

Abstract
The modification of protein and non-protein thiols by oxidants including hydrogen peroxide (H2O2), peroxynitrite anion (ONOO−) and hypochlorous acid (HOCl) is well documented. Using an aromatic thiol, 5-thio-2-nitrobenzoic acid, and biologically-relevant oxidants, we have identified higher oxidation states of sulfur including the sulfonic acid derivative and the disulfide S-oxide, a thiosulfinate, by HPLC and mass spectrometry. The initial reaction of ONOO− with 5-thio-2-nitrobenzoic acid yielded a transient red intermediate, the sulfenate anion. The red intermediate was observed when ONOO− and H2O2 were used to oxidize 5-thio-2-nitrobenzoic acid and it persisted for several seconds at pH 7. HOCl oxidized the disulfide, 5,5′ dithiobis(2-nitrobenzoic acid) to the corresponding sulfonic acid and no additional products were detected. Using this system, we can directly compare the thiol-oxidizing abilities of several oxidants. Because 5-thio-2-nitrobenzoic acid is the product of the reaction of Ellman’s reagent with protein thiols, a detailed study of its stability in biological matrices where oxidants may be generated is warranted.
Keywords: thiol, peroxynitrite, reactive sulfur species, sulfenic acid, thiosulfinate, sulfonic acid
INTRODUCTION
Numerous recent studies show that the function of many proteins depends on cysteine oxidation state.[1, 2] In addition to an “on/off” switch between thiol and disulfide, more subtle regulation depends on sulfur oxidation state and modifications by nitric oxide and glutathione.[3] Like phosphorylation, the reversible, covalent addition of nitric oxide or glutathione alters protein structure and consequently, interactions with downstream signaling targets.[4, 5]
Our interest in protein thiol redox function prompted us to develop a system whereby we can directly compare the thiol-oxidizing ability of three biologically relevant oxidants: peroxynitrite anion (ONOO−), hydrogen peroxide (H2O2) and hypochlorous acid (HOCl). These oxidants are produced in variable quantities and in different cell types in vivo. ONOO− is generated by the diffusion-controlled reaction of nitric oxide (NO) with superoxide anion (O2−).[6, 7] H2O2 is a byproduct of multiple enzymatic and nonenzymatic reactions in vivo.[1] For example, molecular oxygen is reduced to water in mitochondria; however, some partial reduced oxygen species such as O2− can escape from the electron transport chain yielding H2O2 via dismutation. HOCl is produced by the enzyme myeloperoxidase (MPO), a heme protein predominantly expressed in neutrophils.[8] MPO is the only enzyme capable of generating HOCl as physiological chloride concentrations. Of note, Heinecke and coworkers recently reported that MPO is expressed by neurons in Alzheimer’s disease brain; thus the effect of HOCl on protein and non-protein thiols not specific to neutrophils is warranted.[9]
Oxidation of cysteine thiols to the sulfenic acid (RSOH) by ONOO− is widely accepted.[6, 10] In most cases, a cysteine sulfenic acid reacts with an adjacent cysteine thiol on a protein or with a thiol such as the tripeptide glutathione (GSH) to generate a protein disulfide or a mixed disulfide. Detectable sulfenic acids have been observed in several enzyme classes, notably in mammalian and bacterial peroxiredoxins.[11] Poole and coworkers reported that H2O2 oxidizes an active site cysteine thiol in AphC, a bacterial peroxiredoxin, to a stable sulfenic acid that can be trapped with dimendone or 4-chloro-7-nitrobenzofurazan.[12] In AphC, the single cysteine is isolated from other protein or small molecule thiols and therefore the sulfenic acid does not undergo further reaction. Alternatively, the reaction of thiols with HOCl initially yields a sulfenyl chloride intermediate, RSCl, which reacts with a second thiol to form a disulfide.[8]
Thiol metabolites generated by biological oxidants are of interest because protein regulation via the reversible thiol/disulfide redox couple is now widely accepted as a mechanism of cellular signaling. Also over-oxidation of protein thiols to non-reducible forms is believed to represent a type of oxidative stress that could contribute to neurodegeneration in diseases such as Alzheimer’s and Parkinson’s.[13]
We chose 5-thio-2-nitrobenzoic acid (TNB) as our thiol substrate based on its: 1) spectral properties; 2) water solubility; and 3) chemical stability. The TNB/5,5′ dithiobis(2-nitrobenzoic acid) (DTNB-Ellman’s reagent) system is well known and has been used extensively to quantitate free thiols in proteins.[14] TNB, a product of the reaction of DTNB with free thiols, absorbs strongly at 412 nm allowing for facile detection of micromolar concentrations of thiols.
None of the stable oxidation products formed from TNB absorb in the visible range. Because TNB is the product of the reaction of Ellman’s reagent with protein thiols, a detailed study of its stability in biological matrices where oxidants may be generated is warranted.
