

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease of motor neurons (MNs) that causes paralysis. Some forms of ALS are inherited, caused by mutations in the superoxide dismutase-1 (SOD1) gene. The mechanisms of human mutant SOD1 (mSOD1) toxicity to MNs are unresolved. Mitochondria in MNs might be key sites for ALS pathogenesis, but cause-effect relationships between mSOD1 and mitochrondiopathy need further study. We used transgenic mSOD1 mice to test the hypothesis that the mitochondrial permeability transition pore (mPTP) is involved in the MN degeneration of ALS. Components of the multi-protein mPTP are expressed highly in mouse MNs, including the voltage-dependent anion channel, adenine nucleotide translocator (ANT), and cyclophilin D (CyPD), and are present in mitochondria marked by manganese SOD. MNs in pre-symptomatic mSOD1-G93A mice form swollen megamitochondria with CyPD immunoreactivity. Early disease is associated with mitochondrial cristae remodeling and matrix vesiculation in ventral horn neuron dendrites. MN cell bodies accumulate mitochondria derived from the distal axons projecting to skeletal muscle. Incipient disease in spinal cord is associated with increased oxidative and nitrative stress, indicated by protein carbonyls and nitration of CyPD and ANT. Reducing the levels of CyPD by genetic ablation significantly delays disease onset and extends the lifespan of G93A-mSOD1 mice expressing high and low levels of mutant protein in a gender-dependent pattern. These results demonstrate that mitochondria have causal roles in the disease mechanisms in MNs in ALS mice. This work defines a new mitochondrial mechanism for MN degeneration in ALS.
Keywords: adenine nucleotide translocase, cristae remodeling, mutant SOD1, nitration, porin, ppif, voltage-dependent anion channel
Introduction
ALS is a progressive, severely disabling fatal neurological disease in humans characterized by weakness, muscle atrophy, spasticity, and eventual paralysis and death generally within 3 to 5 years after symptoms begin (Rowland and Shneider, 2001). The cause of the spasticity, paralysis and morbidity is progressive degeneration and loss of upper motor neurons (MNs) in cerebral cortex and lower MNs in brainstem and spinal cord (Rowland and Shneider, 2001; Price et al., 2006). More than 5600 people in the US are diagnosed with ALS each year (ALS Association, www.alsa.org). The triggering events for MN degeneration are not understood and why MNs are selectively vulnerable in ALS is unclear. The molecular pathogenesis of most forms of ALS is understood poorly, contributing to the lack of effective mechanism-based therapies to treat this disease (Martin and Liu, 2004). Two forms of ALS exist: sporadic and familial (Rowland and Shneider, 2001). The majority of ALS cases are sporadic with no known genetic component, except for missense mutations in TAR-DNA binding protein (Kabashi et al., 2007). Aging is a strong risk factor for ALS because the average age of onset is 55 (ALS Association, www.alsa.org). Familial forms of ALS (fALS) have autosomal dominant or autosomal recessive inheritance patterns and make up ∼10% or less of all ALS cases (Price et al., 2006). ALS-linked mutations occur in genes encoding SOD1 (ALS1), Alsin (ALS2), senataxin (ALS4), vesicle associated membrane protein (VAMP/synaptobrevin)-associated protein B (ALS8), dynactin, and TAR-DNA binding protein (Price et al., 2006; Schymick et al., 2007).
Mutations in the SOD1 gene account for ∼20% of all fALS cases (∼2% of all ALS cases) (Deng et al., 1993; Rosen et al., 1993). SOD1 (also known as copper/zinc SOD) is a metalloenzyme of 153 amino acids (∼16 kDa) that binds one copper ion and one zinc ion per subunit. SOD1, functioning as a ∼32 kDa non-covalently linked homodimer, is responsible for the detoxification and maintenance of intracellular superoxide anion (O2-) concentration in the low femtomolar range by catalyzing the dismutation of O2- to molecular oxygen and hydrogen peroxide (O2- + O2- + 2H+ → H2O2 + O2)(McCord and Fridovich, 1969). SOD1 is ubiquitous (intracellular SOD concentrations are typically ∼10-40 μM) in most tissues and possibly greater in neurons (Rakhit et al., 2004). SOD1 mutants appear to gain a toxic property or function, rather than having diminished O2- scavenging activity (Deng et al., 1993; Borchelt et al., 1994; Yim et al., 1996). A common mutation in SOD1 is the substitution of glycine by alanine at position 93 (G93A). Gurney et al were the first to develop transgenic (tg) mice that express this mutant form of human SOD1 linked to fALS (Gurney et al., 1994; Dal Canto et al., 1994). These mice are used widely as an animal model of ALS (Bendotti and Carri, 2004; Martin and Liu, 2004; Cozzolino et al., 2008; Turner and Talbot, 2008). Effects of this human mutant gene on mice are profound. Hemizygous tg mice expressing high copy number of the G93A variant of mutant SOD1 (mSOD1) become completely paralyzed and die at ∼16-18 weeks of age (Gurney et al., 1994). MNs in mice expressing G93Ahigh-mSOD1 undergo prominent degeneration; about 70% of lumbar MNs are eliminated by end-stage disease (Martin et al., 2007). MNs in these mice do not degenerate with the classic morphology of apoptosis (Bendotti et al 2001; Martin and Liu, 2004; Martin et al., 2007). The MN degeneration seen in these mice more closely resembles a prolonged necrotic-like cell death process involving early-occurring mitochondrial damage, cellular swelling, and dissolution (Martin et al., 1998; Martin and Liu, 2004; Martin et al., 2007). Mitochondrial microvacuolar damage in MNs emerges by 4 weeks of age in G93A mice with high expression (Bendotti et al 2001; Martin et al., 2007). Thus, mitochondria might be primary sites of pathogenesis in mSOD1 mice (Wong et al., 1995; Kong and Xu et al., 1998; Jaarsma et al., 2001; Higgins et al., 2002; Higgins et al., 2003; Martin et al., 2007). Human SOD1 mutants associate with spinal cord mitochondria and can aggregate with Bcl-2 (Liu et al., 2004; Pasinelli et al., 2004). mSOD1 (and low-mobility species) binding to mitochondria has been reported to be spinal cord selective and age-dependent (Vande Velde et al., 2008). Human SOD1 mutants can also shift mitochondrial redox potential when expressed in cultured cells (Ferri et al., 2006).
