

Abstract
Previous studies demonstrated that maternal cocaine administration caused a significant decrease in protein kinase C epsilon (PRKCE) abundance in the left ventricle and an increase in susceptibility of the heart to ischemic injury in adult male offspring. The present study tested the hypothesis that epigenetic modification has a key role in cocaine-mediated programming of cardiac Prkce gene repression. Pregnant Sprague-Dawley rats were administered saline or cocaine (30 mg/kg/day i.p.) from Days 15 to 21 of gestational age, and hearts of 3-mo-old adult offspring were studied. Cocaine exposure significantly decreased Prkce mRNA levels in the left ventricle of male but not female offspring. CpG dinucleotides identified in Bhlhb2, Pparg, E2f, and Egr1 binding sites at the Prkce gene promoter were densely methylated in males and females and were unaffected by cocaine exposure. In contrast, methylation of CpGs in the two Sp1 binding sites (−346 and −268) was low and was significantly increased by cocaine exposure in male offspring. In females, methylation of the Sp1 binding site at −268 but not −346 was increased. Reporter gene assays showed that both Sp1 binding sites had a strong stimulatory role in Prkce gene activity. Methylation of the Sp1 binding sites significantly decreased SP1 binding to the Prkce promoter. Cocaine exposure did not affect nuclear SP1 protein levels but decreased the SP1 binding affinity to its binding site at −268. The results demonstrate an epigenetic mechanism of DNA methylation in programming of cardiac Prkce gene repression, linking fetal cocaine exposure and pathophysiological consequences in the heart of adult male offspring in a gender-dependent manner.
Keywords: developmental biology, DNA methylation, epigenetic, fetal programming, gender dimorphism, gene regulation, kinases, rats, transcription factor SP1
Maternal cocaine administration results in fetal programming of Prkce gene repression in the left ventricle of adult male offspring in a gender-dependent manner.
INTRODUCTION
Recent results of large epidemiological studies [1–6] have indicated that in utero adverse stimuli during pregnancy cause fetal programming of gene expression patterns and increase the risk of ischemic heart disease in adulthood. Cocaine abuse among women of childbearing age is prevalent in the United States, and offspring born to mothers with a history of cocaine abuse have a high incidence of congenital cardiovascular malformations, including abnormalities of ventricular structure and function, arrhythmias, and intracardiac conduction abnormalities, which persist beyond the period of exposure to cocaine. Other recent studies [7–12] in a rat model demonstrated that maternal cocaine administration during gestation increased apoptosis in the term fetal heart, caused cardiac remodeling with myocyte hypertrophy in the left ventricle during postnatal development, and increased susceptibility of the heart to ischemia-reperfusion injury in adult male offspring in a gender-dependent manner.
The mechanisms whereby fetal cocaine exposure causes an increase in vulnerability of ischemic injury in the heart of adult offspring are not clear. Among other mechanisms, protein kinase C epsilon (PRKCE) has a pivotal cardioprotective role during cardiac ischemia-reperfusion injury [13–15]. Findings in a Prkce knockout mouse model have demonstrated that Prkce expression is not required for cardiac function under normal physiological conditions but that PRKCE activation is necessary and sufficient for acute cardioprotection during cardiac ischemia-reperfusion [16]. Maternal cocaine administration caused a significant decrease in PRKCE protein levels in the left ventricle of adult male offspring [10], suggesting in utero epigenetic modification and programming of Prkce gene repression in the heart. DNA methylation is a common mechanism for epigenetic modification of gene expression patterns and occurs at cytosines of the dinucleotide sequence of CpG [17]. Methylation in promoter regions is generally associated with repression of transcription, leading to long-term shutdown of the associated gene. Methylation of CpG islands in gene promoter regions alters chromatin structure and transcription. Similarly, methylation of CpG dinucleotides within transcription factor binding sites affects transcription [18]. The present study provides evidence in a rat model that fetal cocaine exposure causes a significant increase in methylation status of CpG dinucleotides in non-CpG islands at the proximal promoter region of the Prkce gene, resulting in decreased binding of sequence-specific transcription factors to the promoter and in reduced Prkce gene expression in the left ventricle of adult male offspring in a gender-dependent manner.
MATERIALS AND METHODS
Experimental Animals
A previous study [10] demonstrated that maternal cocaine administration from Days 15 to 21 of gestational age significantly decreased PRKCE protein abundance in the left ventricle and increased its susceptibility to ischemia-reperfusion injury in adult offspring. To further determine the molecular mechanisms of Prkce gene repression in the left ventricle, the same animal model of fetal cocaine exposure was investigated in the present study. Time-dated pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Portage, MI) and were randomly divided into the following two groups: 1) saline administration control and 2) cocaine administration (15 mg/kg i.p. twice daily at 1000 h and 1600 h) from Days 15 to 21 of gestational age, as described previously [10]. While the intraperitoneal route closely resembles intranasal administration in humans in the kinetics and plasma cocaine levels attained [19], mitigates the stress caused by tissue necrosis that often occurs with subcutaneous injection of cocaine, and avoids the difficulty of intravenous injection in rats, it is not a common method of cocaine abuse in pregnant women. The animals were allowed to give birth naturally, and no fetal loss in the control and cocaine-exposed groups was observed. Pups were weaned and separated by gender at 21 days after birth. The offspring were given food and water ad libitum and were subjected to no further treatment before euthanasia. Hearts were isolated from 3-wk-old and 3-mo-old offspring from six litters in each group. All procedures and protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Western Blot
Left ventricles were homogenized in ice-cold lysis buffer composed of 20 mM Hepes, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM ethylenediaminetetraacetic acid, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 2 μg/ml of aprotinin, and 10 μg/ml of leupeptin, followed by incubation on ice for 30 min. The homogenates were ultrasonicated and centrifuged at 20 000 × g for 30 min at 4°C. To determine nuclear SP1 levels, nuclear extracts were prepared from hearts using the CellLytic nuclear extraction kit from Sigma (St. Louis, MO). Samples with equal quantities of protein were loaded onto 12% SDS-polyacrylamide gel and separated by electrophoresis at 100 V for 1 h. Proteins were then transferred onto Immobilon-P membranes (Millipore Corporation, Billerica, MA) and were probed with the primary antibodies of PRKCE and SP1 (Santa Cruz Biotechnology, Santa Cruz, CA). After incubation with horseradish peroxidase-conjugated secondary antibody (Amersham, Arlington Heights, IL), proteins were visualized with an enhanced chemiluminescence reagent, and the blots were exposed to Hyperfilm (Amersham). Results were quantified using the Kodak electrophoresis documentation and analysis system and Kodak ID image analysis software (Eastman Kodak, Rochester, NY).