Although numerous studies reported the oxidation of protein thiols by ONOO−, HOCl and H2O2, few studies compare all three oxidants directly. Our methodology is straightforward with a 12 minute isocratic HPLC separation of TNB oxidation products produced at physiological pH.
EXPERIMENTAL
Materials
All chemicals were reagent grade and were from Sigma-Aldrich or Fisher. Peroxynitrite anion was synthesized from acidic H2O2 and NaNO2 as described.[15] ONOO− (10 ml) was passed through a column containing 2 g solid MnO2 to remove unreacted H2O2. ONOO− was stored at −80 ° C and its concentration was determined by diluting an aliquot in 0.4 M NaOH and measuring absorbance at 302 nm (ε302 = 1670 M−1 cm−1). The concentration of HOCl/OCl- was also determined from its absorbance at 292 nm (ε292 = 350 M−1 cm−1 for OCl−).[16]
HPLC conditions
Reaction products were analyzed by reverse phase HPLC on a Zorbax 300SB C18 column (4.6 × 150 mm, 5 μm) and were eluted isocratically at 0.5 ml/min in 45% acetonitrile/55% water containing 0.1% TFA. Run times were 12 minutes and reactions were monitored at 260 and 320 nm.
Preparation of TNB
TNB was prepared from DTNB as described by with some modification.[17] DTNB (0.5 g) in 25 ml 0.5 M Tris-HCl pH 8.8 was treated with 2.5 ml β-mercaptoethanol. The pH of the solution was adjusted to 1.5 with 6 M HCl. Orange crystals of TNB formed after 6–8 hours at 4 ° C. The crystals were filtered and washed with cold 0.1 M HCl. The purity of TNB was 99.5% as determined by HPLC with detection at 320 nm. Solid TNB was stable at RT indefinitely.
Reactions of TNB with oxidants
Reactions containing TNB and oxidants were prepared in 0.10 M phosphate buffer (PB) pH 7.0 unless otherwise noted. A fresh stock solution of TNB was prepared and its concentration determined from its absorbance at 412 nm (ε412 = 13,600 M−1 cm−1). For a typical reaction, TNB was diluted to 1 mM in PB pH 7.0 and the oxidant was added in a small volume (1–5 μl) to achieve the desired concentration in a total volume of 200 μl. For ONOO− and HOCl reactions, the reaction time was 5 min at RT whereas for H2O2, samples were incubated for up to 30 min. The reactions were diluted with 10% H3PO4 prior to HPLC analysis (10 nmol TNB injected).
Oxidation of DTNB with mCPBA
DTNB (100 mg, 0.252 mmol) was dissolved in THF or a 1:1 mixture of THF/CH2Cl2 to 0.1M and cooled to −78 ° C. m-chloroperbenzoic acid (mCPBA) (0.247 mmol) dissolved in 1 ml THF was added dropwise to the DTNB solution over 10 minutes. After one hour at −78 ° C, the reaction was warmed to 0 ° C and left for an additional hour. No remaining mCPBA was detected by TLC. Reaction products were analyzed by HPLC or mass spectrometry. The solvent was evaporated under vacuum and the solid material was stored at −80 ° C.
Oxidation of DTNB with performic acid
Performic acid reagent was prepared by mixing 0.1 ml 30% H2O2 and 0.9 ml 88% formic acid as described.[18] DTNB (100 nmol) was treated with 100 μl performic acid for 3 hours at 4 ° C. Water (200 μl) was added and the reaction was evaporated to dryness under vacuum in a Speed-Vac concentrator. The reaction products were analyzed by HPLC, UV-Visible spectroscopy and mass spectrometry.
Mass spectrometry of TNB reaction products
TNB reaction products were detected by electrospray ionization mass spectrometry using a Hewlett-Packard Series 1100 instrument in negative ion mode. One microliter of sample in 45% acetonitrile containing 0.1% TFA was injected.
RESULTS
The oxidation products formed by the reaction of TNB with ONOO− at pH 7.0 were separated by reverse-phase HPLC and initially monitored at 320 nm. Five peaks were detected including unreacted TNB, the disulfide DTNB, a very polar product that eluted at 3.6 minutes (unknown 1) and a product that eluted at 6.4 minutes between TNB and DTNB (Figure 1A - unknown 2). A fifth peak eluting just before TNB at 4.6 minutes was present as a minor contaminant in the original TNB preparation. Its area did not change in response to oxidants. When the reaction was monitored at 260 nm, the peak areas of the unidentified products eluting at 3.6 and 6.4 minutes were much larger indicating that 320 nm is not their absorbance maximum. (Figure 1B) We continued to use 320 nm as our detection wavelength because all reactants and products absorb at 320 nm but not all absorb at 260 nm in the HPLC elution solvent.
Figure 1.