Although mitochondrial toxicity of mSOD1 has been implicated in the pathogenesis of ALS, cause-effect relationships between mitochondrial damage and disease initiation and/or progression are still vague. This relationship needs to be discerned because new mechanism-based treatments for ALS could hinge on this knowledge. In the present study we demonstrate that mitochondrial abnormalities are related causally to the disease process in ALS mice through the mitochondrial matrix protein cyclophilin D (CyPD) that regulates the mitochondrial permeability transition pore and necrotic cell death forms (Baines et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005).
Materials and methods
Mice
The ALS mice studied here were tg mice expressing human mSOD1. Two different mSOD1 mouse lines were studied. One mouse line (B6SJL-TgN[SOD1-G93A]1Gur, G1H line, stock #002726, The Jackson Laboratory, Bar Harbor, ME) had a high copy number of human G93A mutant SOD1 allele (∼20 copies) and a rapid disease onset. We also used G93A-mSOD1 mice with reduced transgene copy number and a much slower disease progression, B6SJL-Tg(SOD1-G93A)dl1Gur/J mice (The Jackson Laboratory, stock #002300). Controls were tg mice expressing the normal human SOD1 gene (B6SJL-TgN[SOD1]2Gur, The Jackson Laboratory, stock # 002297) and non-tg mice studied at the same ages as the mutants. CyPD null mice are described elsewhere (Baines et al., 2005). The institutional Animal Care and Use Committee approved the animal protocols.
Western blotting
The presence of CyPD, ANT, and VDAC in mouse CNS was evaluated by immunoblotting. Non-tg mice (n=4), ppif-/- mice (n=4), human wtSOD1 tg mice (n=4), and human G93Ahigh-mSOD1 at different stages of disease, including pre-symptomatic, early- to mid-symptomatic, and end-stage (n=8), were deeply anesthetized with sodium pentobarbital, decapitated, and the brain and spinal cord were removed quickly and placed in ice-cold Hanks buffer. The forebrain was dissected from the brainstem and all samples were frozen quickly on dry ice. Forebrain, brainstem, and spinal cord samples were used to prepare soluble protein and mitochondrial-enriched membrane protein fractions by subcellular fractionation using a confirmed protocol (Martin et al., 2003). Protein fractions were subjected to SDS-PAGE and immunoblotting using enhanced chemilumesence (ECL) detection as described (Martin et al., 2003). Antibodies to mPTP proteins were purchased from commercial sources. For CyPD a mouse monoclonal antibody (Mitoscience, clone E11AE12BD4) was used. For ANT, a mouse monoclonal antibody (Mitoscience, clone 5F51BB5AG7) and a rabbit polyclonal antibody (Santa Cruz, H-188) were used. For VDAC two mouse monoclonal antibodies (Mitoscience, clone 20B12AF2 and Calbiochem, clone 89-173/016) were used. ANT and VDAC antibodies were not isoform specific.
Immunohistochemistry
mSOD1 tg mice at pre-symptomatic (n= 6) and symptomatic stages (n=10), wtSOD1 tg mice (n=6) and non-tg mice (n=6) were deeply anesthetized and perfusion-fixed using 4% paraformaldehyde (PF). After perfusion-fixation, spinal cords remained in situ for 4 hours before they were removed from the vertebral column and then placed in 20% glycerol for cryoprotection before frozen-sectioning for immunohistochemistry. Transverse serial symmetrical sections (40 μm) through lumbar spinal cord were cut using a sliding microtome and stored individually in 96-well plates.
We determined the in situ localizations of mPTP proteins in MNs as done for other mitochondrial proteins (Martin and Liu, 2002; Martin et al., 2007). We studied VDAC, ANT, and CyPD. Antibodies to these proteins were characterized by western blotting to determine specificity in tissue extracts of wildtype and gene knockout mice (CyPD). Additional negative controls for antibody specificity were incubating sections in the primary antibody diluent without primary antibody with all other steps similar. For single labeling proteins were detected using a standard immunoperoxidase method with diaminobenzidine (DAB) as chromogen.