Real-Time RT-PCR
RNA was extracted from left ventricles using TRIzol reagents (Invitrogen, Carlsbad, CA). Prkce mRNA levels were determined by real-time RT-PCR using the iCycler Thermal cycler (BioRad, Hercules, CA). Specific PKCε primers were 5′-GCGAAGCCCCTAAGACAAT-3′ (forward) and 5′-CACCCCAGATGAAATCCCTAC-3′ (reverse). Real-time RT-PCR was performed in a final volume of 25 μl. Each reaction mixture consisted of 600 nM concentrations of primers, 33 U of Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI), and iQ SYBR Green Supermix (BioRad) containing the following: 0.625 U of Taq polymerase; 400 μM concentration each of deoxy-ATP, deoxycytidine triphosphate, deoxyglutamyl transpeptidase, and deoxythymidine triphosphates; 100 mM KCl; 16.6 mM ammonium sulfate; 40 mM Tris-HCl; 6 mM MgSo4; SYBR Green I; 20 nM fluorescein; and stabilizers. RT-PCR was performed under the following conditions: 42°C for 30 min and then 95°C for 15 min, followed by 50 cycles of 95°C for 20 sec and 52°C for 1 min. Gapdh was used as an internal reference, and serial dilutions of the positive control were performed on each plate to create a standard curve. The PCR was performed in triplicate, and threshold cycle numbers were averaged.
Sequencing of the Promoter Region of the Rat Prkce Gene
The rat Prkce gene is located on chromosome 6. It was previously shown that cocaine exposure changes the methylation status of the putative Ap1 binding site at the proposed promoter of the Prkce gene in the fetal heart [20]. Since then, rat chromosome 6 genomic sequences have been updated, and examination of the updated sequences of 5′ upstream of the Prkce transcriptional start site reveals no Ap1 binding site. In addition, the rat Prkce promoter in the GenBank/RefSeq is missing sequences from nucleotides 7945999 to 7946320. Primers flanking the gap region upstream of the Prkce transcriptional start site were designed and synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). Genomic DNA isolated from rat left ventricles was used as a PCR template, and amplified DNA fragments were cloned using pDrive Cloning Vector (Qiagen, Studio City, CA) and sequenced.
Quantitative Methylation-Specific PCR
DNA was isolated from left ventricles using a GenElute Mammalian Genomic DNA Mini-Prep kit (Sigma), denatured with 2 N NaoH at 42°C for 15 min, and treated with sodium bisulfite at 55°C for 16 h, as previously described [21]. DNA was purified with a Wizard DNA cleanup system (Promega) and resuspended in 120 μl of H2o. The bisulfite-modified DNA was used as a template for real-time fluorogenic methylation-specific PCR [22, 23]. Bisulfite treatment of DNA converts cytosines to uracils. However, methylated cytosines at CpG dinucleotides are not converted. Specific primers were designed to amplify the target regions of interest with unmethylated CpG dinucleotides by detecting uracils and those with methylated CpG dinucleotides by detecting cytosines. Primers and probes designed for detecting methylated and unmethylated promoter regions of interest are listed in Table 1. All oligonucleotide primers and probes were synthesized by Integrated DNA Technologies, Inc. Gapdh was used as an internal reference gene. Real-time methylation-specific PCR was performed using the iQ SYBR Green Supermix with iCycler real-time PCR system (BioRad). Data are presented as the percentage of methylation of the regions of interest (methylated CpG/ [methylated CpG + unmethylated CpG] × 100%).
TABLE 1.
Sequences of quantitative methylation-specific PCR primers.

Electrophoretic Mobility Shift Assays
Nuclear extracts were prepared from left ventricles as already described. The oligonucleotide probes of unmethylated and methylated Sp1 and Mtf1 binding sequences from the rat Prkce promoter region were synthesized by Integrated DNA Technologies, Inc. and are listed in Table 2. Probe labeling and electrophoretic mobility shift assays were performed using a biotin 3′ end-labeling kit and a LightShift chemiluminescent electrophoretic mobility shift assay kit per the manufacturer's protocol (Pierce Biotechnology, Rockford, IL). Dot blot and series of controls were performed to ensure sufficient labeling and successful shift. Binding reactions were performed in 20 μl containing 50 fmol of oligoprobes, 1× binding buffer, 1 μg of polydeoxyinosinic acid and polydeoxycytidylic acid, and either 1 μg of nuclear extracts or 0.2 μg of purified SP1 (Promega) or MTF1 (Abnova Inc., Taipei, Taiwan, China). In competition studies, unlabeled oligonucleotide probes were added in 100-fold molar excess before the addition of the biotin-labeled probes. For supershift assays, 2 μg of SP1 and MTF1 antibodies (Santa Cruz Biotechnology) was added and further incubated for 1 h at 4°C. DNA-protein complexes were resolved with 6% nondenaturing polyacrylamide minigels (29:1 cross-linking ratio). Nylon membranes were used for transferring DNA-protein complex, followed by UV cross-linking to the membrane. After applying chemiluminescent substrate, the membranes were exposed to Kodak x-ray film (Eastman Kodak) for autoradiography.
TABLE 2.
Oligonucleotide sequences for electrophoretic mobility shift assay.

Chromatin Immunoprecipitation Assays
Left ventricles were minced and fixed with 1% formaldehyde. Cross-linking was stopped by the addition of glycine to a final concentration of 125 mM. Chromatin extracts were prepared and sonicated to produce DNA fragments between 100 and 500 bp in length. Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP-IT kit from Active Motif (Carlsbad, CA). Both positive and negative controls provided by the kit were performed along with the experimental samples. Antibody-pulled chromatin extracts were used as templates for PCR, and DNA from an aliquot of nonprecipitated lysates was used as a template for total input. Two sets of primers flanking the two Sp1 binding sites at −346 and −268 of the rat Prkce promoter were used. These included 5′-ACCATTTCCTCTCGACATGC-3′ (forward) and 5′-AGATTTCAACCCGGATCCTC-3′ (reverse) and 5′-AGAGGATCCGGGTTGAAATC-3′ (forward) and 5′-CTCACCTACCTTTCCGAAACA-3′ (reverse), which yielded products of 117 bp and 116 bp in length, respectively. The PCR amplification products were visualized on 1% agarose gel stained with ethidium bromide. To quantify the PCR amplification, 45 cycles of real-time PCR were performed with 3-min initial denaturation, followed by 95°C for 30 sec, 54°C for 30 sec, and 72°C for 30 sec, using the iQ SYBR Green Supermix with iCycler real-time PCR system.
Reporter Gene Assays
A 1941-bp fragment of the rat Prkce promoter region spanning −1941 to −1 bp relative to the transcriptional start site of Prkce was amplified by PCR and inserted into pDrive Cloning Vector. The KpnI-HindIII fragment flanking the Prkce promoter region was then inserted into the luciferase reporter gene plasmid, pGL3 (Promega), to yield the full-length promoter-reporter plasmid denoted as pPrkce1941k-Luc. Five 5′-deletion mutants were also constructed, including 1163, 444, 302, 241, and 106 bp of Prkce gene 5′ flanking sequences. All promoter construct sequences were confirmed using DNA sequencing analyses. Cell transfection was performed using rat embryonic heart-derived myogenic cell line H9C2 [24]. H9C2 cells were obtained from American Type Culture Collection (Rockville, MD) and were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum and 22 mM glucose. High-glucose medium increased the expression of Prkce in H9C2 cells [25]. H9C2 cells were seeded in six-well plates (2 × 106 cells/plate) and transiently cotransfected with 1 μg of promoter-reporter vector along with 0.05 μg of internal control pRL-SV40 vector using Tfx-20 transfection reagents for eukaryotic cells (Promega) according to the manufacturer's instructions. After 48 h, firefly and Renilla reniformis luciferase activities in cell extracts were measured in a luminometer using a dual-luciferase reporter assay system (Promega). The truncated promoter activities were then calculated by normalizing the firefly luciferase activity to R. reniformis luciferase activity.