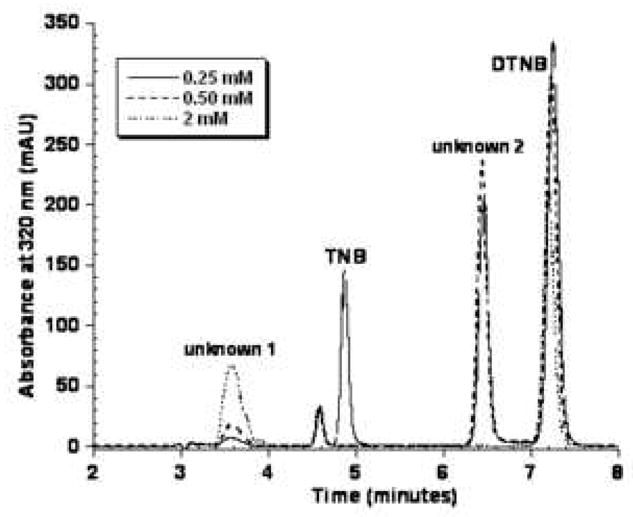


Figure 1A. Representative chromatogram of the products obtained from the reaction of 1 mM TNB with varying concentrations of ONOO− in 0.1 M sodium phosphate pH 7.0. Samples were analyzed by reverse-phase HPLC with detection at 320 nm as described in the Experimental section.
Figure 1B. Detection of TNB and its oxidation products by reverse-phase HPLC at 260 and 320 nm.
Figure 1C. UV-Visible spectra of TNB/ONOO− reaction products. Products were collected after HPLC separation and scanned in 45% acetonitrile containing 0.1% TFA.
Figure 1C shows the UV-Visible spectra of DTNB, the polar product tentatively identified as the sulfonic acid RSO3H (unknown 1) and the putative thiosulfinate, RSS(O)R labeled unknown 2 in Figure 1A.
When 2 mM ONOO− was added to 1 mM TNB, no peak was observed at 6.4 minutes (Figure 1A). Rather, the peak at 3.6 minutes increased considerably (unknown 1) and the DTNB peak decreased in area. This suggests excess ONOO− is capable of further oxidizing both the 6.4 minute product and DTNB to form the species eluting at 3.6 minutes.
To aid in identifying the unknown products, the TNB/ONOO− reaction was subsequently treated with DTT to determine if either unidentified product were reducible. When excess DTT was added, the 6.4 minute peak disappeared, as well as the DTNB peak but the peak area for the 3.6 minute peak did not change. The 6.4 minute peak was collected, treated with DTT and re-injected to yield only TNB.
Based on this finding and its polarity, the 6.4 minute peak (unknown 2 in Figure 1A) was tentatively assigned as the thiosulfinate, RSS(O)R, also referred to as the disulfide S-monoxide.
Further evidence to support this was the color change that was observed when ONOO− was first added to the TNB solution. A solution of TNB in neutral phosphate buffer is yellow whereas DTNB is colorless. Following addition of 1 mM ONOO− to 1 mM TNB and rapid vortexing, a transient red color was observed. At pH 7.0, the lifetime was roughly two seconds at best; however at higher pH values tested (up to pH 8.8), the stability of the red intermediate increased until it persisted for up to 30 seconds. A report by Blakeley and coworkers describes a transient red color with absorbance maximum at 492 nm when DTNB is treated with 3.0 M NaOH.[19] They attribute this color change to the formation of the TNB-derived sulfenate ion, RSO−. Their finding is consistent with our observations given that a sulfenic acid, RSOH, is formed by the reaction of ONOO− with thiols.[6, 10] The TNB-derived sulfenic acid is colorless whereas the sulfenate ion is red. We estimate that the pKa for the TNB-derived sulfenic acid is near pH 7 since no red color is detected for the TNB/ONOO− reaction at lower pH values of 5 or 6 but is detected at pH 7 and higher.
Controls were performed in which TNB solutions at pH values ranging from 5 to 9 were treated with 10 equivalents of NaNO2. This control was performed to rule out the possibility that the transient red color was due to S-nitrosation of the TNB thiol. No color change was observed at any of the pH values tested. The UV-visible spectra of the TNB solutions were obtained before and after NaNO2 treatment and no changes were observed.
Using several different ONOO− preparations, the reaction of TNB with ONOO− was studied in closer detail. In our synthesis of ONOO−, H2O2 is used as a starting material. If the ONOO− preparation is not treated to remove H2O2, then the product profile is different. Figure 2A shows that when H2O2 and peroxide-depleted ONOO− are added in equal amounts, 0.5 mM each to 1 mM TNB, the product profile changed. The DTNB peak increased and the tentative thiosulfinate RSS(O)R decreased in area. The area of the 3.6 minute peak does increase slightly though it is not readily apparent at 320 nm. Even though more total oxidant was added, some TNB remained and the yield of the 6.4 minute peak decreased.
Figure 2.