Immunofluoresence was used for dual labeling to colocalize mPTP proteins with the mitochondrial marker manganese SOD (or SOD2) using a rabbit polyclonal antibody (Assay Design). Immunoreactivity was visualized with species-specific secondary antibodies (all raised in goat) conjugated to Alexa Fluor 488 or Alexa Fluor 594 (Invitrogen Corporation, Carlsbad, CA). Spinal cord sections were imaged using a 100× oil immersion objective (1.3 numerical aperture lens) mounted on a Zeiss LSM 510 confocal microscope. All sections were imaged under identical conditions and analyzed using identical parameters.
Electron microscopy (EM)
Tg human mSOD1 at pre-symptomatic stages of disease (n=4) and wtSOD1 mice (n=4) were evaluated for mitochondrial ultrastructural pathology in spinal cord ventral horn using conventional EM. Mice were deeply anesthetized and perfusion-fixed using 2% PF and 2% glutaraldehyde. Lumbar spinal cords were dissected carefully to isolate ventral horn tissue columns that were processed en bloc. Sample were rinsed in phosphate buffer, placed in 2% osmium tetroxide for two hours, dehydrated, and embedded in plastic. Semithin sections (1 μm) stained with 1% toluidine blue were screened for areas of interest, and then thin sections (gold interference color) were cut on an ultramicrotome (Sorvall, Norwalk, CT), contrasted with lead citrate and uranyl acetate, and viewed with a JEOL 100S electron microscope.
Immunoprecipitation and detection of nitrated proteins
Immunoprecipitation and western blot analysis of spinal cord homogenates from mSOD1 and wtSOD1 mice were used to identify peroxynitrite-mediated nitration of CyPD and ANT. Protein (100 μg) from mitochondria-enriched membrane fractions from mouse spinal cord was immunoprecipitated using 5 μg of monoclonal nitrotyrosine antibody (Upstate, Lake Placid, NY) as described previously (Golden et al., 2003; Martin et al., 2007). After immunocapture the samples were subjected to SDS-PAGE and electroelution for western blot detection of CyPD and ANT using ECL.
Protein oxidation assay
To detect oxidative modification of proteins in mSOD1 mouse spinal cord we used an OxyBlot protein oxidation detection kit (Chemicon) for carbonyl groups (aldehydes and ketones). Aliquots of equal amounts of protein (10 μg) were denatured with 10% SDS, then derivatized to 2-4 dinitrophenylhydrazone (DNP) by a reaction with 2-4 dinitrophenylhydrazine (DNPH) for exactly five minutes. This reaction allows for a chemical conjugation of the DNPH to the carbonyl group of the protein side chain to create a hydrazone moiety that can be immunodetected. The reaction was quenched with a neutralizing solution (6% SDS), and a characteristic color change to brown followed. Samples were then placed on ice, and loaded onto 10% gels for SDS-PAGE. Proteins with conjugated DNP residues where detected with polyclonal rabbit antibody DNP used at a concentration of 1:150. Proteins were then visualized goat anti-rabbit HRP-conjugated antibody and ECL.
Mitochondrial imaging in vivo
Mitochondria were visualized in MNs of wtSOD1 and G93Ahigh-mSOD1 tg mice in vivo using in situ fluorescent dye imaging. MN mitochondria within the preterminal axons and synaptic terminals were labeled using the cell-permeable, fixable mitochondrian selective probe (MitoTracker Red CM-H2XRos, Molecular Probes, Eugene, OR). Stock solution was prepared by dissolving the dye powder in anhydrous DMSO to a final concentration of 1 mM. Working solution of tracer was prepared in Influx (Molecular Probes) pinocytic cell-loading reagent. MNs were loaded in vivo by unilateral injection of probe into the gastrocnemius (3 injection sites within the gastrocnemius; 2 μl/injection, 100 nM-200 nM) to label lumbar MNs (n= 4/genotype/probe). Mitotracker is endocytosed at the neuromuscular junction and transported retrogradely (Martin et al., 2005). Mice were killed 4 days later by deep anesthesia and perfusion-fixation with 4% PF. The spinal cord was removed and cryoprotected in 20% glycerol overnight. Lumbar spinal cord sections were cut (40 μm) serially using a sliding microtome, and every 10th section was mounted on a glass slide and coverslipped with antifade buffer. Sections were viewed with a Deltavision deconvolution microscope (Applied Precision, Inc, Issaquah, WA) using a 100× oil immersion objective lens. Deconvolution (DECON) is the process of relocating signal scatter and out-of-focus information present in digital images so that the image is clarified and its data is restored quantitatively. Mitochondria in age-matched wtSOD1 and mSOD1 mouse (n=4/genotype) MNs were imaged in ∼50 neurons, cut through the approximate cell center as judged by the nucleus, in each mouse. Image processing for relative fluorescence intensity, reflected by the average-integrated intensity of pixels, was done using Velocity software.
Mouse crossing experiments
Mice homozygous for the CyPD targeted mutation are viable, with normal growth and appearance, and are fertile (Baines et al., 2005). Mitochondria isolated from heart of CyPD-/- mice are devoid of CyPD, resistant to mitochondria swelling and mPT, and are protected from mitochondrial Ca2+ overload and oxidative stress (Baines et al., 2005). We crossed G93A-mSOD1 mice (high and low expressers) to ppif-/- mice to test the hypothesis that CyPD, and possibly the mPTP, has a role in MN cell death caused by mSOD1. Mice with targeted deletions of both CyPD alleles were crossed to G93A mice and F1 offspring positive for the G93A transgene were crossed with F1 siblings lacking the transgene. The F2 generation therefore yielded progeny carrying all PCR-confirmed CyPD genotypes (ppif+/+, ppif+/-, and ppif-/-) with or without the G93A mSOD1 transgene. These mice were maintained for 3 generations and followed longitudinally.