Statistical Analysis
Data are expressed as mean ± SEM. Experimental number (n) represents offspring from different dams. Statistical significance (P < 0.05) was determined by ANOVA, followed by Neuman-Keuls post hoc testing.
RESULTS
Maternal Cocaine Administration Suppresses Cardiac Prkce Expression
To determine whether fetal cocaine exposure alters Prkce gene expression in the left ventricle during postnatal development, PRKCE protein and mRNA levels in the left ventricle were measured by Western blot analysis and quantitative real-time RT-PCR, respectively, in 3-wk-old and 3-mo-old offspring. In male offspring, prenatal cocaine exposure resulted in significant decreases in mRNA and protein abundance of PRKCE in the left ventricle at both developmental ages (Fig. 1). In contrast, neither mRNA nor protein levels of PRKCE were significantly altered by fetal cocaine exposure in female offspring at both ages.
FIG. 1.
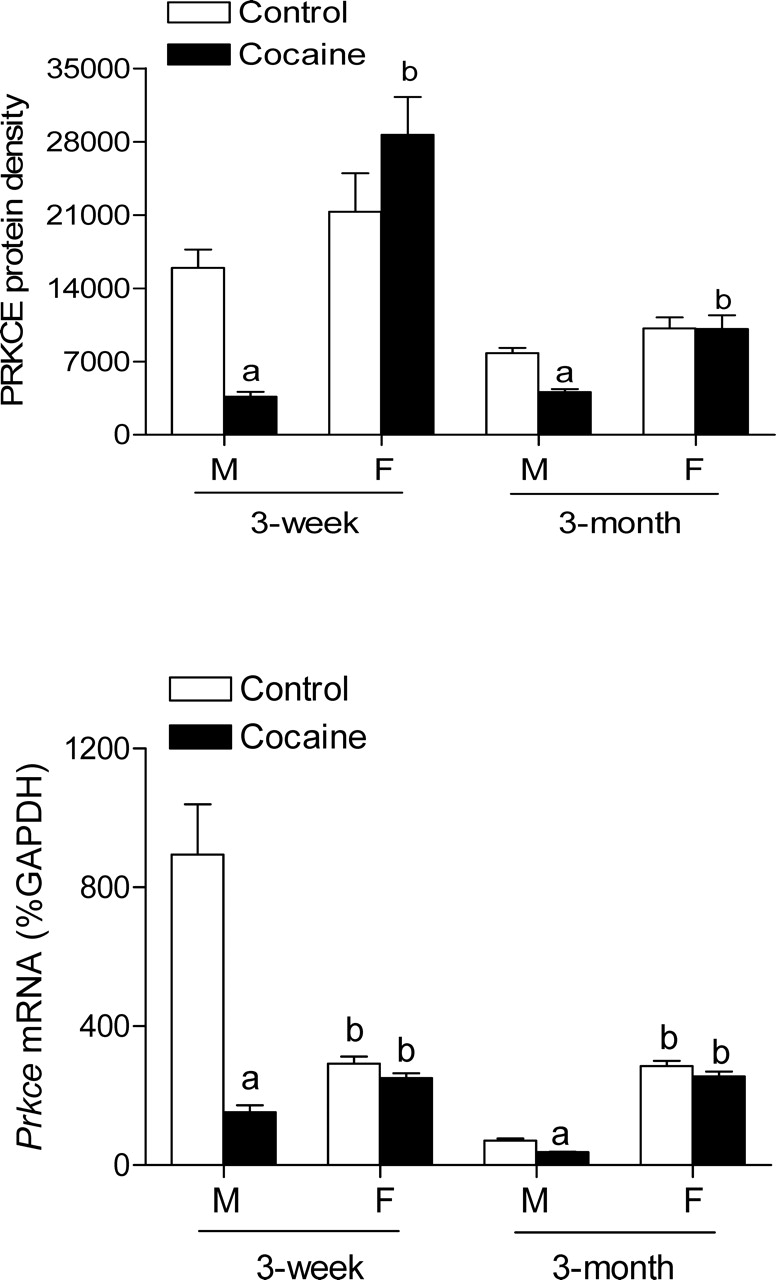
Effect of prenatal cocaine exposure on Prkce mRNA and protein abundance in the left ventricle. Time-dated pregnant Sprague-Dawley rats were administered saline or cocaine (30 mg/kg/day) from Days 15 to 20 of gestational age, and left ventricles were obtained from 3-wk-old and 3-mo-old male (M) and female (F) offspring. Prkce mRNA and protein abundance were determined using quantitative real-time RT-PCR and Western blot analysis, respectively. Data are mean ± SEM, n = 6. Data were analyzed by two-way ANOVA for each age, with cocaine exposure as one factor and gender as the other. aP < 0.05, cocaine vs. control; bP < 0.05, female vs. male.
Sequencing of the Prkce Gene Regulatory Region
Sequencing analysis reveals that nucleotides from 7945649 to 7946320 in rat chromosome 6 in GenBank/RefSeq do not exist. The sequence of the regulatory region consisting of 1786 bp upstream of the rat Prkce transcriptional start site is shown in Figure 2 (GenBank accession number EU882734). The rat Prkce gene promoter lacks a TATA-like element and contains high CpG levels. The following eight putative transcription factor binding sites containing CpG dinucleotides in their core binding sequences were identified: Bhlhb2 (class B basic helix-loop-helix protein 2) at −1723, Pparg (peroxisome proliferator-activated receptor gamma) at −1688, E2f (E2F transcription factor) at −1621, Egr1 (early growth response protein 1) at −1008, Mtf1 (metal regulatory transcription factor 1) at −603, Sp1 (transcription factor Sp1) at −346, Sp1 at −268, and Mtf1 at −168 (Figs. 2 and 3).
FIG. 2.

Sequence of the rat Prkce gene regulatory region. The updated sequence of the Prkce promoter (GenBank accession number EU882734) revealed that the GenBank/RefSeq rat chromosome 6 sequence from nucleotides 7945649 to 7946320 did not exist. Arrows indicate the direction of transcription or mRNA. The regulatory region of the rat Prkce gene lacks a TATA-like element and contains high CpG levels. Predicted transcription factor binding sites with CpG dinucleotides are in boldface, including Bhlhb2, Pparg, E2f, Egr1, Mtf1, Sp1, Sp1, and Mtf1.
FIG. 3.
