

Abstract
Barnase is one of the few protein models that has been studied extensively for protein folding. Previous studies led to the conclusion that barnase folds through a very stable submillisecond intermediate (≈3 kcal/mol). The structure of this intermediate was characterized intensively by using a protein engineering approach. This intermediate has now been reexamined with three direct and independent methods. (i) Hydrogen exchange experiments show very small protection factors (≈2) for the putative intermediate, indicating a stability of ≈0.0 kcal/mol. (ii) Denaturant-dependent unfolding of the putative intermediate is noncooperative and indicates a stability less than 0.0 kcal/mol. (iii) The logarithm of the unfolding rate constant of native barnase vs. denaturant concentrations is not linear. Together with the measured rate (“I” to N), this nonlinear behavior accounts for almost all of the protein stability, leaving only about 0.3 kcal/mol that could be attributed to the rapidly formed intermediate. Other observations previously interpreted to support the presence of an intermediate are now known to have alternative explanations. These results cast doubts on the previous conclusions on the nature of the early folding state in barnase and therefore should have important implications in understanding the early folding events of barnase and other proteins in general.
Barnase folds in multiple kinetic phases at pH 6.3 and 25°C (1). A submillisecond burst phase is observed (2). Approximately 80% of the molecules then fold in a fast single exponential phase with a folding rate constant kf of ≈13 s−1. The other 20% folds slowly because of cis–trans isomerization of prolines (1). In the earlier studies of the folding pathway of barnase, Fersht and coworkers (1) concluded that the major kinetic folding phase represented the folding from a stable submillisecond intermediate to the native state. This conclusion was based on the following experimental results. (i) There was a loss of CD signal within the submillisecond burst phase (2), consistent with a fast intermediate. (ii) The log kf (folding rate constant) as a function of denaturant concentrations is not linear (1), as illustrated in the chevron plot in Fig. 1, consistent with an on-pathway intermediate. (iii) An H/D pulse-labeling experiment revealed strong protections for some amide protons within 6 ms, suggesting a very stable intermediate (3). (iv) The stability for the intermediate was calculated to be 3.2 kcal/mol, taken as the difference between the equilibrium unfolding free energy for native barnase (ΔGNU) and the free energy for the major folding phase, ΔGuapp, calculated as −RTln(ku/kf) (1). Here, ku is the unfolding rate constant extrapolated from the right limb of the chevron plot assuming a linear relationship between log ku and denaturant concentrations (see illustrations in Fig. 1).
Figure 1.

Chevron curves of wild-type barnase at 25°C and pH 6.3 (solid circles) and pH 7.23 in the presence of 0.1 M Na2SO4 (open circles). The solid curve is the best fit of the observed rate constants to a three-state model with Eqs. 1–3. It yields the following values: KIU = 0.009; mIU = 4.05 M−1; kIN = 11.9 s−1; mIN = 2.63 M−1; kNI = 8.6 × 10−6 s−1; mNI = 2.80 M−1; mNU (sum of mIU, mIN, and mNI) = 9.49 M−1; and mIU/mNU = 0.43. The dashed line illustrates the linear extrapolation of unfolding rate constant log ku. It should be noted that the ku in water, obtained with guanidinium chloride (GdmCl) and linear extrapolation, is 10 times smaller than the value obtained with urea (1).
Recent progress in protein-folding studies questioned these conclusions. (i) The burst phase signal lost within submilliseconds may simply represent the conformational readjustment of the unfolded state to lower denaturant concentrations as suggested for cytochrome c and ribonuclease A (4, 5). (ii) Alternative interpretations such as movement of the transition state ensemble can explain curved chevron plots, as proposed for Arc repressor (6) and U1A (7). (iii) A more recent hydrogen-exchange (HX) pulse-labeling experiment could not reproduce the earlier barnase results and failed to detect any stable intermediate (8). (iv) A native-state HX experiment also failed to reveal the postulated intermediate (9). (v) The correct extrapolation of log ku may curve downward at low denaturant concentrations, as has been shown for several proteins including Arc repressor (6), U1A (7), protein L (10), and CI2 (11). This nonlinear behavior would increase the calculated ΔGuapp for the major folding phase and decrease the calculated stability left over for the putative intermediate. These alternative interpretations and new experimental results suggest that the earlier conclusion needs to be reexamined by using more direct methods. Such a reexamination is important, because barnase has been taken as a classical illustration of how proteins fold, and its intermediate has been characterized intensively with a protein-engineering approach (12). In particular, a definite conclusion on the postulated intermediate of barnase will help to understand the ability and limitations of both the protein-engineering and the native-state HX approaches (1, 13). This article reports a reexamination of these issues.