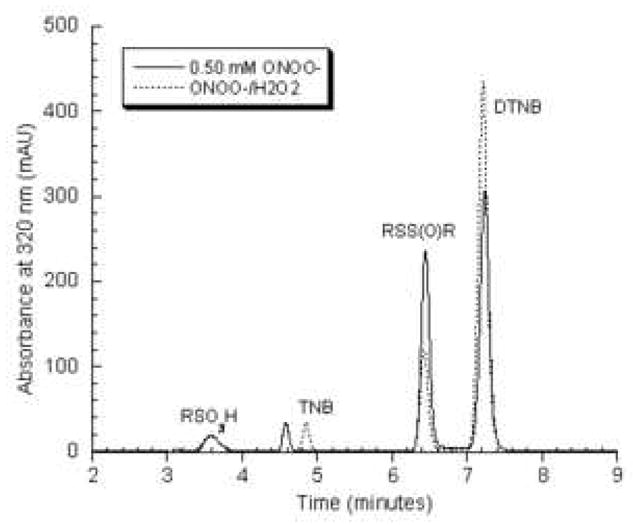

Figure 2A. Representative chromatogram of the reaction of 1 mM TNB with 0.5 mM ONOO− (solid line) or 1 mM TNB treated with 0.5 mM ONOO− and 0.5 mM H2O2 (dashed line) at pH 7.0. TNB was treated with oxidants for 5 minutes at room temperature before samples were diluted in 10% H3PO4 and analyzed by reverse-phase HPLC.
Figure 2B. Stability of the thiosulfinate product in 0.1 M sodium phosphate buffer pH 7.0 or in 10% H3PO4. Samples of TNB treated with 0.5 mM ONOO− either analyzed immediately or were incubated at room temperature for 24 hours and then analyzed by HPLC with detection at 320 nm.
The stability of the TNB/ONOO− reaction products was assessed by incubating the reactions either in phosphate buffer or 10% H3PO4 overnight at room temperature. Samples are diluted with 10% H3PO4 to protonate the TNB-derived oxidation products prior to HPLC analysis. Figure 2B shows that the area of the 3.6 minute peak did not change in either case. For the sample stored in buffer overnight, the area of the putative thiosulfinate at 6.4 minute decreased by 21% and the area of the DTNB peak increased proportionately. Since the molar absorptivity values of DTNB and the putative thiosulfinate are different, the change was not directly quantitated. The TNB/ONOO− reaction that was stored in acid overnight showed no peak at 6.4 minutes and a more substantial increase in DTNB area was detected. Of note, ONOO− reacts rapidly with thiols and any excess ONOO− will decompose in solution to form nitrate ion within seconds. Thus, we were not concerned about further reaction of TNB oxidation products with ONOO−, but rather with the degradation of the reaction products over time.
To confirm the identity of the 6.4 minute peak observed in the ONOO−/TNB reaction as the thiosulfinate, we chose to prepare an authentic standard by treating DTNB with m-chloroperobenzoic acid, mCPBA. Several groups have reported that mCPBA oxidizes disulfides to the corresponding thiosulfinate.[20, 21] We performed this reaction several times and analyzed the reaction products by electrospray ionization mass spectrometry in negative ion mode. A representative HPLC chromatogram of the DTNB/mCPBA reaction products is shown in Figure 3A. Rather than cleanly converting DTNB to the thiosulfinate, a number of products were detected. The broad peak eluting at 3.6 minutes gave a mass (M-1) of 246 which is consistent with the sulfonic acid (Scheme 1). The products at 5.2 and 6.2 minutes in Figure 3A had masses (M-1) of 427 and 411 corresponding to the thiosulfonate (RSS(O2)R and the desired thiosulfinate. Multiple attempts to modify the reaction conditions for the oxidation of DTNB by mCPBA including solvent, temperature and time, did not yield a clean synthesis of the thiosulfinate, RSS(O)R.
Figure 3.


Figure 3A. HPLC separation of products formed by the reaction of DTNB with mCPBA. DTNB was treated with mCPBA as described in the Experimental section. The molecular masses of the labeled products were confirmed by mass spectrometry.
Figure 3B. Co-injection of the DTNB/mCPBA reaction (solid line) and an aliquot of 1 mM TNB treated with 0.5 mM ONOO− at pH 7.0 (same as in Figure 1A). Samples were diluted in 10% H3PO4 and analyzed immediately by reverse-phase HPLC with detection at 320 nm.
Scheme 1.

Structures of TNB, DTNB and oxidation products
To confirm that the 6.4 minute peak detected in the TNB/ONOO− reactions (Figures 1A and 1B) was the thiosulfinate, we performed a co-injection of the DTNB/mCPBA reaction, containing the known thiosulfinate, with the TNB/ONOO− reaction. In Figure 3B, we show an overlay of the same DTNB/mCPBA product profile presented in Figure 3A with a sample containing both the DTNB/mCPBA and a sample that also contains an aliquot of 1 mM TNB treated with 0.5 mM ONOO− (as in Figure 1B). The peak that gave a mass of 411 at 6.2 minutes in Figure 3A overlays exactly with the peak generated in the TNB/ONOO− reaction.
