

Abstract
The therapeutic potential of cytolytic peptides is plagued by nonspecificity and enzymatic degradation. We report the first stable incorporation of melittin (a 26 amino acid amphipathic peptide) into an outer lipid monolayer of perfluorocarbon nanoparticles. Melittin binds avidly to the nanoparticles (dissociation constant ~3.27 nM) and retains its pore-forming activity after contact-mediated delivery to model bilayer membrane (liposomes) thereby demonstrating the effectiveness of perfluorocarbon nanoparticles as unique nanocarriers for cytolytic peptides.
Melittin is a water-soluble cationic amphipathic 26 amino acid α-helical peptide (Figure 1A) derived from the venom of the honeybee Apis Mellifera.1 The basis of melittin action is a physical and chemical disruption of membrane structures that leads to cell lysis.2,3 Melittin (and other such cytolytic peptides) are attractive candidates for cancer chemotherapy for two reasons: (1) cancer cells are less likely to develop resistance to a membrane pore former and (2) the combination of a chemotherapeutic drug (e.g., doxorubicin, paclitaxel) together with melittin could be synergistic, thereby reducing the required therapeutic dose of either one.4 Although the potential applicability of melittin (and other cytolytic peptides) as a cancer chemotherapeutic agent has long been recognized, the rapid degradation of the peptide in blood and its nonspecific cellular lytic activity pose significant challenges. Recent studies have highlighted differences in the action of melittin on lipid membranes with varied chemical compositions that may render it more lethal to cancer cells, especially when coupled with the use of D-amino acid constituents that would prevent its cleavage by serum peptidases.5 The membrane disrupting action of cytolytic peptides in traditional bilayer systems such as liposomes6 has proved to be a major hurdle in achieving cytolytic peptide delivery to tumor cells. The need to employ novel drug delivery systems has been highlighted in a recent review on the potential utility of cytolytic peptides for cancer therapeutics.7 Melittin thus represents one of a large number of membrane-active peptides that have been difficult to employ as a therapeutic agent.
Figure 1.
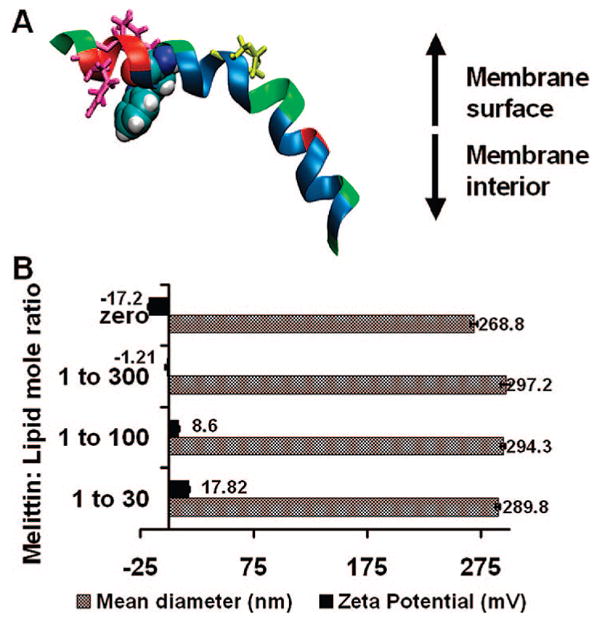
Characterization of melittin-loaded nanoparticles. (A) Structure of melittin: tryptophan, blue; proline, yellow; arginine and lysine, hot pink. (B) Mean hydrodynamic diameter and zeta potential of nanoparticles before and after incorporation of melittin.