Materials and Methods
Materials.
Proteins and buffer solutions were prepared as described (8).
Folding and HX Competition Experiments.
The folding-HX competition experiments (14) were performed with a Biologic QFM-4 (Grenoble, France; dead time ≈1 ms). Folding of denatured barnase (≈10 mg/ml) in 3.20 M deuterated GdmCl solution (pD 6.3, 10 mM Mes/D2O) was initiated by a 20-fold dilution with a buffer solution (pH 6.3, 50 mM Mes/H2O) at 25°C. Reactions were allowed to proceed for several minutes before pH was adjusted to 5.0 with 0.5 M HOAc. Solutions (50 ml) were concentrated, and buffer was exchanged with a 10% (vol/vol) D2O and 10 mM NaOAc solution with Amicon cells. The same experiment was performed at pH 7.23 (0.2 M K2HPO4/0.1 M Na2SO4). The 1H–15N heteronuclear sequential quantum correlation NMR spectra were collected on a Bruker AMX 500 spectrometer (Billerica, MA) and processed with felix (version 97, Biosym Technologies, San Diego). Peak intensities were used to measure the proton occupancies with amide protons Q104, R110, and W94 (4NH) averaged as internal references. Calculations of exchange rate constants, kex, and protection factors (Pf = kint/kex) used Eq. 5 with 8 s−1 for kf and intrinsic exchange rate constant, kint, based on peptide models (15).
Kinetic and Equilibrium CD and Fluorescence.
Stopped-flow CD experiments were performed with the Biologic SFM4. Up to 30 kinetic traces with a 1.0-cm path-length observation cell were averaged for each data point. Kinetic runs were initiated by mixing denatured barnase (pH 1.6 or 9.4 M urea) with folding buffer (pH 6.3, 50 mM Mes) with a final protein concentration of 14.6 μM (extinction coefficient 25,900 M−1⋅cm−1 at 280 nm). Stopped-flow fluorescence was performed similarly at ≈3 μM protein concentration.
Three-State Chevron Curve Fitting.
Chevron curves were fitted according to Eqs. 1–3 assuming U and I are in rapid equilibration, following Raschke and Marqusee (16).
 |
1 |
and
 |
2 |
 |
3 |
where KIUH2O is the unfolding equilibrium constant of the intermediate in water; kINH2O and kNIH2O are the values of the corresponding microscopic rate constants in the absence of denaturant; and m‡IN and m‡NI are the coefficients for the denaturant dependence of the microscopic rate constants.
Intermediate Melting Curve Fitting.
The unfolding equilibrium constant of the intermediate KIU was obtained by assuming a linear dependence of log KIU on urea concentration and fitting the standard Santoro–Bolen equation (17) with floating parameters for baselines. The complete equation used for the fitting is
 |
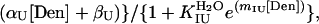 |
4 |
where I is the value of the CD signal; KIUH2O is the unfolding equilibrium constant of the intermediate in water; αI, βI, αU, and βU are the parameters of the baselines for the intermediate (I) and unfolded (U) states, respectively; and mIU is the coefficient for the denaturant dependence of the ln KIU. All curve fittings were carried out with sigmaplot (SPSS, Chicago).
Results
Principles for Measurement of the Stability of the Early Folding State from HX Protection Factors.