The 6.4 minute peak was collected from HPLC separation of a TNB/ONOO− reaction was collected and analyzed promptly by mass spectrometry to confirm a mass (M-1) of 411. Two peaks were detected, both 411 confirming the identity of the thiosulfinate and 395 corresponding to DTNB. We attribute the DTNB peak at 395 to the presence of trifluoroacetic acid (TFA) in the HPLC elution solvent. We had previously shown that acid treatment converted the 6.4 minute peak to DTNB (Figure 2B). Based on all the data presented, we conclude that the 6.4 minute peak detected in our TNB/ONOO− reactions is the thiosulfinate.
Another interesting observation is that the thiosulfonate, RSS(O2)R, eluting at 5.2 minutes, is quite unstable and the peak area decreased by ~50% in the co-injection. By comparing peak area data, we conclude that the thiosulfonate was converted to the sulfonic acid eluting at 3.6 minutes.
We treated DTNB with performic acid, an oxidant used routinely to oxidize cysteine in peptides to cysteic acid, the corresponding sulfonic acid.[18] This reagent is used to ensure that disulfides will not re-form in protein and peptide samples. HPLC analysis of DTNB oxidized with performic acid yielded only the 3.6 minute peak, the sulfonic acid. The UV spectrum of the resulting sulfonic acid showed an absorbance maximum at 260 nm. By scanning known amounts of the sulfonic acid produced from DTNB, we determined a molar absorptivity of approximately 7000 M−1 cm−1 at 260 nm for the TNB-derived sulfonic acid in either acid or neutral phosphate buffer. The molar absorptivity at 320 nm, the wavelength used in most of our HPLC studies, was much lower with values of 1100 and 2090 M−1 cm−1 in acid and phosphate buffer, respectively. The only literature report of a TNB-derived sulfonic acid reported nearly identical molar absorptivity values.[22]
The reaction of TNB with additional oxidants, including H2O2 and HOCl, was also examined in this study. H2O2 has been used as the oxidant to generate a protein sulfenic acid in the bacterial hydroperoxide reductase AphC; thus, we were interested to see if the TNB sulfenic acid would be generated in sufficient concentration to observe the TNB thiosulfinate as we did for ONOO−.[12] TNB (1 mM) was treated with concentrations of H2O2 ranging from 0.1 – 5 mM and at several pH values ranging from 5 – 8.5. At all concentrations and pH values tested, the only TNB oxidation product was DTNB. No thiosulfinate was observed. This finding implies that the reaction of H2O2 with TNB was too slow to generate a sufficient concentration of the sulfenic acid for dimerization to the thiosulfinate (Scheme 2). This is not unexpected given that the rate of reaction of H2O2 with thiols is several orders of magnitude slower than that of ONOO− with thiols.
Scheme 2.

Thiol oxidation by ONOO− and subsequent thiosulfinate formation
When up to 80 equivalents of H2O2 were added to TNB at pH 8.0, a transient red-orange color was observed. The transient color never reached the same intensity as the red color of the sulfenate, RSO−, observed with ONOO− as the oxidant. However, this finding is important because it supports the mechanism of thiol oxidation by H2O2 as proceeding through formation of the sulfenic acid, RSOH, following by subsequent reaction with a second thiol to form the disulfide and H2O.
Lastly, HOCl was used as the thiol oxidant. At all concentrations of HOCl and pH values tested, no thiosulfinate product was observed. Though the reaction of HOCl with thiols is more rapid than that of H2O2, the mechanism of oxidation is different. The initial reaction of HOCl with a thiol generates the sulfenyl chloride, RSCl, not the sulfenic acid.[8] Thus the thiosulfinate cannot form.
When excess HOCl (2 mM) was added to 1 mM TNB, the sulfonic acid product was detected at 3.6 minutes in addition to DTNB. This finding suggests that HOCl is a strong enough oxidant to react with DTNB, the initial product of the reaction, to form the sulfonic acid in a manner similar to performic acid.
The observation of RSO3H formation with excess HOCl led us to treat the disulfide DTNB with 10 equivalents of each oxidant. The chromatogram in Figure 4 shows that DTNB was oxidized completely to the sulfonic acid by HOCl and no additional products were detected at 320 nm or at 260 nm (data not shown). Excess ONOO− oxidized only 15% of the starting DTNB to the sulfonic acid. For the DTNB/H2O2 reaction, there was no decrease in the DTNB peak area relative to control. Thus, using the common reagent DTNB, we can directly compare HOCl and ONOO− with regard to their ability to oxidize a disulfide to the sulfonic acid.
Figure 4.