Previously, we have shown the effectiveness of perfluorocarbon (PFC) nanoparticles for molecular imaging and site-specific drug delivery in various pathological disorders including, but not limited to, atherosclerosis and cancer.8–11 These nanoparticles are formulated as an oil-in-water emulsion such that the hydrophobic perfluorooctyl bromide (PFOB) nanoparticles are dispersed in water stabilized by a surfactant lipid monolayer (98 mol % egg lecithin and 2 mol % dipalmitoylphosphatidylethanolamine, DPPE). We hypothesized that membrane-active peptides might constitute attractive candidates for incorporation onto the nanodroplet monolayer that might take full advantage of the delivery mechanism proposed in this work. Because of the hydrophobicity and lipophobicity of the perfluorocarbon core that militates against effective pore formation and particle destruction by peptides, the presence of a lipid membrane that can harbor peptide amphiphiles such as melittin, and the ease of decoration with targeting ligands attached via lipid anchors,12,13 we propose that these perfluorocarbon vehicles may represent an ideal carrier for therapeutic cytolytic peptides. In this study, we formulate melittin-carrying perfluorocarbon nanoparticles and confirm the incorporation of melittin by surface plasmon resonance (SPR), tryptophan fluorescence, and circular dichroism (CD) spectroscopy. Finally, we demonstrate how melittin can move from the nanoparticle monolayer onto a model lipid bilayer (e.g., liposome) across a hemifusion stalk and effect its biological pore-forming action.
To formulate melittin-loaded nanoparticles, the perfluorocarbon nanoparticles were synthesized by sonication and microfluidization and the stabilization achieved by using 98 mol % egg lecithin and 2 mol % dipalmitoylphosphatidylethanolamine. Melittin (in selected amounts in phosphate-buffered saline) was then added to the preformed nanoparticles, and the residual free (unbound) melittin washed out by centrifugation at 1000 rpm for 10 min. The amount of unbound melittin was quantified by measuring the tryptophan fluorescence. Depending on the amount of melittin added, the melittin-loaded nanoparticles exhibited lipid:melittin ratios varying from 1000 to 30. We monitored the mean particle diameter and the zeta potential with varying lipid: melittin ratios (Figure 1B). No change was manifest in the mean diameter (~290 nm) of the PFC nanoparticles after addition of melittin, but as expected, with increasing melittin to lipid ratios, the zeta potential of the nanoparticles increased from −17.2 mV (without melittin) to 17.82 mV (lipid:melittin ratio 30). Further, unlike bilayer liposomes that clearly show the typical semilunar crescents and disruption after melittin incorporation (Figure 2A,B), the structural integrity of melittin-carrying perfluorocarbon nanoparticles was confirmed by transmission electron microscopy with the visible monolayer lipid staining being unchanged before and after addition of melittin (Figure 2A,B). The schematic depiction of interaction of melittin with liposomes leading to pore formation and its stable insertion in the lipid monolayer of PFC nanoparticles is shown in Figure 2C.
Figure 2.
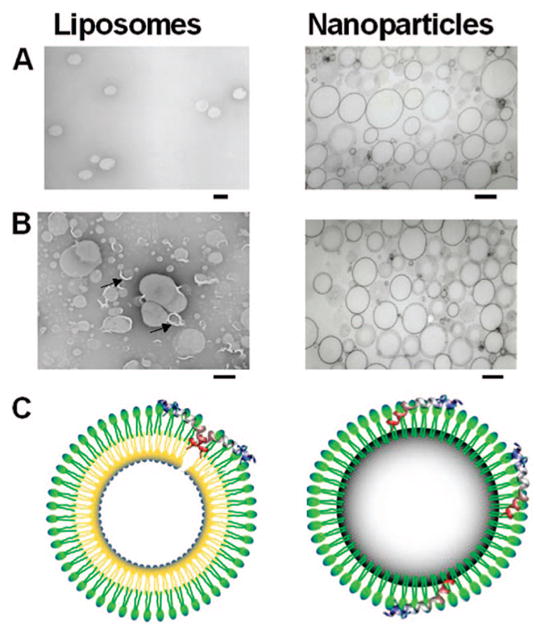
Stable insertion of melittin in PFC nanoparticles. Transmission electron micrographs of liposomes and perfluorocarbon nanoparticles of identical lipid compositions before (A) and after (B) incorporation of melittin. Scale bars correspond to 200 nm. Arrows point to disruptions of the liposome membrane. (C) A schematic of the proposed structure of PFC nanoparticle with the melittin inserted in a monolayer of phospholipids stabilizing the individual nanoparticle structure. Also shown is a diagram of a bilayer liposome disrupted due to melittin insertion.