At the end-stage of disease, G93A-ppif+/+, G93A-ppif+/-, and G93A-ppif-/- were weighed, deeply anesthetized, and perfusion-fixed using 4% PF. Tissue handling was the same as describe above. Transverse serial symmetrical sections (40 μm) through spinal cord were cut using a sliding microtome and stored individually in 96-well plates. A subsample of sections was stained with cresyl violet for Nissl staining and immunohistochemically for VDAC to assess spinal cord pathology and morphology of remaining MNs.
Data analysis
Group means and variances were evaluated statistically by one-way ANOVA and a Newman-Keuls post-hoc test.
Photography and figure construction
The original images used for figure construction were generated using digital photography. Digital images were captured as TiF files using a SPOT digital camera and SPOT Advanced software (Diagnostic Instruments) or a Nikon digital camera (DXM1200) and ACT-1 software. Images were altered slightly for brightness and contrast using ArcSoft PhotoStudio 2000 or Adobe Photoshop software without changing the content and actual result. Figure composition was done using CorelDraw 9 software with final figures being converted to TiF files. Files of composite figures were adjusted for brightness and contrast in Adobe Photoshop.
Results
Localization of mPTP proteins in mouse spinal MNs
Western blotting of mouse CNS extracts was done to screen for specific antibodies to mPTP proteins. CyPD was detected as a single band with a monoclonal antibody at ∼20 kDa in spinal cord (Fig. 1A), consistent with expectation (Johnson et al., 1999). This reactive band was absent in protein extracts of brain and spinal cord from ppif-/- mice (Fig. 1A). CyPD was found at similar levels in spinal cord and forebrain but was slightly greater in amount in brainstem (Fig. 1B). Both ANT and VDAC were detected with monoclonal and polyclonal antibodies at ∼33-35 kDa (Fig. 1B,C), consistent with expectations (Yu et al., 1995; Graham et al., 1997). ANT levels appeared highest in brainstem followed by spinal cord and then forebrain (Fig. 1B). VDAC levels were similar in spinal cord, brainstem, and forebrain (Fig. 1C).
Figure 1.
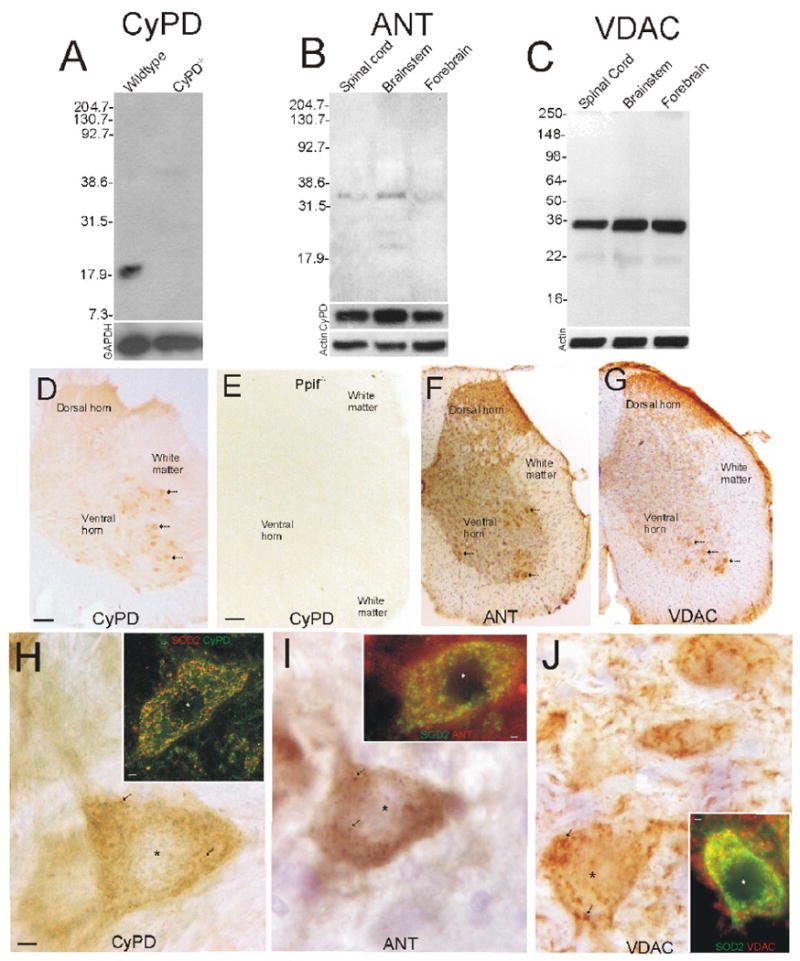
Expression and localization of mPTP proteins in non-tg mouse spinal cord.
A. Full-length blot demonstrating mono-specific detection of CyPD at ∼18-20 kDa in mitochondrial-enriched membrane fractions of mouse spinal cord. This immunoreactive band is not present in spinal cord of ppif-/- mice. Molecular weight markers (in kDa) are indicated at left. Blots were re-probed for glyceraldehyde-phosphate dehydrogenase (GAPDH) as a protein loading control.
