative trait loci for thermotolerance phenotypes in Drosophila melanogaster. Heredity 96 232–242. [DOI] [PubMed] [Google Scholar]
- Nelson, R. J., and S. Chiavegatto, 2001. Molecular basis of aggression. Trends Neurosci. 24 713–719. [DOI] [PubMed] [Google Scholar]
- Nelson, R. J., G. E. Demas, P. L. Huang, M. C. Fishman, V. L. Dawson et al., 1995. Behavioral abnormalities in male mice lacking neuronal nitric oxide synthase. Nature 378 383–386. [DOI] [PubMed] [Google Scholar]
- Nuzhdin, S. V., E. G. Pasyukova, C. L. Dilda, Z. B. Zeng and T. F. Mackay, 1997. Sex-specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 94 9734–9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin, S. V., and S. G. Reiwitch, 2000. Are the same genes responsible for intra- and interspecific variability for sex comb tooth number in Drosophila? Heredity 84 97–102. [DOI] [PubMed] [Google Scholar]
- Parks, A. L., K. R. Cook, M. Belvin, N. A. Dompe, R. Fawcett et al., 2004. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36 288–292. [DOI] [PubMed] [Google Scholar]
- Pasyukova, E. G., C. Vieira and T. F. Mackay, 2000. Deficiency mapping of quantitative trait loci affecting longevity in Drosophila melanogaster. Genetics 156 1129–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiwitch, S. G., and S. V. Nuzhdin, 2002. Quantitative trait loci for lifespan of mated Drosophila melanogaster affect both sexes. Genet. Res. 80 225–230. [DOI] [PubMed] [Google Scholar]
- Roberts, A. E., T. Araki, K. D. Swanson, K. T. Montgomery, T. A. Schiripo et al., 2007. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat. Genet. 39 70–74. [DOI] [PubMed] [Google Scholar]
- Rujescu, D., I. Giegling, L. Mandelli, B. Schneider, A. M. Hartmann et al., 2008. NOS-I and -III gene variants are differentially associated with facets of suicidal behavior and aggression-related traits. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B 42–48. [DOI] [PubMed] [Google Scholar]
- Ryder, E., M. Ashburner, R. Bautista-Llacer, J. Drummond, J. Webster et al., 2007. The DrosDel deletion collection: a Drosophila genomewide chromosomal deficiency resource. Genetics 177 615–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia, M., L. A. Pennacchio, C. Zhao, K. K. Yadav, V. Fodale et al., 2007. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 39 75–79. [DOI] [PubMed] [Google Scholar]
- Vieira, C., E. G. Pasyukova, Z. B. Zeng, J. B. Hackett, R. F. Lyman et al., 2000. Genotype-environment interaction for quantitative trait loci affecting life span in Drosophila melanogaster. Genetics 154 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrontou, E., S. P. Nilsen, E. Demir, E. A. Kravitz and B. J. Dickson, 2006. fruitless regulates aggression and dominance in Drosophila. Nat. Neurosci. 9 1469–1471. [DOI] [PubMed] [Google Scholar]
- Wayne, M. L., J. B. Hackett, C. L. Dilda, S. V. Nuzhdin, E. G. Pasyukova et al., 2001. Quantitative trait locus mapping of fitness-related traits in Drosophila melanogaster. Genet. Res. 77 107–116. [DOI] [PubMed] [Google Scholar]
- Wolf, M., G. S. Van Doorn, O. Leimar and F. J. Weissing, 2007. Life-history trade-offs favour the evolution of animal personalities. Nature 447 581–584. [DOI] [PubMed] [Google Scholar]
- Wood, D. S., M. Zeviani, A. Prelle, E. Bonilla, G. Salviati et al., 1987. Is nebulin the defective gene product in Duchenne muscular dystrophy? N. Engl. J. Med. 316 107–108. [PubMed] [Google Scholar]