We employed surface plasmon resonance to determine the release rate of melittin from PFC nanoparticles. Previously the interaction of melittin with liposomes has been studied by surface plasmon resonance, and the binding to the membrane was seen to precede the formation of a membrane pore.14 Surface plasmon resonance studies were carried out
where M and L denote melittin and lipid, respectively. The first stage of the reaction included the rapid association of melittin with the nanoparticles. The ka1 was 1.16 (±0.017) × 105 (M s)−1 and kd1 was 0.0563 (±0.0004) s−1. For the second stage of the reaction, ka2 was 0.104 (±0.0007) s−1 and kd2 was 0.007 (±0.0001) s−1. The dissociation constant for the entire process was therefore 3.27 nM. This was much lower when compared with reported values for melittin dissociation constant from bilayered PC/cholesterol and PE/PG membranes (~1.5 μM),14 representing the increased affinity of melittin for association with PFC nanoparticles under these conditions. The electron microscopy and surface plasmon resonance data together demonstrate that melittin can interact with amphiphile-stabilized perfluorocarbon emulsions without the formation of a disruptive transmembrane pore complex.
The characterization of melittin binding to the monolayer of nanoparticles was undertaken using circular dichroism to delineate the secondary structure of nanoparticle-associated melittin. When melittin binds to a bilayer membrane, it adopts an α-helical configuration.17,18 We compared the CD spectra of nanoparticle-bound and free-melittin to determine whether similar changes in the secondary structure of melittin occurred upon binding to the nanoparticles. For peptides and proteins, absorption in the spectral region below 240 nm is due principally to the peptide bond; a weak but broad n to π transition centered around 220 nm and a more intense π to π* transition around 190 nm. When melittin is associated with the nanoparticle lipid monolayer, a double negative peak is observed: one at 220 nm and other at 208 nm characteristic of a α-helical configuration (Figure 3B).
Figure 3.
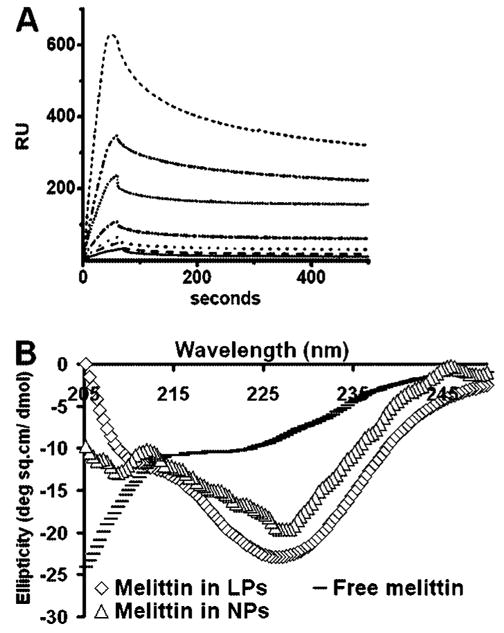
Characterization of melittin binding to nanoparticles. (A) Binding of melittin to nanoparticles immobilized on the surface of a hydrophilic L1 chip. Sensorgrams show the kinetics of binding of melittin to lipid monolayers of perfluorocarbon nanoparticles coated on a Biacore L1 chip. Melittin concentrations (from top to bottom) were 1000, 500, 250, 100, 50, 25, and 15 nM. (B) Far-UV CD spectra of melittin in PBS, bilayered liposomes (LPs), and in monolayered PFOB nanoparticles (NPs) at 25 °C at a lipid/melittin ratio of 80. Note the double negative peak (208 and 228 nm) for melittin inserted in nanoparticle lipid monolayer characteristic of a α-helical conformation.