B. Full-length blot demonstrating mono-specific detection of ANT at ∼33 kDa in mitochondrial-enriched membrane fractions of mouse spinal cord, brainstem, and forebrain. Molecular weight markers (in kDa) are indicated at left. Blots were reprobed for CyPD and actin.
C. Full-length blot demonstrating mono-specific detection of VDAC at ∼35 kDa in mitochondrial-enriched membrane fractions of mouse spinal cord, brainstem, and forebrain. Molecular weight markers (in kDa) are indicated at left. Blots were reprobed for actin as a protein loading control.
D. Immunohistochemical localization of CyPD in mouse spinal cord. Antibody binding was visualized using immunoperoxidase with DAB as chromogen to give brown signal. A hemisection of lumbar spinal cord is shown. MNs in ventral horn are strongly immunopositive (arrows) for CyPD. Faint neuropil immunoreactivity is present in the dorsal horn and ventral horn and low immunolabeling is in white matter. Scale bar (same for F,G) = 100 μm.
E. Immunostaining for CyPD in ppif-/- mouse spinal cord is negative. Scale bar = 80 μm.
F and G. Immunohistochemical localization of ANT (F) and VDAC (G) in mouse spinal cord. Antibody binding was visualized as in D but with cresyl violet counterstaining. A hemisection of lumbar spinal cord is shown. MNs in ventral horn are strongly immunopositive for ANT and VDAC (arrows) and neuropil immunoreactivity is present in the dorsal horn and ventral horn. Immunolabeling is in white matter is low.
H-J. Localization of CyPD (H), ANT (I) and VDAC (J) in mouse spinal MNs. Immunoreactivities for CyPD and ANT were seen as diffuse labeling in the cytoplasm and as discrete particles (arrows). VDAC immunoreactivity was seen as larger structures in the cytoplasm. The nucleus is marked by the asterisk. Dual immunofluorescent staining for CyPD, ANT, or VDAC and the mitochondrial marker SOD2 and confocal or epifluorescence microscopy (insets) shows co-localization of CyPD, ANT, and VDAC with SOD2 (seen as yellow-orange) in MNs, but there is also non-mitochondrial pools. Scale bar in H (same for I and J) = 7 μm. Scale bars in insets = 1.5 μm.
Immunohistochemistry, done by immunoperoxidase and immunofluoresence, showed enrichment of mPTP proteins in wildtype mouse spinal MNs. CyPD was concentrated in large cells in ventral horn (Fig. 1D,H), while only faint staining was seen in other regions of spinal cord (Fig. 1D). No CyPD immunoreactivity was seen in ppif-/- mouse spinal cord (Fig. 1E). ANT and VDAC were also concentrated in large cells in ventral horn (Fig. 1F,G,I,J). The VDAC localization pattern was different from CyPD and ANT patterns in that it was also present in superficial layers of dorsal horn (Fig. 1G) and it also had a prominent neuropil localization (Fig. 1J). At greater magnification, CyPD immunoreactivity in MN cell bodies was seen as fine discrete dots and as diffuse cytoplasmic labeling (Fig. 1H). Dual labeling for CyPD and the mitochondrial marker SOD2 and confocal microscopy showed a high amount of CyPD localization to mitochondria and also a non-mitochondrial pool of CyPD (Fig. 1H, inset). ANT immunoreactivity in MN cell bodies was seen also as fine discrete dots and as diffuse cytoplasmic labeling (Fig. 1I). ANT was found in mitochondrial and non-mitochondrial pools (Fig. 1I, inset). VDAC immunoreactivity within the cytoplasm of MN cell bodies was seen as fine dots, large clumps, and long strands (Fig. 1J). VDAC immunoreactivity was also near or associated with the plasma membrane of MNs (Fig. 1J) and was also seen associated with processes within the neuropil (Fig. 1J). Dual labeling for VDAC and the mitochondrial marker SOD2 showed mitochondrial and non-mitochondrial localizations of VDAC within MNs (Fig. 1J, inset).
CyPD in SOD1 tg mice
The localization of CyPD in spinal cord of tg mice expressing human wtSOD1 and G93A-mSOD1 was examined by immunohistochemistry. As in non-tg mice (Fig. 1D), CyPD was enriched in MNs of wtSOD1 and mSOD1 tg mice (Fig. 2A,C). CyPD immunoreactivity in MNs in wtSOD1 tg mice was seen as fine discrete particles and diffuse cytoplasmic pools (Fig. 2B). The spinal cord neuropil of wtSOD1 tg mice contained very sparse CyPD immunoreactivity (Fig. 2B). The CyPD localization pattern in MNs of pre-symptomatic and symptomatic mSOD1 mice differed markedly from that seen in wtSOD1 tg mice (Fig. 2C). CyPD immunoreactivity in MNs of mSOD1 tg mice was seen as fine and large particles and clumps in the cytoplasm (Fig. 2D,C). In highly swollen MNs, CyPD was present at the cell periphery and clustered around the nucleus (Fig. 2E). In the neuropil of mSOD1 mice CyPD was seen often as fine dots and larger circular profiles (Fig. 2D,E). Western blots of mitochondrial membrane-enriched fractions isolated from spinal cord did not show any major differences in the amount of CyPD in mSOD1 tg mice at pre-symptomatic and early symptomatic stages of disease compared to wtSOD1 tg mice (Fig. 2F), but at end-stage disease CyPD levels appeared lower than control (Fig. 2F).