The stability of the early folding state of barnase can be determined based on the protection factors of the slowly exchanging amide protons, because 13 amide protons of barnase exchange with solvent protons only through global unfolding (9, 18). Fersht and coworkers have shown: (i) the averaged ΔGHX values for these protons are in excellent agreement with the ΔGNU values measured by differential scanning calorimetry (18), where ΔGHX is defined as −RTln(kex/kint); (ii) the changes of the global unfolding free energy (ΔΔGNU) are the same as ΔΔGHX between wild-type barnase and the mutants for the 13 amide protons (18); and (iii) the HX rates of these amide protons are accelerated at very low concentrations of GdmCl (9). These results require that no exchange occurs for the 13 amide protons through any structural fluctuations in N or any intermediate state, other than the fully unfolded state (9, 18). Thus, the Pf measured after the intermediate is formed for these protons should reflect the stability (I to U) of the proposed intermediate state with the relationship between ΔGIU = −RTln[1/(Pf − 1)] (19). For example, if the proposed folding intermediate of barnase has a stability of ≈3.0 kcal/mol as suggested previously (1), all of the 13 amide protons would be protected by a factor of >100 in the intermediate.
The Protection Factors of the Amide Protons in the Early Folding State of Barnase Measured by Competition HX.
To explore the stability of the proposed intermediate, we directly measured HX rate constants of amide protons in the early folding state of barnase by using the competition HX method (14). In this experiment, unfolded barnase in D2O and denaturant was diluted with H2O solution to initiate folding and HX reactions. Folding was allowed to proceed for several minutes to completion. In the native state, many amide protons are protected from exchange for weeks, which allows the fractions of the 1H [Pocc(H)] exchanged during the HX-folding process to be measured by using NMR from folded barnase. Because the stability of barnase decreases significantly, and the chevron plots became less curved at pHs above 7.5 (20), we performed competition experiments at two different pHs: 6.3 and 7.23. These experiments used the following relationship among kex, kf, and Pocc(H) for barnase when the competition proceeded until native proteins were formed completely (14):
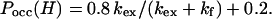 |
5 |
The factors 0.8 and 0.2 represent the fraction of the trans (fast folding) and cis (slow folding) proline isomers in the unfolded state, as determined by Matouschek et al. (1, 12). If one measures Pocc(H) and kf, then kex can be calculated (Eq. 5), and Pf can be obtained (Pf = kint/kex). At pH 6.3 and 25°C, 3 (K19, S50, and S91) of the 13 amide protons have large kint (13, 10, and 9 s−1, respectively), which is comparable to the kf (8 s−1) of barnase at 0.16 M GdmCl. Therefore, the kex of these protons can be measured accurately in the competition experiment. The Pf values for the three amide protons K19, S50, and S91 were 1.7, 1.5, and 1.1, respectively. These numbers are typical of amide protons in unfolded polypeptide chains (15).
The same competition experiment was performed at a slightly higher pH (7.23) in the presence of 0.1 M Na2SO4. The stabilizing salt ensures that the proposed intermediate, if it exists, should be more stable. This further stabilization is illustrated by the right shift of the folding limb of the chevron plot (Fig. 1). At pH 7.23, the HX protection of many additional amide protons can be measured directly. All of the amide protons that could be identified and resolved in the two-dimensional 1H–15N correlation spectrum (21) have Pf values between 0.5 and 2.5, except for one close to 4.5 (Fig. 2). Similar results were obtained in the presence of 0.1 M Na2SO4 at 0.0 and 0.36 M urea and at higher concentrations of Na2SO4 (up to 0.4 M, unpublished results). Importantly, the 13 amide protons are not protected more than others. The average Pf for the slowly exchanging amide protons is about 2. This value leads to a KIU of 1.0 and a ΔGIU value of ≈0.0 kcal/mol.
Figure 2.

HX protection factors in the early folding state. The filled bars are the Pf values of the 13 amide protons. The asterisks indicate the averaged Pf measured at both pH 6.3 and 7.23. Others in open bars are at pH 7.23.
Noncooperative Melting of the Early Folding State by Urea.