Previous spectroscopic studies of the interaction of melittin with liposome bilayer membranes have demonstrated that the tryptophan at position 19 embeds in the membrane and physically responds to membrane association.19 We used the intrinsic tryptophan fluorescence (TRP19) in melittin (excitation, 280 nm; emission, 355 nm) to study the nature of the melittin interaction with the lipid monolayer of perfluorocarbon nanoparticles. Typically melittin undergoes a blue shift (or a change in emission from 350 to 345 nm) upon insertion into lipid bilayers due to the hydrophobic membrane environment.20 No blue shift was manifest when the melittin in solution incorporated into the lipid monolayer of the nanoparticles. Surprisingly, the fluorescence of nanoparticle-associated melittin was completely quenched (Figure 4A). Because tryptophan fluorescence quenching is not generally a feature of peptide interaction with membranes, we tested the special role of the PFOB core as a mechanism for the observed fluorescence quenching upon integration into nanoparticles. Bromine-containing molecules are known to quench the fluorescence of tryptophan either by (1) heavy atom collisional quenching that requires contact between the tryptophan and bromine or (2) Forster energy transfer, because brominated molecules exhibit significant absorption at the wavelength of tryptophan emission.21–23 By substituting a perfluoro-15-crown-5-ether core for the PFOB in the nanoparticles, the effect of bromine on the fluorescence spectra of melittin was eliminated. The crown ether core nanoparticles no longer quenched the melittin fluorescence, indicating the specific role of PFOB in the quenching of melittin tryptophan fluorescence (Figure 4B). Intermolecular quenching only occurs at short distances (the Forster distance at which quenching is 50% efficient is 8 Å).23 Though the nanoparticle environment may extend this distance,24 the tryptophan residue at position 19 must be located well within the lipid monolayer in the tail region close to the brominated core of the nanoparticle. This specific interaction could indeed in part explain the nanomolar dissociation constants calculated from surface plasmon resonance experiments.
Figure 4.
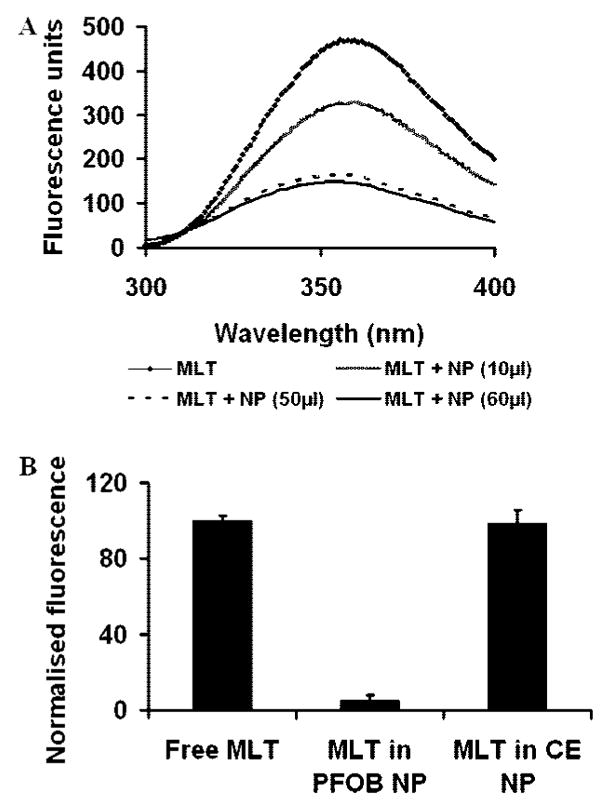
Interaction of melittin tryptophan with PFC nanoparticles. (A) Fluorescence emission spectra showing the quenching of endogenous tryptophan fluorescence of melittin (MLT, 20 μM) upon insertion into the lipid monolayer of nanoparticles (NP) with a PFOB core (10 μL, 3.21 μM PFOB; 50μL, 16.03 μM PFOB; 60μL, 19.24 μM PFOB). (B) Percentage quenching of melittin tryptophan fluorescence with different perfluorocarbon cores in nanoparticles: MLT, melittin; CE, perfluoro-15-crown 5-ether. Melittin concentration, 20 μM. The lipid/melittin ratio was 80.
These results confirm that melittin in this lipid monolayer environment assumes a α-helical conformation similar to that reported for melittin in bilayer membranes. Also, the melittin interacts with the hydrophobic brominated perfluorocarbon core of the nanoparticles and exists as a stably integrated component of the monolayer membrane in its α-helical form. This finding is significant because it indicates that melittin embedded in the nanoparticle lipid monolayer in its expected α-helical conformation might transfer readily to a cellular bilayer membrane during the interaction between the amphipathic-stabilized nanoparticles and a targeted cellular bilayer.