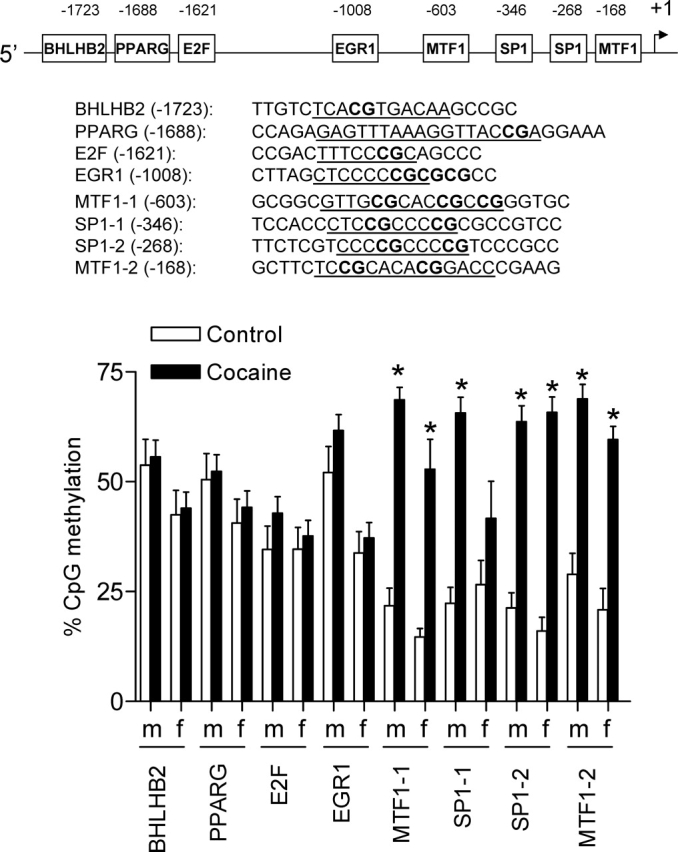
Effect of prenatal cocaine exposure on CpG methylation in the Prkce promoter in the left ventricle. Time-dated pregnant Sprague-Dawley rats were administered saline or cocaine (30 mg/kg/day) from Days 15 to 20 of gestational age, and left ventricles were obtained from 3-mo-old male (m) and female (f) offspring. DNA was extracted and treated with sodium bisulfite. The modified DNA was used as a template for real-time fluorogenic methylation-specific PCR. Primers for the Prkce promoter regions of interest are listed in Table 1. Gapdh was used as an internal reference gene. Real-time methylation-specific PCR was performed using the iQ SYBR Green Supermix with iCycler real-time PCR system. Data are mean ± SEM, n = 6. Data were analyzed by two-way ANOVA for each binding site, with cocaine exposure as one factor and gender as the other. The underlining indicates the binding sequences. *P < 0.05, cocaine vs. control.
Maternal Cocaine Administration Increases Prkce Promoter Methylation in the Heart
To determine the methylation status of CpG dinucleotides in the eight putative binding sites identified, quantitative methylation-specific PCR was performed after bisulfite treatment of DNA isolated from left ventricles of 3-mo-old offspring. Figure 3 shows the methylation pattern of CpG dinucleotides in the eight binding sites of the Prkce gene regulatory region in the heart. In males and females, methylation levels were high in the binding sites of Bhlhb2, Pparg, E2f, and Egr1 (∼40%–50%) compared with those of Sp1 and Mtf1 (∼20%) in the control hearts. Prenatal cocaine exposure had no significant effects on CpG dinucleotide methylation in Bhlhb2, Pparg, E2f, or Egr1 binding sites in male or female left ventricles. In male offspring, fetal cocaine exposure resulted in significant increases in methylation levels of Mtf1 at −603, Sp1 at −346, Sp1 at −268, and Mtf1 at −168. In female offspring, cocaine exposure caused significant increases in methylation levels of Mtf1 at −603, Sp1 at −268, and Mtf1 at −168 but not Sp1 at −346.
Involvement of the Sp1 and Mtf1 Binding Sites in Prkce Promoter Activation
Given the finding that methylation levels in the putative Sp1 and Mtf1 binding sites were significantly increased by cocaine exposure, the involvement of the Sp1 and Mtf1 binding sites in the regulation of Prkce promoter activity was determined. The activities of 5′-deletion mutants were evaluated in the rat embryonic ventricular myocyte cell line H9C2. The level of basal full-length promoter activity obtained with pPrkce1941-Luc was approximately 10-fold greater than that obtained with vector alone in the high-glucose medium. Promoter activity fell by 28% with deletion from −444 to −302, which contains the Sp1 binding site at −346, and by another 31% with deletion from −302 to −241, which contains the Sp1 binding site at −268 (Fig. 4), indicating a strong stimulatory role of SP1 binding to both Sp1 binding sites at −346 and −268 in the regulation of Prkce promoter activity. In contrast, deletion of the Mtf1 binding site at −603 or −168 had no significant effects on Prkce promoter activity.
FIG. 4.

Role of the Sp1 binding sites in Prkce promoter activity. Full-length and 5′-truncated Prkce promoter-reporter gene constructs were transiently cotransfected with pRL-SV40-driven R. reniformis luciferase in a rat embryonic heart-derived myogenic cell line (H9C2). After 48 h, firefly and R. reniformis luciferase activities in cell extracts were measured using a dual-luciferase reporter assay system. The truncated promoter activities were then calculated by normalizing the firefly luciferase activity to R. reniformis luciferase activity. Data are mean ± SEM, n = 4. Data were analyzed by one-way ANOVA. aP < 0.05, compared with pPrkce444-luc; bP < 0.05, compared with pPrkce302-luc.
To further evaluate the nuclear proteins binding to the putative Sp1 and Mtf1 elements of the Prkce promoter at −603, −346, −268, and −168, electrophoretic mobility shift assays were performed. Incubation of nuclear extracts from rat left ventricles with double-stranded oligonucleotide probes encompassing the putative Sp1 binding sites at −346 and −268 resulted in the appearance of a major DNA-protein complex (Fig. 5A). Supershift analyses showed that the SP1 antibody caused supershifting of the DNA-protein complex in both Sp1 binding sites. In contrast, although nuclear extracts caused the shift of both oligonucleotide probes encompassing the putative Mtf1 sites at −603 and −168, addition of the MTF1 antibody failed to produce the supershift (data not shown), suggesting that an unknown protein in nuclear extracts binds to the putative Mtf1 binding sequence. Western analyses using the MTF1 antibody demonstrated the existence of MTF1 protein in rat left ventricle nuclear extracts (data not shown).
FIG. 5.