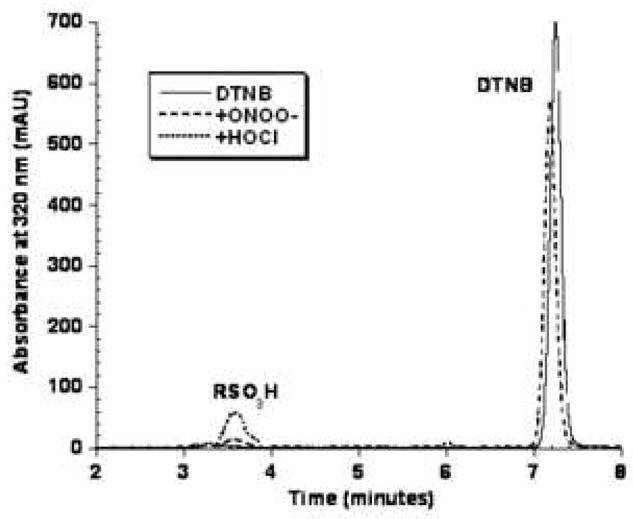
Representative chromatogram of the reaction of 0.5 mM DTNB in 0.1 M sodium phosphate pH 7.0 treated with 5 mM each of ONOO− or HOCl for 5 minutes. Samples were diluted in 10% H3PO4 and analyzed by reverse-phase HPLC with detection at 320 nm.
To further investigate the TNB/ONOO− reaction, we varied the pH of the reaction buffer. This is particularly important in this study because many of the chemical species involved have pKa values in the physiological range. For example, the pKa of peroxynitrous acid (ONOOH) is 6.8 and is believed to be the thiol oxidant, not ONOO−(see Scheme 2).[15] A thiolate, RS−, is generally thought to be more readily oxidized than a thiol RSH.[6] The reported pKa of TNB is 4.53.[14] The pKa of the transient TNB-derived sulfenic acid, RSOH, is clearly in the physiological range since its conjugate base, RSO−, has the intense red color that we observe at pH values above 7. Based on these values, we were interested to see if the TNB/ONOO− product profiles varied as a function of pH. When 1 mM TNB was treated with 0.25 or 0.5 mM ONOO− in phosphate buffers ranging from pH 5 to pH 8, all reactions yielded similar amounts of thiosulfinate and DTNB as the predominant products with a small amount of sulfonic acid detected. There was no clear trend observed over the pH range tested; therefore, we did not expand out of that pH range.
Because the molar absorptivity values of TNB and its oxidation products at 320 nm are not identical, we cannot directly quantitate the product distribution for a given reaction. However, we did quantitate the yield of DTT-reducible vs non-reducible products at several pH values by measuring absorbance at 412 nm, attributed to remaining TNB, before and after DTT addition. Both DTNB and the thiosulfinate are reduced to TNB by DTT whereas the sulfonic acid is not. The results are summarized in Table 1. As the concentration of ONOO− increased, the yield of DTT-reducible products decreased at all pH values. When 1 mM TNB was treated with 2 mM ONOO−, the predominant product comprising nearly 70% of products was the sulfonic acid. ONOO−, added in excess is very efficient at generating the sulfonic acid from the thiol TNB whereas ONOO− does not oxidize the disulfide DTNB to the sulfonic acid as efficiently as HOCl (Figure 4).
Table 1.
Percentage of DTT-reducible products as a function of pH and ONOO− concentration.
| % DTT-reducible products (DTNB + thiosulfinate) | |||
|---|---|---|---|
| [ONOO−] | pH 6.0 | pH 7.0 | pH 8.0 |
| 0.25 mM | 95 ± 2% | 96 ± 3% | 96 ± 3% |
| 0.50 mM | 91 ± 3% | 89 ± 4% | 89 ± 3% |
| 2.0 mM | 31 ± 4% | 32 ± 3% | 33 ± 4% |
Following ONOO− addition, an aliquot of the reaction was analyzed by UV/Visible spectroscopy to quantitate remaining TNB at 412 nm. Subsequently DTT (5 mM) was added to the original TNB/ONOO− reaction and TNB was quantitated again. Control samples did not contain oxidant but were treated with DTT in a comparable manner. Data represents the average of two independent experiments.
DISCUSSION
ONOO− is a strong oxidant, derived from NO and superoxide anion, that is capable of oxidizing several amino acids in proteins including cysteine and tyrosine. Given its biological relevance, considerable effort has been directed at understanding ONOO− reactivity in isolated systems and in more complex biological matrices. Because TNB is a product of the reaction of Ellman’s reagent with protein thiols, we are interested in its stability in biological systems where oxidants such as ONOO−, H2O2 or HOCl may still be present. Subsequent oxidant of the TNB product could potentially skew quantitation of protein thiols.