To be an effective carrier of therapeutic agents, the nanoparticles must transfer melittin (or any other drug) rapidly in an active form to the target bilayer membranes. To begin to understand the process involved in this transfer, we first sought to elucidate the mechanism of interaction of lipid monolayered PFOB nanoparticles with bilayered liposomes. We hypothesized that the nanoparticle lipid monolayer and the outer lipid layer of the liposomes would form a hemifusion stalk. This would lead to the movement of membrane-active peptides like melittin from the nanoparticle lipid monolayer to the target membrane. Also, the fact that melittin is already embedded in it is natural α-helical configuration in the nanoparticle monolayer would aid in its transfer to the liposomes. To visualize the interaction between nanoparticles and 200 nm liposomes, we employed deep etch platinum replica electron microscopy. To obtain images of the complex between nanoparticles and liposomes, these complexes were concentrated by centrifugation. Panels A and B of Figure 5 show a nanoparticle and a liposome visualized by platinum replica after freeze fracture at 170 K.25 The PFOB of the nanoparticle core does not take the platinum replica well and is therefore is poorly visualized (Figure 5A). However disruption of the outer membrane leaflet exposes no second membrane leaflet underneath. This is distinct from the typical bilayer structure of our liposome preparation in Figure 5B. The centrifuged preparation of nanoparticles and liposomes was composed primarily of structures similar to those in panels C and D of Figure 5. The structures are polar and manifest a large range of smooth versus ruffled structures. Clearly the fracture of the smooth component shows a typical interfacial fracture plane representing the liposome bilayer and the ruffled structure is the remnant of the nanoparticle.
Figure 5.
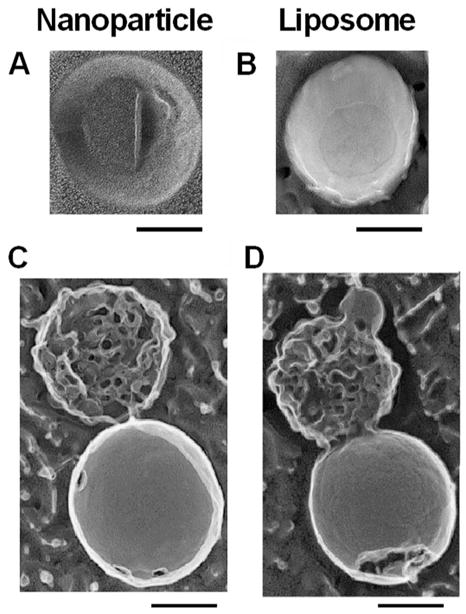
Interaction of PFC nanoparticles with liposomes. Deep etch platinum replicas of nanoparticles (A), liposomes (B), and the nanoparticle–liposome complex structures (C and D). The PFOB appears resistant to forming a complete platinum replica and spread in a layer on the support. We fractured the preparations of the nanoparticles and the liposomes. The liposome in D clearly shows the fracture along the interfacial bilayer plane. The nanoparticle does not display a classical fracture but rather displays a torn monolayer with the underlying PFOB not visualized by the platinum. The complexes between nanoparticles and liposomes were isolated and concentrated by centrifugation. This preparation contained many complex structures like the ones seen in panels C and D. Scale bars correspond to 100 nm.
We assessed the functional activity of melittin bound to nanoparticles by determining pore formation upon transfer to liposomes.26–29 We used uniform unilamellar liposomes prepared as described in the Supporting Information to define a melittin/lipid activation scale for the formation of the melittin pore. This strategy permitted us to compare the pore activation by melittin when transferred in free form from solution as compared to its bound form on the nanoparticles. We prepared PFOB nanoparticles with 0.35 nmol of melittin per milligram of nanoparticles. When these nanoparticles were mixed with liposomes containing carboxyfluorescein at quenching concentrations, we observed a rapid dequenching of the fluorescence that resulted from melittin pore formation in the liposomes (Figure 6A). When devoid of bound melittin, the nanoparticles produced no dequenching.
Figure 6.