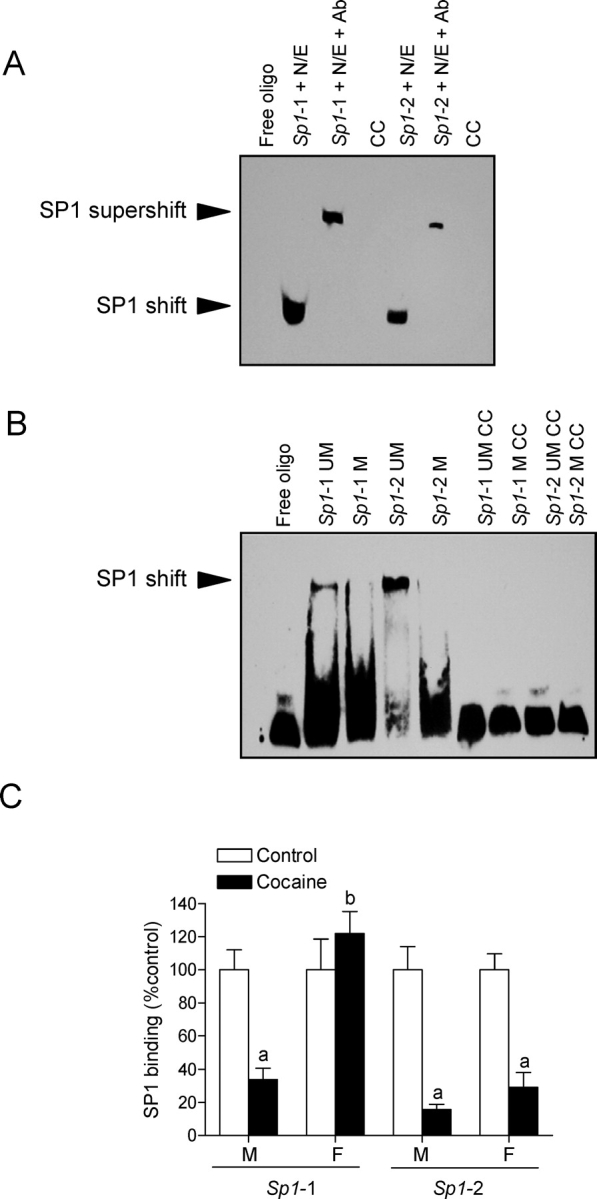
SP1 binding to unmethylated and methylated Sp1 binding elements. A) Nuclear extracts (N/E) from rat left ventricles were incubated with double-stranded oligonucleotide probes containing the Prkce gene consensus Sp1 binding motif at −346 (Sp1-1) and −268 (Sp1-2), in the absence or presence of the SP1 antibody (Ab). Cold competition (CC) was performed with unlabeled competitor oligonucleotide at a 100-fold molar excess. Free oligo, no nuclear extracts, or recombinant SP1 was added. B) Recombinant SP1 was incubated with double-stranded oligonucleotide probes containing unmethylated (UM) or methylated (M) CpG dinucleotides at the consensus Sp1 binding motif at −346 (Sp1-1) and −268 (Sp1-2). Cold competition was performed with unlabeled competitor oligonucleotide at a 100-fold molar excess. C) Time-dated pregnant Sprague-Dawley rats were administered saline or cocaine (30 mg/kg/day) from Days 15 to 20 of gestational age, and left ventricles were obtained from 3-mo-old male (M) and female (F) offspring. Chromatin extracts prepared from left ventricles were sonicated to produce DNA fragments between 100 and 500 bp in length. Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP-IT kit according to the manufacturer's protocol using anti-SP1 antibody. Antibody-pulled chromatin extracts were used as templates for quantitative real-time PCR using primers flanking the two Sp1 binding elements at −346 and −268 of the rat Prkce promoter. Data are mean ± SEM, n = 6. Data were analyzed by two-way ANOVA for each binding site, with cocaine exposure as one factor and gender as the other. aP < 0.05, cocaine vs. control; bP < 0.05, female vs. male.
Methylation of the Sp1 Binding Sites Inhibits SP1 Binding
To determine whether methylation of the Sp1 binding sites in the Prkce promoter inhibits SP1 binding, electrophoretic mobility shift assays were performed with methylated and unmethylated oligonucleotide probes containing the Sp1 binding sites at −346 and −268. As shown in Figure 5B, recombinant SP1 bound and shifted the double-stranded unmethylated Sp1 oligonucleotides at both sites but failed to cause the shift of the double-stranded methylated Sp1 oligonucleotides. To verify whether cocaine-mediated increases in methylation of the Sp1 binding sites can inhibit SP1 binding to the Prkce promoter in vivo in the context of intact chromatin, we performed ChIP assays using the SP1 antibody. Figure 5C shows the binding of SP1 to both Sp1 elements at −346 and −268 in the promoter region of the Prkce gene in intact chromatin in vivo in the left ventricle of 3-mo-old offspring. Quantitative real-time PCR demonstrated that fetal cocaine exposure caused marked decreases of 66% and 85% in SP1 binding to the Sp1 binding sites at −346 and −268, respectively, in the left ventricle of male offspring. In females, prenatal cocaine exposure resulted in a 70% decrease in SP1 binding to the site at −268 but had no effect on its binding to the site at −346.
Effect of Maternal Cocaine Administration on SP1 Abundance and Activity in the Left Ventricle
To determine whether prenatal cocaine-induced decreases in SP1 binding in adult male hearts were due to effects on SP1 abundance, Western blot analyses were performed using the SP1 antibody. No significant differences were observed in SP1 protein levels in nuclear extracts isolated from the left ventricle of adult male offspring administered saline or cocaine before birth (Fig. 6A). The binding affinity of SP1 to the unmethylated Sp1 binding sites was determined in competition studies performed in pooled nuclear extracts from adult male left ventricles with increasing ratios of unlabeled/labeled oligonucleotides encompassing the Sp1 binding sites at −346 and −268. As shown in Figure 6, B and C, prenatal cocaine exposure had no significant effect on SP1 binding to the site at −346 in nuclear extracts of adult left ventricle but decreased its binding affinity to the site at −268.
FIG. 6.