In this study, we show that ONOO− is capable of generating, at substoichiometric concentrations, a detectable sulfenic acid/sulfenate RSO(H) as evidenced by a transient red color. The sulfenic acid was formed in sufficient quantity that the dimerization to form thiosulfinate, RSS(O)R, competed with the reaction of sulfenic acid with unreacted TNB.(Scheme 2) This confirms conclusively that ONOO− oxidizes thiols to a sulfenic acid. The reaction of TNB with H2O2 also yielded RSO(H) at pH 7 but 80 equivalents of oxidant were required. This is not unexpected given that the rate constant for the reaction of thiols with H2O2 is roughly three orders of magnitude lower than that of ONOO− with thiols.[6] Though HOCl is an efficient thiol oxidant, it does not generate the sulfenic acid/sulfenate but proceeds via the sulfenyl chloride.[8]
Because of the color change from yellow to red, TNB could be used to screen oxidants for their ability to form RSO(H) in aqueous solution and at neutral pH. Sulfenic acid chemistry has been limited because few stable small molecule sulfenic acids can be generated in solution. In addition to TNB, we have done some studies with thionitropyridine (TNP). The sulfenate formed from TNP and ONOO− is much more stable than the TNB-derived sulfenate and persists in solution for minutes rather than seconds.
In addition to sulfenic acid chemistry, our work provides a method to generate stable aromatic thiosulfinates at neutral pH and in aqueous solution. Whereas mCPBA oxidation of DTNB yielded both the thiosulfinate and the thiosulfonate, ONOO− oxidation of TNB generated thiosulfinate but no thiosulfonate (Figure 3A). The thiosulfinate generated from TNB was fairly stable with a 21% decrease in peak area after 24 hour incubation in phosphate buffer pH 7.0 (Figure 2B). However, our work shows that the TNB-derived thiosulfinate is not stable in acidic solution and only the disulfide DTNB was detected after 24 hours in 10% phosphoric acid (Figure 2B).
Radi and coworkers first described the oxidation of thiols by ONOO− in 1991.[6] Their pH studies showed that the protonated thiol, RSH, is oxidized by ONOO− whereas H2O2 preferentially oxidizes the thiolate RS−. Whereas biological thiols such as cysteine and GSH have pKa values of 8.3 and 9, respectively, the pKa of TNB is 4.53.[14] Thus, in our experiments, at all pH values tested, it is the thiolate that predominates in solution. Because the pKa of ONOO(H) is 6.8, we anticipated a change in product profile over the pH range of 5–8. However, there was no significant difference in the distribution of products when TNB was reacted with 0.5 equivalents of ONOO− (data not shown). Table 1 shows that pH did not affect the combined yield of DTNB and thiosulfinate (DTT-reducible products).
According to Radi and coworkers, ONOO− reacts with RSH by a direct two-electron oxidation to yield RSOH and nitrite.[6] A thiolate, like TNB, is more readily oxidized by one electron to the corresponding thiyl radical by ONOOH. A more recent manuscript by Cadenas and coworkers shows initial oxidation of a thiol to the thiyl radical and subsequently to RSOH.[10] Regardless of the pH and the possible distribution of reactive intermediates in solution, the product profile from the reaction of TNB with ONOO(H) did not change significantly.
The thiosulfinate is also susceptible to further oxidation by ONOO− or H2O2 to form the sulfonic acid RSO3H (Figure 2A). Those oxidations, coupled with reaction with molecular oxygen, leads to RSO2H and RSO3H formation as described in several recent papers.[23, 24]
Though many aromatic sulfonic acids are commercially available, the TNB-derived sulfonic acid is not. Thus, oxidation of DTNB by HOCl is an excellent path to form the sulfonic acid with no contaminating side products. Likewise, whereas a protein sulfenic acid and subsequent oxidation states have no distinct spectral properties, herein we described 1) a red sulfenate, 2) a sulfonic acid with a strong absorbance at 260 nm (ε~ 7000 M−1 cm−1 at neutral pH) and 3) a thiosulfinate that also has a maximum absorbance at 260 nm. The starting materials, TNB and/or DTNB, are readily available and water soluble over a broad pH range. Lastly, we have developed a short and efficient 12 minute separation of products by HPLC using isocratic solvent conditions.
Acknowledgments
The authors acknowledge support from the National Institute of Neurological Disorders and Stroke (R15-NS38885 to LML) and the Jeffress Memorial Trust (Grant # J-670 to LML). Special thanks to Professor M.G. Finn and Yeon-Hee Lim of the Scripps Research Institute.