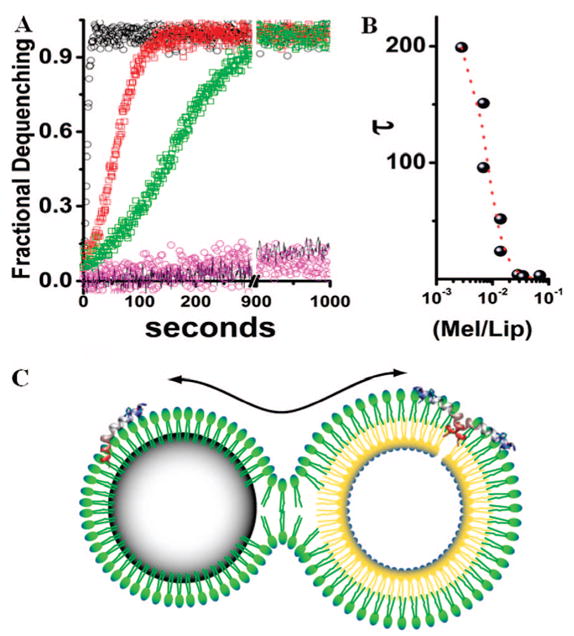
Activity of nanoparticle-bound melittin on liposomes. (A) A comparison of pore activation by melittin transferred to liposomes from solution or from the surface of nanoparticles measured by dequenching times due to release of carboxyfluorescein from liposome core. Black circles are pore activation from 200 nM free melittin, t1/2 = 2.8 s. Red circles are pore activation from the 200 nM melittin bound to 40 mg of nanoparticles, t1/2 = 21 s. Green squares are pore activation by melittin transferred to liposomes from 20 mg nanoparticles with 200 nM melittin, t1/2 = 58 s. Also shown is the fluorescence dequenching in the presence of the nanoparticles without melittin (pink) and the dequenching of incubated liposomes (black line). See text for the interpretation of the melittin kinetics. (B) The concentration dependence of melittin as a function of the ratio of melittin to lipid versus the half-time for dequenching. (C) Schematic of the proposed hemifusion model between a nanoparticle and a liposome. The continuity of the emulsion monolayer and the outer leaflet of the liposome permits the transfer of cargo from the PFC nanoparticle to the bilayer membrane leading to formation of a pore and subsequent release of carboxyfluorescein from the liposome core.
The time constant for pore activation in liposomes was determined with the use of 0.8 mg of the nanoparticles containing 1.4 nmol of melittin. This aliquot of the treated nanoparticles suspended in the assay volume represented a total melittin concentration of 2.8 μM which was completely bound to the nanoparticles. Because the melittin is confined to the nanoparticles, the effective melittin concentration depends upon the nanoparticle concentration. Therefore 2.8 μM melittin in the assay volume corresponded to 10 fM lytic particles at a mean particle diameter of 289.8 nm where the ratio of moles of lytic particles to lipid is 0.00026. The time constant for 50% dequenching (t1/2) is ~200 s. In the assay system the same ratio of free melittin molecules to lipid exhibits a 10-fold longer time constant ~2000 s. Considered in this way it is clear that the nanoparticle bound melittin is fully active. In fact, when transferred from the nanoparticles, this mode of delivery enhances melittin transfer to liposomes by approximately 12-fold per milligram of lytic particles. Thus, under conditions when no free melittin can be detected in solution, the melittin-loaded nanoparticles initiated dramatic pore activation in unilamellar liposomes. Although the liposomes did not contain a specific target receptor for the nanoparticles, we mixed them at concentrations where particle diffusion yielded a measurable rate of pore activations as shown in Figure 6A. Under these conditions the concentration of lytic particles determines the transfer to liposomes and the subsequent pore activation. This manipulation, coupled with the defined experimental system where there is only one target for delivery (i.e., liposomes) allowed us to study the kinetics of melittin pore activation. Accordingly, we postulate that when melittin is delivered by the nanoparticles, its activity is not diminished. Furthermore, because melittin multimers are required to form membrane pores, the melittin–nanoparticle complex must deliver multiple active melittin peptides to any single liposome target. Moreover, the speed of this delivery is facilitated by the high fluidity that the PFOB core generates in the stabilizing amphiphile monolayer on the nanoparticles that augments rapid mixing of components.30,31 These properties significantly improve the efficacy of PFOB emulsion-based nanoparticles as therapeutic vehicles for contact-mediated transfer of such peptides and other selected therapeutic agents. As shown schematically in Figure 6B, the close apposition of the nanoparticle monolayer and a liposome bilayer is proposed to result in the formation of a hemifusion stalk. The lipids and cargo can then mix by diffusion across this stalk. The process of mixing of membrane components across hemifusion structures is a well-described consequence of fusion of bilayer membranes in both liposomes and physiological membranes.32,33 We suggest that the transferred melittin molecules form a pore in the liposome bilayer that leads to discharge of carboxyfluorescein from the liposomal core, dequenching, and rapid destruction of the liposome itself, which would be characteristic of its action on other types of bilayered membrane structures, such as living cells.