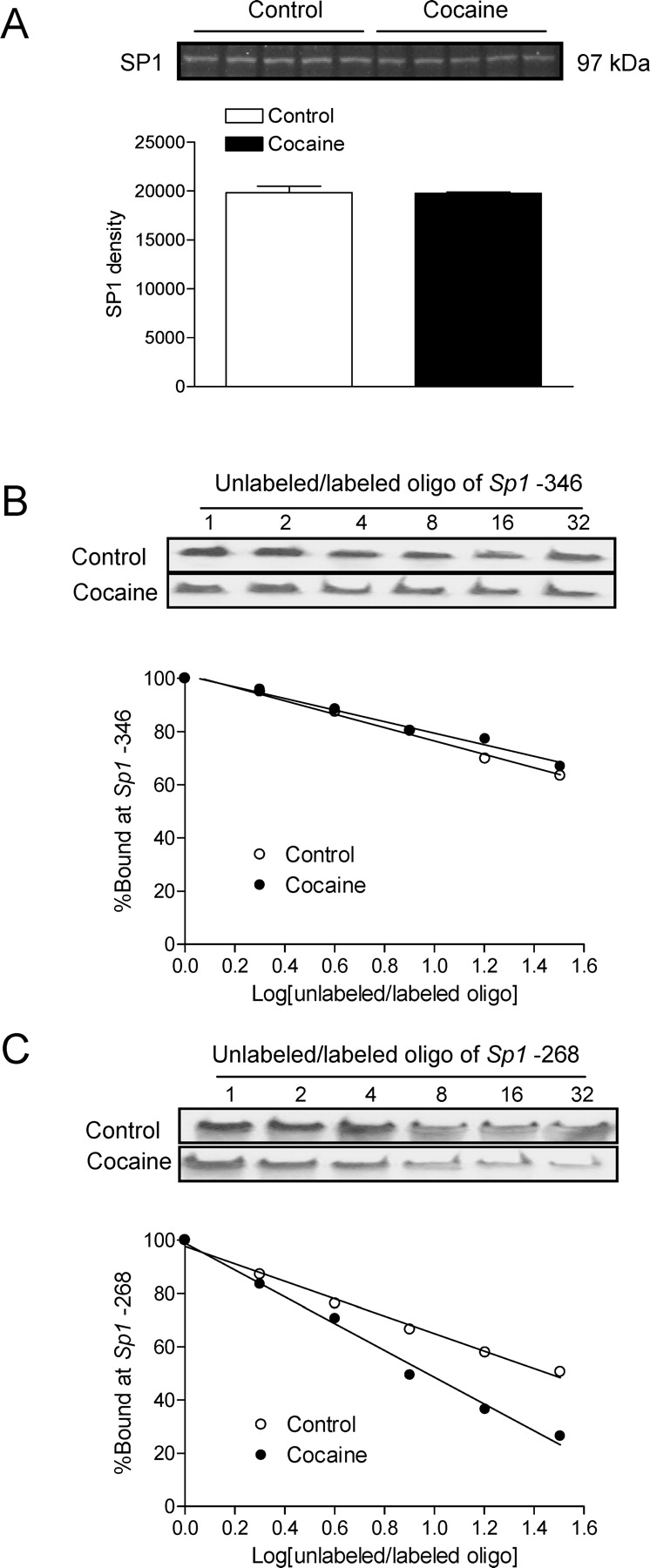
Effect of prenatal cocaine exposure on SP1 abundance and activity in the left ventricle. Time-dated pregnant Sprague-Dawley rats were administered saline or cocaine (30 mg/kg/day) from Days 15 to 20 of gestational age, and left ventricles were obtained from 3-mo-old male offspring. A) SP1 protein abundance in nuclear extracts prepared from left ventricles was determined by Western blot using the SP1 antibody. Data are mean ± SEM, n = 5. B and C) Pooled nuclear extracts from left ventricles were incubated with labeled double-stranded oligonucleotide probes containing the Prkce gene consensus Sp1 binding motif at −346 and −268 in the presence of 1, 2, 4, 8, 16, and 32 folds of the unlabeled oligonucleotides.
DISCUSSION
The present study shows that prenatal cocaine exposure results in decreased expression of Prkce mRNA and protein in the left ventricle of adult male but not female offspring. This gender-dependent difference in Prkce gene expression was also observed in 3-wk-old offspring before sexual maturity. While this result suggests that the gender-specific effects of prenatal cocaine exposure on Prkce expression in the heart of offspring may not be primarily mediated by sex hormones developed postnatally, future studies are needed to investigate further the mechanisms of the gender dichotomy observed herein and to test for a role of sex steroid hormones in male and female gonadectomized rats. Alternatively, potential differences in steroid hormone receptor expression between male and female fetuses may contribute to the gender differences observed in fetal programming, although the fetuses are exposed to the same levels of hormones in utero. Accumulating evidence suggests that, in addition to sex-defining steroids, differences exist between genetically male (XY) and female (XX) cells in determining an “ischemia-sensitive” phenotype [26]. Previous studies [27–33] have demonstrated consistent gender differences in adult offspring, with males often being more susceptible than females on various measures to the effects of prenatal cocaine exposure in the rat. Similarly, in animal models of intrauterine undernutrition, gender dimorphism manifesting as severity of hypertension among adult offspring has been observed [34]. The pathophysiological significance of the gender-dependent decrease in PRKCE levels in the male heart was demonstrated by the findings that prenatal cocaine exposure increased susceptibility of the heart to ischemia-reperfusion injury in adult male but not female offspring [10].
Although PRKC has been demonstrated to be important in regulating the expression of many other genes, studies of mechanisms in the regulation of Prkc gene expression itself are extremely limited. The finding of a persistent decrease in Prkce mRNA levels among adult male offspring after fetal exposure to cocaine suggests an epigenetic modification and in utero programming of Prkce gene repression in the heart. The present study demonstrates that the rat Prkce gene promoter lacks a TATA-like element and contains high CpG levels, suggesting a rigorous regulation by methylation. DNA methylation is a chief mechanism in epigenetic modification and is generally associated with transcription repression, leading to long-term shutdown of the associated gene. Epigenetic mechanisms are essential for development and differentiation and allow an organism to respond to the environment through changes in gene expression [18, 35, 36]. It has been shown that methylation patterns can be altered in cancer, in aging, and by diet [18, 37–40]. Whereas it is likely that methylation of CpGs outside of transcription factor binding sites can affect promoter activity, the present study focused on the methylation of recognition sequences of putative transcription factors, including those for Bhlhb2, Pparg, E2f, Egr1, Mtf1, and Sp1, which contain CpG dinucleotides in their core binding sites. Four of them, Bhlhb2, Pparg, E2f, and Egr1, are heavily methylated in the control hearts of both genders, suggesting that they may not be actively involved in the regulation of Prkce gene expression in the left ventricle. Consistently, deletion of these sites did not significantly affect Prkce promoter activity.
The finding that MTF1 antibody failed to produce the supershift of nuclear extracts binding to the two putative Mtf1 sites at −603 and −168 suggests that an unknown protein other than MTF1 in the rat heart binds to the putative Mtf1 binding sequence. Although it is intriguing that methylation of the putative Mtf1 sites was increased in cocaine-exposed animals among male and female offspring, deletion of these sites did not significantly affect Prkce promoter activity, suggesting their minimum effects in the regulation of Prkce gene expression in the left ventricle. In contrast, the antiserum to SP1 caused supershifting of the DNA-protein complex, resulting from binding of nuclear extracts from the left ventricle with the double-stranded oligonucleotide probes containing the putative Sp1 element in the Prkce gene at −346 and −268, indicating that they are the Sp1 binding sites in the Prkce gene in rat hearts. The functional significance of the Sp1 binding sites in the regulation of Prkce gene activity was demonstrated further by the finding that deletion of the Sp1 binding sites significantly decreased Prkce promoter activity.
The present study demonstrated that methylation levels of the Sp1 binding sites were low in control left ventricles and were significantly increased in cocaine-exposed left ventricles, suggesting an important role of the Sp1 binding sites in epigenetic modification of Prkce gene expression in the heart. Although transcriptional regulation by DNA methylation is often observed in CpG islands located around the promoter region via the sequence-nonspecific and methylation-specific binding of inhibiting methylated CpG-binding proteins [38, 41], DNA methylation of sequence-specific transcription factor binding sites can alter gene expression through changes in the binding affinity of transcription factors by altering the major groove structure of DNA to which the DNA-binding proteins bind [42–44]. The finding that the Sp1 probes with methylated CpG dinucleotides at the core of the consensus Sp1 elements in the Prkce promoter abolished the binding of SP1 indicates that CpG methylation in non-CpG islands and sequence-specific Sp1 binding sites can directly inhibit the DNA binding of SP1, resulting in down-regulation of Prkce gene expression in the heart. Although it was suggested that SP1 binding might not be affected by DNA methylation [45], recent studies [40, 46] have demonstrated that methylation of a recognition sequence of the Sp1 binding site diminishes its DNA-binding activity.