The stability of the early folding state of barnase was also explored directly by using CD and fluorescence. For barnase, the strongest CD signal in the far UV region at wavelength of 230 nm provides the information of secondary structures (2). The fluorescence signal, excited at 280 nm and collected above 310 nm, reflects the environment of the tryptophan residues in barnase. We measured the amplitude of these signals for the early folding state from the kinetic traces for barnase folding at 10 ms as a function of urea concentration by using a stopped-flow apparatus. By 10 ms, all of the burst phase intermediate is formed, and almost none has gone forward to N (kf ≈ 13 s−1). The urea dependence of the amplitude therefore reflects how the proposed intermediate of barnase unfolds by denaturant at pH 6.3 and 25°C. Two examples of the kinetic traces are shown in Fig. 3 A and B. The values measured (shown in Fig. 3 C and D) did not show a sigmoid type of cooperative transition, although small deviations from linearity are observable. These results are very similar to the behavior of nonfolding cytochrome c fragments (4) and the unfolded, disulfide-broken state of ribonuclease A (5), suggesting that the early folding state of barnase may not be a discrete folding intermediate. The change in CD and fluorescence signals within submilliseconds may simply represent the conformational readjustment of the unfolded state under different solvent conditions. When the CD values were fitted to a two-state cooperative unfolding model with floating parameters for both pretransition and posttransition baselines (see Eq. 4), we obtained a value of 2.6 for KIU (= [U]/[I]), indicating that the putative intermediate state is even less stable than the unfolded state by 0.6 kcal/mol. This value is expected from an incomplete transition curve (Fig. 3C).
Figure 3.

Noncooperative unfolding of the postulated intermediate. Kinetic traces generated by the stopped-flow CD at 0.94 M urea (A) and fluorescence in water (B) at pH 6.3 and 25°C. The values at 10 ms of folding as indicated by the arrows at different urea concentrations were plotted in C (solid circles) and D (solid circles). The global unfolding of barnase is shown with the dotted line. The curved solid line is the fitting curve with a two-state model with parameters for both pretransition and posttransition baselines (see Materials and Methods). The dashed line represents the posttransition baseline. Mdeg, millidegree.
Direct Observation of Nonlinear Behavior of log ku vs. GdmCl Concentrations.
If the postulated intermediate is not as stable as proposed previously or does not exist, one expects that ΔGNU, measured by equilibrium melting, will be equal to −RTln(ku/kf). This expectation is consistent with measured rates (Fig. 1) only if log ku is downward curved at lower denaturant concentrations. A reanalysis of the previous native-state HX results by Clarke and Fersht (9) indicated that this nonlinear behavior is indeed the case (Fig. 4).
Figure 4.

Downward deviation of the log ku at 30°C and pD 6.8 (pDread + 0.4) in D2O. The open and filled squares are rate constants measured by using stopped-flow fluorescence under the conditions used in the native-state HX experiments (9). The open and filled circles represent the HX rate constants of the slowly exchanging amide protons measured at GdmCl concentrations from 0.0 to 0.51 M and from 0.73 M to 1.20 M, respectively (9). The convergence of the exchange rate constants of the 13 amide protons at above 0.51 M GdmCl is indicative of EX1 exchange mechanism. The solid straight line represents the linear fitting of the filled circles with a slope of 2.425 and an intercept of −5.924. The dashed line is the linear fitting to the log ku at higher GdmCl concentrations.
Because the 13 slowly exchanging amide protons can exchange only through global unfolding, their exchange process (in D2O) can be written as in reaction scheme (6) with the intermediate state and the unfolded state included in the effective unfolded state Ueff (U + I). This treatment is because proton signals were monitored only in the native state to measure the HX rates (see detailed discussions in ref. 22 about HX process for a three-state model).
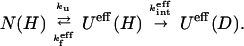 |
6 |
Here, kinteff and kfeff are the effective exchange rate constant and the effective rate constant of generating the native state, respectively. The measured exchange rate constant kex can be written as kex = ku kinteff/(kfeff + kinteff). It follows that kex = ku if kinteff ≫ kfeff (EX1 mechanism) and kex = ku/kfeff = Ku if kinteff ≪= kfeff (EX2 mechanism; see discussions in ref. 22). Simply stated, equilibrium HX experiments provide a means to measure the unfolding rate constant ku when HX occurs through EX1 mechanism and the global equilibrium unfolding constant Ku when HX occurs through an EX2 mechanism.