To summarize, our goal in this study was to design, formulate, characterize, and evaluate the utility of stable high-payload perfluorocarbon particles as nanovehicles for melittin delivery. However, the formulation of peptide-delivery agents is attended by unique challenges; chief among these is the preservation of the complex peptide/protein structure throughout the formulation process that is required to retain biological activity. In this light, our procedure for the addition of melittin as a final step in the assembly of melittin-loaded perfluorocarbon nanoparticles is an attractive proposition for formulation because the peptide itself is not subject to pH changes, sonication, or emulsification, all of which represent harsh processes that can destroy fragile peptides. Our results indicate that perfluorocarbon nanoparticles retain their structural integrity even after addition of melittin. This characteristic is in stark contrast to the effect of melittin on other lipid-based drug delivery vehicles, i.e., liposomes. Because of its very high surface tension, the hydrophobic and lipophobic perfluorocarbon core not only prevents disintegration of the nanoparticle after melittin insertion but may actually contribute to the stability and very slow dissociation of melittin from the stabilizing monolayer. Moreover, the unique ability of perfluorocarbon nanoparticles to form hemifusion stalks with model bilayer membranes allows delivery of cytolytic peptides directly to the target membrane. We anticipate that particles with this or a similar core type may represent a preferred vehicle for use as a carrier that effects direct cell membrane delivery of amphipathic peptide agents in vivo.
Supplementary Material
Acknowledgments
This work was supported in part by the US National Institutes of Health Grants R01 HL 073646 and U54 CA119342 to Samuel Wickline and an American Heart Association predoctoral fellowship to Neelesh Soman (0515446Z).
Footnotes
Supporting Information Available: A description of materials and methods. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tosteson MT, Tosteson DC. Biophys J. 1981;36(1):109–116. doi: 10.1016/S0006-3495(81)84719-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee MT, Chen FY, Huang HW. Biochemistry. 2004;43(12):3590–3599. doi: 10.1021/bi036153r. [DOI] [PubMed] [Google Scholar]
- 3.Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. Biophys J. 2001;81(3):1475–1485. doi: 10.1016/S0006-3495(01)75802-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hui L, Leung K, Chen HM. Anticancer Res. 2002;22(5):2811–2816. [PubMed] [Google Scholar]
- 5.Papo N, Seger D, Makovitzki A, Kalchenko V, Eshhar Z, Degani H, Shai Y. Cancer Res. 2006;66(10):5371–5378. doi: 10.1158/0008-5472.CAN-05-4569. [DOI] [PubMed] [Google Scholar]
- 6.Bechinger B. J Membr Biol. 1997;156(3):197–211. doi: 10.1007/s002329900201. [DOI] [PubMed] [Google Scholar]
- 7.Hoskin DW, Ramamoorthy A. Biochim Biophys Acta. 2008;1778(2):357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmieder AH, Winter PM, Caruthers SD, Harris TD, Williams TA, Allen JS, Lacy EK, Zhang H, Scott MJ, Hu G, Robertson JD, Wickline SA, Lanza GM. Magn Reson Med. 2005;53(3):621–627. doi: 10.1002/mrm.20391. [DOI] [PubMed] [Google Scholar]
- 9.Wickline SA, Neubauer AM, Winter PM, Caruthers SD, Lanza GM. Magn Reson Imaging. 2007;25(4):667–680. doi: 10.1002/jmri.20866. [DOI] [PubMed] [Google Scholar]