Figure 2.
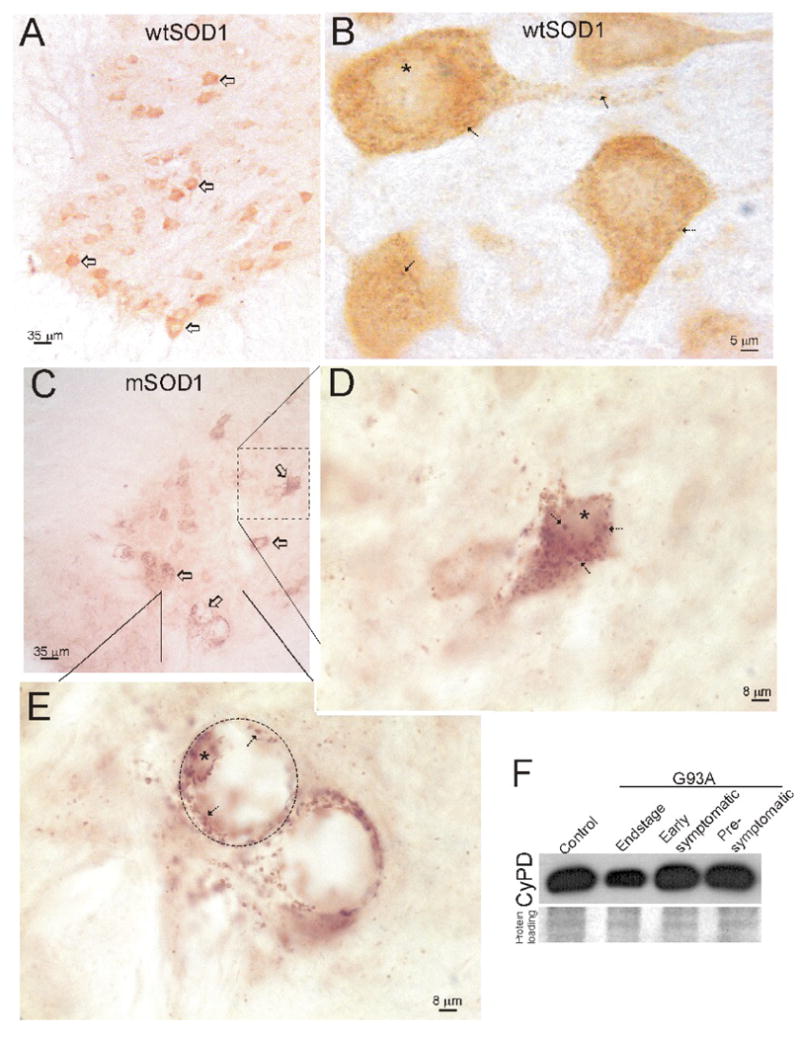
Expression and localization of CyPD in tg human wtSOD1 and G93Ahigh-mSOD1 mouse spinal cord.
A. Spinal MNs (open arrows) in tg wtSOD1 mice have high immunoreactivity for CyPD compared to the surrounding neuropil and white matter.
B. In MNs of tg wtSOD1 mice, CyPD immunoreactivity is seen as discrete dots in the cytoplasm (arrows) of the cell body and proximal dendrites and diffusely in cytoplasm. Asterisk marks nucleus.
C. Spinal MNs (open arrows) in tg G93Ahigh-mSOD1 (early symptomatic shown here) mice have high immunoreactivity for CyPD compared to the surrounding neuropil and white matter matter.
D. In mSOD1 mouse MNs at early symptoms the CyPD-positive particles within the cytoplasm appear to swell.
E. In subsets of MNs showing near end-stage degeneration with severe swelling of the cell body (hatched lines delineate MNs) due to a slow necrotic-like process (Martin and Liu, 2004; Martin et al., 2007), CyPD immunoreactivity is marginated to the periphery of the cell (arrows) and decorates the nuclear surface (asterisk).
F. Western blots of mitochondrial membrane-enriched fractions of spinal cord of G93Ahigh-mSOD1 mice at pre-symptomatic and early symptomatic disease stages reveal levels of CyPD similar to tg wtSOD1 control, but at end-stage disease CyPD levels appeared slightly lower than control.
To identify the subcellular locations of CyPD in wtSOD1 and mSOD1 tg mouse MNs confocal microscopy was used to evaluate SOD2 and CyPD/SOD2 immunolabeled spinal cord sections. SOD2 immunoreactivity in MNs of wtSOD tg mice had a pattern that was completely discrete and seen as round, elliptical, and tubular profiles (Fig. 3A), consistent with a mitochondrial localization (Jensen, 2005). Vast numbers of mitochondria were seen in MNs (Fig. 3A). SOD2 immunoreactivity in MNs of symptomatic G93A-mSOD tg mice was seen mostly as small and large circular profiles (Fig. 3B). Dual labeling for CyPD/SOD2 in wtSOD1 tg mouse MNs showed CyPD in mitochondrial and cytosolic compartments (Fig. 3C) similar to non-tg mouse MNs (Fig. 1H, inset). In pre-symptomatic mice, the mitochondrial network had a very linear and tubular organization with CyPD localized mostly to mitochondria and sparse cytosolic localization (Fig. 3D). Symptomatic mSOD1 mouse MNs showed a near complete localization of CyPD to mitochondria that were swollen (Fig. 3E).