In the native-state HX experiments on barnase at pD 6.8 (pDread + 0.4) and 30°C, it was demonstrated that the 13 slowly exchanging amide protons exchanged with solvent protons by an EX2 mechanism at 0.0 M GdmCl and by an EX1 mechanism at above 0.51 M GdmCl (9). These conclusions were based on the following findings. (i) The HX rate constants of these amide protons were sensitive to pH changes at 0.0 M GdmCl but became independent of pH at GdmCl concentrations higher than 0.51 M GdmCl. (ii) The HX rate constants were different for different amide protons at 0.0 M GdmCl, but they converged to the same value at above 0.51 M GdmCl (see Fig. 4). Therefore, at 0.0 M GdmCl, the ΔGHX for these amide protons should represent the global unfolding free energy ΔGNU. Above 0.51 M GdmCl, the measured kex should represent ku.
Table 1 lists the ΔGHX values for the four slowly exchanging amide protons measured at 0.0 M GdmCl. The averaged value is 10.1 kcal/mol. Fig. 4 shows that the log ku at 0.0 M GdmCl extrapolated from the HX rate constants measured under EX1 mechanism (above 0.7 M GdmCl) is −5.92. Because the directly measured folding rate constant kf is 14.7 s−1, the ΔGuapp [−RTln(ku/kf)] for barnase at 0.0 M GdmCl is calculated to be 9.8 kcal/mol. This number is only 0.3 kcal/mol smaller than the value 10.1 kcal/mol of the averaged ΔGHX for the slowly exchanging amide protons (Table 1).
Table 1.
ΔGHX values for the slowly exchanging amide protons at 0.0 M GdmCl, 30°C, pD 6.8 (pDread + 0.4) in D2O
| Residue number | kex, s−1* | kint, s−1† | ΔGHX, kcal/mol |
|---|---|---|---|
| 19 | 2.83 × 10−7 | 7.3 | 10.3 |
| 50 | 4.27 × 10−7 | 8.5 | 10.1 |
| 52 | 1.63 × 10−7 | 3.0 | 10.1 |
| 98 | 1.77 × 10−7 | 2.8 | 10.0 |
| Average | 10.1 |
It should be noted that Clarke and Fersht (9) obtained higher ΔGHX values. Obviously, they used different kint. An independent check of the kint values can be made easily by using the program on the web site http://dino.fold.fccc.edu:8080/sphere.html.
Values of kex are from Clarke and Fersht (9).
Values of kint are from Bai et al. (15).
If a linear extrapolation is used to obtain a log ku in D2O from the unfolding values of the right limb of the chevron plot (Fig. 4) as done by Matouschek et al. (1), the value of log ku is −4.3. This value leads to a ΔGuapp of 7.6 kcal/mol, which is 2.5 kcal/mol less than the global unfolding free energy 10.1 kcal/mol. The missing stability was postulated by Matouschek et al. (1) to represent the stability of the early intermediate. However, the directly measured nonlinear log ku shown in Fig. 4 explains the missing stability and leaves only 0.3 kcal/mol for the postulated intermediate.
Matouschek et al. (1) initially used an unstable mutant (L14A) to obtain log ku values down to 0.5 M urea at pH 3.0 to justify the linear extrapolation. A linear behavior was observed in the range between 0.5 M and 3.5 M urea for this mutant. However, the slope shown from this linear fitting was clearly larger than that obtained between 3.7 M and 4.7 M urea at pH 6.3 (see figure 2 in ref. 1), consistent with a downsloping behavior. Later reexaminations of log ku at high urea concentrations revealed some nonlinear behavior (23). In a more recent attempt, Dalby et al. (24) measured log ku in a wider range of urea concentrations by extrapolating ku values at higher temperatures to 25°C by using the Eyring equation. The same nonlinear behavior of log ku was confirmed, but they were unable to explore the important region below 2 M urea concentrations. The native-state experiment by Clarke and Fersht (9) that entered the EX1 region now provides unfolding rates at low denaturant that clearly demonstrates the downcurving nature of the chevron unfolding limb at low urea and allows a more accurate extrapolation of ku to zero denaturant, as shown in Fig. 4.