- 10.Hu G, Lijowski M, Zhang H, Partlow KC, Caruthers SD, Kiefer G, Gulyas G, Athey P, Scott MJ, Wickline SA, Lanza GM. Int J Cancer. 2007;120(9):1951–1957. doi: 10.1002/ijc.22581. [DOI] [PubMed] [Google Scholar]
- 11.Winter PM, Neubauer AM, Caruthers SD, Harris TD, Robertson JD, Williams TA, Schmieder AH, Hu G, Allen JS, Lacy EK, Zhang H, Wickline SA, Lanza GM. Arterioscler, Thromb, Vasc Biol. 2006;26(9):2103–2109. doi: 10.1161/01.ATV.0000235724.11299.76. [DOI] [PubMed] [Google Scholar]
- 12.Wickline SA, Neubauer AM, Winter P, Caruthers S, Lanza G. Arterioscler Thromb Vasc Biol. 2006;26(3):435–441. doi: 10.1161/01.ATV.0000201069.47550.8b. [DOI] [PubMed] [Google Scholar]
- 13.Lanza GM, Winter P, Caruthers S, Schmeider A, Crowder K, Morawski A, Zhang H, Scott MJ, Wickline SA. Curr Pharm Biotechnol. 2004;5(6):495–507. doi: 10.2174/1389201043376544. [DOI] [PubMed] [Google Scholar]
- 14.Papo N, Shai Y. Biochemistry. 2003;42(2):458–466. doi: 10.1021/bi0267846. [DOI] [PubMed] [Google Scholar]
- 15.Wiltschi B, Knoll W, Sinner EK. Methods. 2006;39(2):134–146. doi: 10.1016/j.ymeth.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Riess JG. Chem Rev. 2001;101(9):2797–2920. doi: 10.1021/cr970143c. [DOI] [PubMed] [Google Scholar]
- 17.Dempsey CE. Biochim Biophys Acta. 1990;1031(2):143–161. doi: 10.1016/0304-4157(90)90006-x. [DOI] [PubMed] [Google Scholar]
- 18.Toraya S, Nishimura K, Naito A. Biophys J. 2004;87(5):3323–3335. doi: 10.1529/biophysj.104.046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Surewicz WK, Epand RM. Biochemistry. 1984;23(25):6072–6077. doi: 10.1021/bi00320a026. [DOI] [PubMed] [Google Scholar]
- 20.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum Press; New York: 1999. [Google Scholar]
- 21.Berlman IB. J Phys Chem. 1973;77(4):562–567. [Google Scholar]
- 22.Mall S, Sharma RP, East JM, Lee AG. Faraday Discuss. 1998;(111):127–136. doi: 10.1039/a809299k. discussion on pp 137–157. [DOI] [PubMed] [Google Scholar]
- 23.Powl AM, East JM, Lee AG. Biochemistry. 2003;42(48):14306–14317. doi: 10.1021/bi034995k. [DOI] [PubMed] [Google Scholar]
- 24.Bhowmick S, Saini S, Shenoy VB, Bagchi B. J Chem Phys. 2006;125(18):181102. doi: 10.1063/1.2400037. [DOI] [PubMed] [Google Scholar]
- 25.Heuser J. J Cell Biol. 2005;169(2):269–283. doi: 10.1083/jcb.200412169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlesinger PH, Djedovic NK, Ferdani R, Pajewska J, Pajewski R, Gokel GW. Chem Commun (London) 2003;(3):308–309. doi: 10.1039/b211629b. [DOI] [PubMed] [Google Scholar]
- 27.Pajewski R, Djedovic N, Harder E, Ferdani R, Schlesinger PH, Gokel GW. Bioorg Med Chem. 2005;13(1):29–37. doi: 10.1016/j.bmc.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 28.Saito M, Korsmeyer SJ, Schlesinger PH. Nat Cell Biol. 2000;2(8):553–555. doi: 10.1038/35019596. [DOI] [PubMed] [Google Scholar]
- 29.Rex S, Schwarz G. Biochemistry. 1998;37(8):2336–2345. doi: 10.1021/bi971009p. [DOI] [PubMed] [Google Scholar]
- 30.Gerber F, Krafft MP, Vandamme TF, Goldmann M, Fontaine P. Angew Chem, Int Ed. 2005;44(18):2749–2752. doi: 10.1002/anie.200462978. [DOI] [PubMed] [Google Scholar]
- 31.Gerber F, Krafft MP, Vandamme TF, Goldmann M, Fontaine P. Biophys J. 2006;90(9):3184–3192. doi: 10.1529/biophysj.105.077008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leikina E, Chernomordik LV. Mol Biol Cell. 2000;11(7):2359–71. doi: 10.1091/mbc.11.7.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malinin VS, Haque ME, Lentz BR. Biochemistry. 2001;40(28):8292–8299. doi: 10.1021/bi010570r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.