Figure 3.
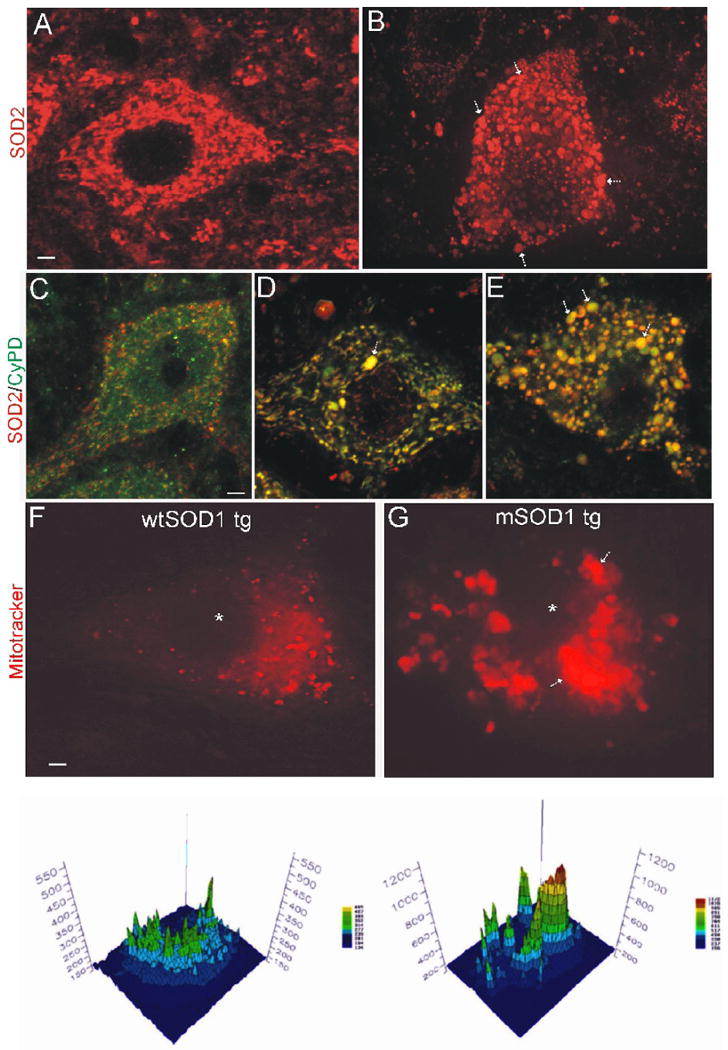
Formation of megamitochondria in MNs of mSOD1.
A and B. Confocal z-stack composite images showing the immunofluorescent detection of SOD2 (red), used to mark specifically mitochondria, in lumbar spinal MNs of tg wtSOD1 (A) and G93Ahigh-mSOD1 (B) mice at 10 weeks of age. The nucleus is the dark center. Vast numbers of mitochondria are present in MNs. In control MNs, mitochondria are small (∼0.3-0.5 μm in diameter) and pleiomorphic (round, ellipitical or filamentous). There is no detection of a non-mitochondrial pool. Mitochondria in MNs of mSOD1 mice become essentially all round. Some of these mitochondria are tiny (∼0.1 μm in diameter), suggesting a fragmentation or fission process (see Fig. 4D,E), while other mitochondria are huge (>2 μm in diameter) indicating swelling. There is no detection of a non-mitochondrial pool of SOD2 in mSOD1 mouse MNs indicating no disruption of mitochondria with release of matrix constituents. Scale bar in A (same for B) = 2.3 μm.
C, D, and E. Immunofluorescent localization of SOD2 (red) and CyPD (green) in lumbar spinal MNs of tg wtSOD1 (C) and G93Ahigh-mSOD1 (D, E) mice at 8 or 10 weeks of age. Colocalization of SOD2 and CyPD is seen as yellow-orange. In control MNs (C), CypD is present in mitochondria and in a non-mitochondrial cytoplasmic pool (green only). Some mitochondria are not positive for CyPD (red only). In mSOD1 mouse MNs before the formation of many mega-mitochondria (D, arrow), most mitochondria, many of which are seen as long vermiform structures, are positive for CyPD. In mSOD1 mouse MNs when many swollen mega-mitochondria are present (E, arrows), essentially all mitochondria are CyPD positive. In some swollen mitochondria CyPD fills the matrix and the SOD2 is marginated to the periphery of the matrix (green cores with red or orange halos). Scale bar in C (same for D and E) = 4 μm.
F and G. DECON analysis of mitochondrial abnormalities in MNs of pre-symptomatic G93Ahigh-mSOD1 tg mice compared to age-matched control human wtSOD1 tg mice. Mitotracker-labeled mitochondria from the distal axon/terminals in skeletal muscle are red. The nucleus is black profile in center (asterisk). Note the enlargement and clumping of mitochondria in the mSOD1 mouse MN (arrows). Scale bar in F (same for G) = 2 μm. The graphs below each image (generated by measuring pixel fluorescence intensity) are maps of mitochondria within the perikaryon of the actual MNs shown in the upper images. The graphs are projections of the imaged MNs. The empty zone in the center of the graph is the location of the nucleus. The fluorescence intensity was quantified in mid-perikaryal optical sections of MNs (see text). mSOD1 mouse MNs have large aggregates of brightly labeled mitochondria.