The stability of the postulated intermediate measured by different methods and the possible errors estimated are summarized in Table 2.
Table 2.
Stability of the proposed intermediate of wild-type barnase measured by different methods in kcal/mol at pH 6.3 and 25°C
| Direct
methods
|
Indirect methods* | |||
|---|---|---|---|---|
| Urea melting† | Competition HX‡ | Native-state HX§ | ||
| ΔGIU | −0.6 | ∼0.0 | 0.3 | 2.7 to 3.2 |
Values are 2.8 kcal/mol from Dalby et al. (24), 2.7 kcal/mol from the chevron plot fitting that used a three-state model with fixed intermediate and transition state (see Materials and Methods and Fig. 1), and 3.2 kcal/mol from Matouschek et al. (1).
The value was obtained by fitting the CD signals (Fig. 3C) to a two-state model with floating parameters for both pretransition and posttransition baselines (Eq. 4). If KIU is fixed with a value of 0.25 (ΔGIU = 0.8 kcal/mol), the fitting yields an mIU value that is 60% of the mNU value for global unfolding (20). This value is significantly larger than the 43% value obtained from the chevron curve fitting (see Fig. 1), indicating that the ΔGIU is unlikely larger than 0.8 kcal/mol.
This value was obtained assuming an average protection factor of 2 (Fig. 2). Simulation studies that use Eq. 5 show that a ±10% error in Pocc(H) measurement may contribute an error less than a factor of 2 in Pf, which may contribute an uncertainty in ΔGIU by ∼0.4 kcal/mol.
The value was calculated from ΔGuapp – ΔGHX (see text) based on the HX data of Clarke and Fersht (9). Possible uncertainties in kint by a factor of 2 may contribute another error in free energy by 0.4 kcal/mol. Thus, the ΔGIUs determined from both HX experiments are unlikely larger than 0.8 kcal/mol.
Discussion
Taken together, (i) the directly measured small HX protection factors, (ii) the noncooperative unfolding behavior of the putative early folding state, and (iii) the downward shifted unfolding rate constants at very low GdmCl concentrations all argue that a stable folding intermediate with a stability of ≈3 kcal/mol does not exist on the folding pathway of wild-type barnase. The proposed intermediate, if it exists, is unlikely to be more stable than the unfolded state by 0.8 kcal/mol (see footnotes in Table 2 for error estimation).
Uncertainties in the Earlier Characterization of the Intermediate of Barnase.
Matouschek et al. (1, 12) used the protein engineering approach to determine side-chain interactions in the proposed intermediate state of barnase. This approach measures the changes of the unfolding free energy of the intermediate (ΔΔGIU) and the global unfolding free energy (ΔΔGNU) between the wild-type protein and mutants. A φI value, defined as ΔΔGIU/ΔΔGNU, was used to indicate the extent of the interactions between the residue that is mutated and other residues in the intermediate. If the φI value is 1.0, it suggests that the mutated residues interact with other residues in the intermediate state as strongly as in the native state. If the φI value is zero, then the interactions are as weak as in the unfolded state. A fractional φI value represents partial interactions. Because the stability of the intermediate state of wild-type barnase is less than 0.8 kcal/mol, the experimentally measured ΔΔGIU for any mutant should not exceed this value. Thus, it provides a criterion to test the accuracy of the earlier measurements. A large number (more than 40%) of ΔΔGIU values measured in the earlier studies by Matouschek et al. (12) were larger than 0.8 kcal/mol (see table 2 in ref. 12). Some of them were even larger than 2 kcal/mol. These large values suggest that there are large uncertainties in the ΔΔGIU and φI values and in the structure of the intermediate characterized based on them. The large ΔΔGIU values obtained by Matouschek et al. (1, 12) seem to result from the assumption that the measured log ku vs. denaturant concentration, which must be extrapolated to zero denaturant, has similar linear behavior for both wild-type and mutant proteins. That is, the computed φI values incorporate the same (apparently incorrect) assumption that leads to an estimated 3 kcal/mol in stability for the supposed intermediate.
Possible Interpretations for the Curved Chevron Plot of Barnase.