To determine whether the cocaine-mediated increase in methylation of the Sp1 binding sites can inhibit SP1 binding to the Prkce promoter in vivo in the context of intact chromatin, ChIP assays were performed using the SP1 antibody. It was demonstrated that prenatal cocaine exposure significantly decreased the recruitment of SP1 to the Prkce promoter in both sites at −346 and −268 in the left ventricle of adult male offspring. The extent of the decrease in SP1 binding in vivo in intact chromatin was comparable to that of increased methylation of the Sp1 binding sites in the cocaine-exposed hearts. In female offspring, the decreased SP1 binding was observed only in the binding site at −268, consistent with the finding that the increased methylation was shown only in the binding site at −268 in female hearts. Whereas the finding of decreased Prkce mRNA and protein abundance is consistent with increased methylation and decreased SP1 binding to the Prkce promoter in both binding sites at −346 and −268 in the left ventricle of male offspring that were exposed to cocaine before birth, the increased methylation and the decreased SP1 binding in the site at −268 alone in female offspring were not associated with significant decreases in Prkce mRNA and protein levels in the left ventricle of cocaine-exposed animals. This suggests that changes in CpG dinucleotide methylation and SP1 binding in the Sp1 binding site at −268 may not have a key role in epigenetic modification of Prkce expression in the heart. Alternatively, an increase in CpG dinucleotide methylation and a decrease in SP1 binding of the Sp1 binding site at −268 or −346 alone may not be sufficient for Prkce gene repression in the heart, and methylation of both sites may be required. Future studies are needed to develop “site-directed” or “positional” methylation of Prkce promoter-luciferase constructs selectively in the Sp1 binding sites at −268 and −346, which should provide insights into their roles in decreased transcriptional activity of the Prkce gene in the heart.
The present investigation has demonstrated evidence of increased CpG methylation in non-CpG island, sequence-specific transcription factor binding sites of the Prkce gene promoter in the left ventricle of adult offspring that were exposed to cocaine before birth, providing a novel model to evaluate subtle epigenetic modifications of gene expression patterns in fetal programming of cardiac function in response to an adverse intrauterine environment. The collective observations in the present study indicate that an increase in methylation of CpG dinucleotides at the core of the consensus Sp1 elements in the Prkce promoter results in decreased SP1 binding to the Prkce promoter, leading to Prkce gene repression in the left ventricle of male offspring. While the molecular mechanisms that underlie such an alteration in methylation pattern as a result of fetal programming remain to be determined, the concept that adverse intrauterine environmental factors may result in highly specific altered methylation patterns in a gene promoter among adult offspring is supported by a recent finding. In a maternal low-protein diet rat model, it has been shown that fetal undernutrition results in increased expression of the At1b angiotensin receptor gene in the adrenal gland of adult offspring, which is caused by a decrease in methylation of the promoter [40]. As is often the case with novel findings, the present study may raise more questions than it answers. For instance, are the changes mediated by a direct effect of cocaine exposure on the fetus or indirectly through its effect on the mother? Given that cocaine inhibits sympathetic neurotransmitter reuptake, potential neural effects should not be ignored. In addition, is the effect of cocaine exposure unique to the heart or may it occur in other tissues? To what extent are the differentiated cells or the progenitors of the heart involved in cocaine-mediated fetal programming? What are the temporal and dose-dependent effects of fetal cocaine exposure? Undoubtedly, these questions warrant further investigation. It may be difficult to translate the present findings directly into the scenario of human cocaine abuse. However, the suggestion that maternal cocaine abuse may result in altered methylation patterns in a specific gene among offspring provides increased mechanistic understanding that is worthy of investigation in humans. Given that large epidemiological studies [1–6] have indicated a link between in utero adverse stimuli during pregnancy and an increased risk of ischemic heart disease in adulthood, the present work may complement the paucity of human data about long-term cardiovascular consequences of fetal cocaine exposure.
Footnotes
1Supported in part by NIH grants HL82779 (to L.Z.) and HL83966 (to L.Z.) and by Loma Linda University School of Medicine.
REFERENCES
- Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS.Fetal nutrition and cardiovascular disease in adult life. Lancet 1993; 341: 938–941. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME.Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989; 298: 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsen T, Eriksson JG, Tuomilehto J, Osmond C, Barker DJ.Growth in utero and during childhood among women who develop coronary heart disease: longitudinal study. BMJ 1999; 319: 1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ.Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ 1999; 318: 427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon DA, Lithell HO, Vagero D, Koupilova I, Mohsen R, Berglund L, Lithell UB, McKeigue PM.Reduced fetal growth rate and increased risk of death from ischemic heart disease: cohort study of 15 000 Swedish men and women born 1915–29. BMJ 1998; 317: 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CE, Fall CH, Kumaran K, Osmond C, Cox V, Barker DJ.Fetal growth and coronary heart disease in south India. Lancet 1996; 348: 1269–1273. [DOI] [PubMed] [Google Scholar]
- Zhang L, Xiao Y, He J.Cocaine and apoptosis in myocardial cells. Anat Rec 1999; 257: 208–216. [DOI] [PubMed] [Google Scholar]
- Xiao Y, He J, Gilbert RD, Zhang L.Cocaine induces apoptosis in fetal myocardial cells through a mitochondria-dependent pathway. J Pharmacol Exp Ther 2000; 292: 8–14. [PubMed] [Google Scholar]
- Xiao Y, Xiao D, He J, Zhang L.Maternal cocaine administration during pregnancy induces apoptosis in fetal rat heart. J Cardiovasc Pharmacol 2001; 37: 639–648. [DOI] [PubMed] [Google Scholar]
- Bae S, Gilbert RD, Ducsay CA, Zhang L.Prenatal cocaine exposure increases heart susceptibility to ischaemia-reperfusion injury in adult male but not female rats. J Physiol 2005; 565(pt 1):149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Zhang L.Prenatal cocaine exposure increases apoptosis of neonatal rat heart and heart susceptibility to ischemia/reperfusion injury in one-month-old rat. Br J Pharmacol 2005; 144: 900–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Xiao YH, Zhang L.Cocaine induces apoptosis in fetal rat myocardial cells through the p38 MAPK and mitochondrial/cytochrome C pathways. J Pharmacol Exp Ther 2005; 312: 112–119. [DOI] [PubMed] [Google Scholar]
- Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW, Mochly-Rosen D.Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci U S A 2001; 98: 11114–11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping P, Zhang J, Pierce WM, Jr, Bolli R.Functional proteomic analysis of protein kinase C ε complexes in normal heart and during cardioprotection. Circ Res 2001; 88: 59–62. [DOI] [PubMed] [Google Scholar]
- Murriel CL, Mochly-Rosen D.Opposing roles of delta and epsilon PKC in cardiac ischemia and reperfusion: targeting the apoptotic machinery. Arch Biochem Biophys 2003; 420: 246–254. [DOI] [PubMed] [Google Scholar]
- Gray MO, Zhou HZ, Schafhalter-Zoppoth I, Zhu P, Mochly-Rosen D, Messing RO.Preservation of base-line hemodynamic function and loss of inducible cardioprotection in adult mice lacking protein kinase C epsilon. J Biol Chem 2004; 279: 3596–3604. [DOI] [PubMed] [Google Scholar]
- Jones PA, Takai D.The role of DNA methylation in mammalian epigenetics. Science 2001; 293: 1068–1070. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A.Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33(suppl):245–254. [DOI] [PubMed] [Google Scholar]
- Javaid JI, Davis JM.Cocaine disposition in discrete regions of rat brain. Biopharm Drug Dispos 1993; 14: 357–364. [DOI] [PubMed] [Google Scholar]
- Zhang H, Darwanto A, Linkhart TA, Sowers LC, Zhang L.Maternal cocaine administration causes an epigenetic modification of protein kinase C gene expression in fetal rat heart. Mol Pharmacol 2007; 71: 1319–1328. [DOI] [PubMed] [Google Scholar]
- Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB.Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 1996; 93: 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fackler MJ, McVeigh M, Mehrotra J, Blum MA, Lange J, Lapides A, Garrett E, Argani P, Sukumar S.Quantitative multiplex methylation-specific PCR assay for the detection of promoter hypermethylation in multiple genes in breast cancer. Cancer Res 2004; 64: 4442–4452. [DOI] [PubMed] [Google Scholar]
- Hoque MO, Begum S, Topaloglu O, Chatterjee A, Rosenbaum E, Van Criekinge W, Westra WH, Schoenberg M, Zahurak M, Goodman SN, Sidransky DJ.Quantitation of promoter methylation of multiple genes in urine DNA and bladder cancer detection. J Natl Cancer Inst 2006; 98: 996–1004. [DOI] [PubMed] [Google Scholar]
- Kaneda R, Ueno S, Yamashita Y, Choi YL, Koinuma K, Takada S, Wada T, Shimada K, Mano H.Genome-wide screening for target regions of histone deacetylases in cardiomyocytes. Circ Res 2005; 97: 210–218. [DOI] [PubMed] [Google Scholar]
- Kim MH, Jung YS, Moon CH, Jeong EM, Lee SH, Baik EJ, Moon CK.Isoform-specific induction of PKC-ε by high glucose protects heart-derived H9c2 cells against hypoxic injury. Biochem Biophys Res Commun 2003; 309: 1–6. [DOI] [PubMed] [Google Scholar]
- Hurn PD, Vannucci SJ, Hagberg H.Adult or perinatal brain injury: does sex matter? Stroke 2005; 36: 193–195. [DOI] [PubMed] [Google Scholar]
- Molina VA, Wagner JM, Spear LP.The behavioral response to stress is altered in adult rats exposed prenatally to cocaine. Physiol Behav 1994; 55: 941–945. [DOI] [PubMed] [Google Scholar]
- Goodwin GA, Rajachandran L, Moody CA, Francis R, Kuhn CM, Spear LP.Effects of prenatal cocaine exposure on haloperidol-induced increases in prolactin release and dopamine turnover in weanling, periadolescent, and adult offspring. Neurotoxicol Teratol 1995; 17: 507–514. [DOI] [PubMed] [Google Scholar]
- Grimm VE, Frieder B.Differential vulnerability of male and female rats to the timing of various perinatal insults. Int J Neurosci 1985; 27: 155–164. [DOI] [PubMed] [Google Scholar]
- Heyser CJ, Spear NE, Spear LP.Effects of prenatal exposure to cocaine on Morris water maze performance in adult rats. Behav Neurosci 1995; 109: 734–743. [DOI] [PubMed] [Google Scholar]
- Spear LP, Campbell J, Snyder K, Silveri M, Katovic N.Animal behavior models: increased sensitivity to stressors and other environmental experiences after prenatal cocaine exposure. Ann N Y Acad Sci 1998; 846: 76–88. [PubMed] [Google Scholar]
- Wood RD, Spear LP.Prenatal cocaine alters social competition of infant, adolescent, and adult rats. Behav Neurosci 1998; 112: 419–431. [DOI] [PubMed] [Google Scholar]
- Spear LP, Silveri MM, Casale M, Katovic NM, Campbell JO, Douglas LA.Cocaine and development: a retrospective perspective. Neurotoxicol Teratol 2002; 24: 321–327. [DOI] [PubMed] [Google Scholar]
- do Carmo Pinho Franco M, Nigro D, Fortes ZB, Tostes RC, Carvalho MH, Lucas SR, Gomes GN, Coimbra TM, Gil FZ.Intrauterine undernutrition: renal and vascular origin of hypertension. Cardiovasc Res 2003; 60: 228–234. [DOI] [PubMed] [Google Scholar]
- Reik W, Dean W, Walter J.Epigenetic reprogramming in mammalian development. Science 2001; 293: 1089–1093. [DOI] [PubMed] [Google Scholar]
- Reik W, Constancia M, Fowden A, Anderson N, Dean W, Ferguson-Smith A, Tycko B, Sibley C.Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J Physiol 2003; 547: 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB.Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet 1994; 7: 536–540. [DOI] [PubMed] [Google Scholar]
- Jones PA, Laird PW.Cancer epigenetics comes of age. Nat Genet 1999; 21: 163–167. [DOI] [PubMed] [Google Scholar]
- Van den Veyver IB.Genetic effects of methylation diets. Annu Rev Nutr 2002; 22: 255–282. [DOI] [PubMed] [Google Scholar]
- Bogdarina I, Welham S, King PJ, Burns SP, Clark AJL.Epigenetic modification of the rennin-angiotensin system in the fetal programming of hypertension. Circ Res 2007; 100: 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade PA.Methyl CpG-binding proteins and transcriptional repression. Bioessays 2001; 23: 1131–1137. [DOI] [PubMed] [Google Scholar]
- Campanero MR, Armstrong MI, Flemington EK.CpG methylation as a mechanism for the regulation of E2F activity. Proc Natl Acad Sci U S A 2000; 97: 6481–6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WG, Srinivasan K, Dai Z, Duan W, Druhan LJ, Ding H, Yee L, Villalona-Calero MA, Plass C, Otterson GA.Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21Cip1 promoter. Mol Cell Biol 2003; 23: 4056–4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Kitazawa R, Maeda S, Kitazawa S.Methylation adjacent to negatively regulating AP-1 site reactivates TrkA gene expression during cancer progression. Oncogene 2005; 24: 5108–5118. [DOI] [PubMed] [Google Scholar]
- Tate PH, Bird AP.Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev 1993; 3: 226–231. [DOI] [PubMed] [Google Scholar]
- Alikhani-Koopaei R, Fouladkou F, Frey FJ, Frey BM.Epigenetic regulation of 11 beta-hydroxysteroid dehydrogenase type 2 expression. J Clin Invest 2004; 114: 1146–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]