ABBREVIATIONS
- ONOO−
peroxynitrite anion
- HOCl
hypochlorous acid
- H2O2
hydrogen peroxide
- TNB
5-thio-2-nitrobenzoic acid
- DTNB
5,5′ dithiobis(2-nitrobenzoic acid)
- GSH
reduced glutathione
- PB
phosphate buffer
- DTT
dithiothreitol
- mCPBA
m-chloroperoxybenzoic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kiley PJ, Storz G. Exploiting thiol modifications. PLoS Biology. 2004;2:1714–1717. doi: 10.1371/journal.pbio.0020400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baty JW, Hampton MB, Winterbourn CC. Detection of oxidant sensitive thiol proteins by fluorescence labeling and two-dimensional electrophoresis. Proteomics. 2002;2:1261–1266. doi: 10.1002/1615-9861(200209)2:9<1261::AID-PROT1261>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 3.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature Reviews Molecular Cell Biology. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 4.Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mulsch A, Dimmeler S. Nitric oxide inhibits caspase-3 by s-nitrosation in vivo. J Biol Chem. 1999;274:6823–6826. doi: 10.1074/jbc.274.11.6823. [DOI] [PubMed] [Google Scholar]
- 5.Lander HM, Ogiste JS, Pearce SFA, Levi R, Novogrodsky A. Nitric oxide-stimulated guanine nucleotide exchange on ras p21. J Biol Chem. 1995;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- 6.Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite oxidation of sulfhydryls. J Biol Chem. 1991;266:4244–4250. [PubMed] [Google Scholar]
- 7.Radi R. Peroxynitrite reactions and diffusion in biology. Chem Res Toxicol. 1998;11:720–721. doi: 10.1021/tx980096z. [DOI] [PubMed] [Google Scholar]
- 8.Peskin AV, Winterbourn CC. Kinetics of the reactions of hypochlorous acid and amino acid chloramines with thiols, methionine and ascorbate. Free Rad Biol Med. 2001;30:572–579. doi: 10.1016/s0891-5849(00)00506-2. [DOI] [PubMed] [Google Scholar]
- 9.Green PS, Mendez AJ, Jacob JS, Crowley JR, Growdon W, Hyman BT, Heinecke JW. Neuronal expression of myeloperoxidase is increased in Alzheimer’s disease. J Neurochem. 2004;90:724–733. doi: 10.1111/j.1471-4159.2004.02527.x. [DOI] [PubMed] [Google Scholar]
- 10.Han D, Canali R, Garcia J, Aguilera R, Gallager TK, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44:11986–11996. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 11.Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG. Reversing the inactivation of periredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- 12.Ellis HR, Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 13.Pamplona R, Dalfo E, Ayala V, Bellmunt MJ, Prat J, Ferrer I, Portero-Otin M. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. J Biol Chem. 2005;280:21522–21530. doi: 10.1074/jbc.M502255200. [DOI] [PubMed] [Google Scholar]
- 14.Riddles PW, Blakeley RL, Zerner B. Ellman’s reagent: 5,5′-dithiobis(2-nitrobenzoic acid)-a reexamination. Anal Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- 15.Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Meth Enzymol. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 16.Morris JC. The acid ionization constant of HOCl from 5 to 35 degrees. Journal of Physical Chemistry. 1966;70:3798–3805. [Google Scholar]
- 17.Miron T, Rabinkov A, Mirelman D, Weiner L, Wilchek M. A spectrophotometric assay for allicin and alliinase (alliin lyase) activity: Reaction of 2-nitro-thiobenzoate with thiosulfinates. Anal Biochem. 1998;265:317–325. doi: 10.1006/abio.1998.2924. [DOI] [PubMed] [Google Scholar]
- 18.Ozols J. Amino acid analysis. Methods in Enzymology. 1990;182:587–601. doi: 10.1016/0076-6879(90)82046-5. [DOI] [PubMed] [Google Scholar]
- 19.Blakeley RL, Riddles PW, Zerner B. Variable stoichiometry in the decomposition of aromatic disulfides in alkaline solution: on the properties of 3-carboxylate-4-nitrobenzenesulfenate ion. Phosphorus and Sulfur. 1980;9:127–136. [Google Scholar]
- 20.Folkins P, Harpp DN. Clear evidence for the formation of alpha-disulfoxides and other intermediates in the mCPBA oxidation of bridged bicyclic thiosulfinates. J Am Chem Soc. 1991;113:8998–9000. [Google Scholar]
- 21.Hunter R, Caira M, Stellenboom N. Thiolsulfinate allicin from garlic: Inspiration for a new antimicrobial agent. Ann NY Acad Sci. 2005;1056:234–241. doi: 10.1196/annals.1352.011. [DOI] [PubMed] [Google Scholar]
- 22.Silverstein RM, Hager LP. The chloroperoxidase-catalyzed oxidation of thiols and disulfides to sulfenyl chlorides. Biochemistry. 1974;13:5069–5073. doi: 10.1021/bi00722a001. [DOI] [PubMed] [Google Scholar]
- 23.Fontana M, Amendola D, Orsini E, Boffi A, Pecci L. Oxidation of hypotaurine and cysteine sulphinic acid by peroxynitrite. Biochem J. 2005;389:233–240. doi: 10.1042/BJ20041696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quijano C, Alvarez B, Gatti RM, Augusto O, Radi R. Pathways of peroxynitrite oxidation of thiol groups. Biochem J. 1997;322:167–173. doi: 10.1042/bj3220167. [DOI] [PMC free article] [PubMed] [Google Scholar]