The significant curvature in the folding limb of the chevron plot of barnase was concluded to be due to the population of a stable intermediate by Matouschek et al. (1). This interpretation, however, is questionable and contrary to the experimental results presented in this article. Two alternative models have been proposed to account for the curvature in chevron plots. (i) Movement of the transition state ensemble as a function of denaturant alone may be responsible for the curved chevron plot. This model was suggested by Otzen et al. (7) and was used to explain the curved chevron plot of U1A. The curved log ku shown in Fig. 4 seems to be consistent with this model. Movement of the transition state ensemble toward the unfolded state as protein stability increases is also expected by the Hammond postulate and was observed from earlier mutation studies on barnase (25, 26). (ii) The early folding state of barnase may be an ensemble of differently collapsed unfolded states. A redistribution of this ensemble as a function of denaturant concentrations may be responsible for the curvature in the chevron plot of barnase. This model was used to explain the curved chevron plots of several proteins by Parker and Marqusee (27). A combination of i and ii is also possible.
Native-State HX and Protein Engineering Approaches.
Clarke et al. (28) questioned the usefulness of the native-state HX approach as an analytical tool to study folding pathways of proteins. They concluded that the native-state HX approach was not a reliable approach, because it was not able to identify the very stable intermediate of barnase that has been studied fully by the protein-engineering approach and the H/D pulse-labeling methods. Previously, we found that the H/D pulse-labeling experiment by Bycroft et al. (3) could not be reproduced, and our recent H/D pulse-labeling experiment did not detect any stable intermediate (8). The experimental results presented herein, including (i) the small HX protection factors, (ii) the noncooperative melting of the early folding state by urea, and (iii) the downward curved log ku, are all consistent with the native-state HX results, i.e., no stable intermediate seems to exist.
To conclude, the initial burst phase folding of barnase seems to reach the ensemble of forms with some characteristics that one may call the unfolded state under the conditions of the folding experiment (4, 5). Whether particular interactions or structures exist in this ensemble that help to predetermine the folding pathways remains to be seen (29). We show that any such interactions provide no net favorable free energy to the overall ensemble.
Acknowledgments
We thank Drs. S. Walter Englander, Tobin R. Sosnick, Hue Sun Chan, and Werner Klee for critical reading the paper and helpful suggestions, Dr. Joeseph J. Barchi, Jr., for technical assistance on NMR, and Dr. Bob W. Hartley for the expression system of barnase. J.T. is supported by the Japanese Society for the Promotion of Science.
Abbreviations
- GdmCl
guanidinium chloride
- HX
hydrogen exchange
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.190265797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.190265797
References
- 1.Matouschek A, Kellis J T, Jr, Serrano L, Bycroft M, Fersht A R. Nature (London) 1990;346:440–445. doi: 10.1038/346440a0. [DOI] [PubMed] [Google Scholar]
- 2.Oliveberg M, Fersht A R. Biochemistry. 1996;35:2738–2749. doi: 10.1021/bi950967t. [DOI] [PubMed] [Google Scholar]
- 3.Bycroft M, Matouschek A, Kellis J T, Jr, Serrano L, Fersht A R. Nature (London) 1990;346:488–491. doi: 10.1038/346488a0. [DOI] [PubMed] [Google Scholar]
- 4.Sosnick T S, Shtilerman M D, Mayne L, Englander S W. Proc Natl Acad Sci USA. 1997;94:8545–8550. doi: 10.1073/pnas.94.16.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qi P X, Sosnick T R, Englander S W. Nat Struct Biol. 1998;5:882–887. doi: 10.1038/2321. [DOI] [PubMed] [Google Scholar]
- 6.Jonsson T, Waldburger C D, Sauer R T. Biochemistry. 1996;35:4795–4802. doi: 10.1021/bi953056s. [DOI] [PubMed] [Google Scholar]
- 7.Otzen D E, Kristensen O, Proctor M, Oliverberg M. Biochemistry. 1999;38:6499–6511. doi: 10.1021/bi982819j. [DOI] [PubMed] [Google Scholar]
- 8.Chu R A, Takei J, Barchi J J, Jr, Bai Y. Biochemistry. 1999;38:14120–14125. doi: 10.1021/bi991799y. [DOI] [PubMed] [Google Scholar]
- 9.Clarke J, Fersht A R. Folding Des. 1996;1:243–254. doi: 10.1016/S1359-0278(96)00038-7. [DOI] [PubMed] [Google Scholar]
- 10.Yi Q, Scalley M L, Simons K T, Gladwin S T, Baker D. Folding Des. 1997;2:271–280. doi: 10.1016/S1359-0278(97)00038-2. [DOI] [PubMed] [Google Scholar]
- 11.Itzhaki L S, Neira J L, Fersht A R. J Mol Biol. 1997;270:89–98. doi: 10.1006/jmbi.1997.1049. [DOI] [PubMed] [Google Scholar]
- 12.Matouschek A, Serrano L, Fersht A R. J Mol Biol. 1992;224:819–824. doi: 10.1016/0022-2836(92)90564-z. [DOI] [PubMed] [Google Scholar]
- 13.Bai Y, Sosnick T R, Mayne L, Englander S W. Science. 1995;269:192–197. doi: 10.1126/science.7618079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmid F X, Baldwin R L. J Mol Biol. 1979;135:199–205. doi: 10.1016/0022-2836(79)90347-4. [DOI] [PubMed] [Google Scholar]
- 15.Bai Y, Milne J S, Mayne L, Englander S W. Proteins. 1993;17:75–82. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raschke T M, Marqusee S. Nat Struct Biol. 1997;4:298–304. doi: 10.1038/nsb0497-298. [DOI] [PubMed] [Google Scholar]
- 17.Santoro M M, Bolen D W. Biochemistry. 1992;31:4901–4907. doi: 10.1021/bi00135a022. [DOI] [PubMed] [Google Scholar]
- 18.Perrett S, Clarke J, Hounslow A M, Fersht A R. Biochemistry. 1995;34:9288–9298. doi: 10.1021/bi00029a003. [DOI] [PubMed] [Google Scholar]
- 19.Englander S W, Kallenbach N R. Q Rev Biophys. 1984;4:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 20.Pace N C, Laurents D V, Erickson R E. Biochemistry. 1992;31:2728–2734. doi: 10.1021/bi00125a013. [DOI] [PubMed] [Google Scholar]
- 21.Jones D N, Bycroft M, Lubinski M J, Fersht A R. FEBS Lett. 1993;331:165–170. doi: 10.1016/0014-5793(93)80319-p. [DOI] [PubMed] [Google Scholar]
- 22.Bai Y. J Biomol NMR. 1999;15:65–67. doi: 10.1023/a:1008316430724. [DOI] [PubMed] [Google Scholar]
- 23.Matouschek A, Matthews J M, Johnson C M, Fersht A R. Protein Eng. 1994;7:1089–1095. doi: 10.1093/protein/7.9.1089. [DOI] [PubMed] [Google Scholar]
- 24.Dalby P A, Oliveberg M, Fersht A R. J Mol Biol. 1998;276:625–646. doi: 10.1006/jmbi.1997.1546. [DOI] [PubMed] [Google Scholar]
- 25.Matouschek A, Fersht A R. Proc Natl Acad Sci USA. 1993;90:7814–7820. doi: 10.1073/pnas.90.16.7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matthews J M, Fersht A R. Biochemistry. 1995;34:6805–6810. doi: 10.1021/bi00020a027. [DOI] [PubMed] [Google Scholar]
- 27.Parker M J, Marqusee S. J Mol Biol. 1999;293:1195–1210. doi: 10.1006/jmbi.1999.3204. [DOI] [PubMed] [Google Scholar]
- 28.Clarke J, Itzhaki L S, Fersht A R. Trends Biochem Sci. 1998;23:378–381. doi: 10.1016/s0968-0004(97)01087-6. [DOI] [PubMed] [Google Scholar]
- 29.Baldwin R L, Rose G D. Trends Biochem Sci. 1999;24:24–26. doi: 10.1016/s0968-0004(98)01346-2. [DOI] [PubMed] [Google Scholar]