MNs in pre-symptomatic mSOD1accumulate mitochondria from the distal axon
Because the morphology and distribution of mitochondria in MN cell bodies of G93Ahigh-mSOD1 mice become altered early in the disease process, we examined the possibility that mitochondria within the MN cell body accumulate from distal regions of the neuron. Axonal-derived mitochondria in MN perikarya were visualized by denconvolution microscopy (Fig. 3F,G) after their labeling by loading injections of Mitotracker Red into the gastrocnemius muscle, thus only a subset of the total population of mitochondrial are seen in MNs (those derived from the distal axon). As a negative control, a sciatic nerve ligature blocked the accumulation of Mitotracker-stained profiles in the MN cell body (data not shown). Mitotracker labeling is distinct from general retrograde transport of dyes and has an appearance and distribution similar to mitochondria. Mitotracker-labeled, axon-derived mitochondria in MN cell bodies of 10-week-old wtSOD1 tg mice were seen dispersed within the perikaryon, although they tended to be concentrated within a particular somal region (Fig. 3F), possibly corresponding to the axon hillock. The mitochondrial distribution profiles in MNs in 10-week-old mSOD1 tg mice (Fig. 3G) were very different from age-matched wtSOD1 tg mice. MNs in mSOD1 mice contained large swollen and aggregated or clustered mitochondria that were intensely labeled (Fig. 3G) compared to the smaller, dispersed, more weakly labeled mitochondria in control MNs (Fig. 3F). Quantification (mean ± SD) of the perikaryal peak fluorescence of mitotracker-labeled mitochondria revealed significantly higher (p < 0.01) amounts in mSOD1 mouse MNs (1120 ± 320) compared in age-matched wtSOD1 tg mouse MNs (490 ± 130).
The mitochondrial inner membrane is remodeled and the matrix vesiculates in pre-symptomatic mSOD1 mouse spinal cord
EM was used to identify abnormalities in the fine structure of mitochondria in the spinal cord ventral horn of G93Ahigh-mSOD1 mice. Dramatic changes in mitochondrial ultrastructure were observed in the IMM and matrix of numerous mitochondria in dendrites of pre-symptomatic 6- and 8-week-old G93Ahigh-mSOD1 mice (Fig. 4A) but not in 4-week-old G93Ahigh-mSOD1 mice (Fig. 4F) and age-matched wtSOD1 tg mice. The conformation of the cristae was changed from straight infoldings to distorted membranes brought about by swelling of the matrix (Fig. 4A). In mitochondria showing early pathology, microvacuoles within the matrix were discerned (Fig. 4A, center mitochondrion). Eventually the matrix in subsets of mitochondria became highly fragmented into cristae-bound compartments (Fig. 4. A,B). Many profiles of processes seen in cross-section in the neuropil were occupied completely with megamitochondria displaying profound remodeling of cristae and severe matrix vesiculation (Fig. 4B). The OMM remained snug around the mitochondria despite profound IMM changes (Fig. 4A,B). Mitochondria with matrix swelling in pre-symptomatic mice were also found with apparent buds (Fig. 4A, lower right, double arrows).
Figure 4.

Ultrastructural and biochemical changes in G93Ahigh-mSOD1 tg mice are consistent with permeability transition pore activation.
A and B. EM shows major cristae remodeling and matrix swelling and vesiculation (black asterisks) in spinal cord ventral horn dendrites of G93Ahigh-mSOD1 mice. Normal mitochondria are marked by white asterisks. Mitochondria delineated by white-dotted boxes are shown at higher magnification in insets. Mitochondria with vacuoles (A, lower left and right; B, inset) have apparent OMM and IMM contacts (darker spots without narrow space separating the OMM and IMM, hatched arrows). The OMM appears breached at some locations (A, lower left and right, open arrows). Some mitochondria with vesiculation of the matrix have buds extending from the surface (A, lower right, double arrow), suggesting a non-replicative fission or fragmentation process. Some dendrites (B, open arrow) have mitochondria with extreme cristae remodeling and matrix vesiculation. A nearby mitochondrion exhibits early swelling (B, hatched arrow). Scale bars = 0.63 μm (A); 0.3 μm (A, insets); 0.3 μm (B); 0.15 μm (B, inset).
C. Graph showing the estimated number (mean ± SD) of contact sites per mitochondria in spinal cord ventral horn dendrites (identified by presynaptic contacts) of wtSOD1 and G93Ahigh-mSOD1 mice. Asterisk denotes significantly different (p < 0.05).
D. Apparent fragmentation of mitochondria was seen in spinal cord ventral horn dendrites of G93Ahigh-mSOD1 mice. Shown is a cross-sectional profile of a dendrite with four identifiable mitochondria (1-4). All of the mitochondria contain matrix vacuoles. Mitochondrion 4 has fragmented into several smaller mitochondrial buds with vacuoles (open arrows). Scale bar = 4 μm.
E. Apparent fission of mitochondria was seen in spinal MN cell bodies of G93Ahigh-mSOD1 mice. Contact sites between mitochondria are distinct (arrows). One mitochondrion is abnormal with pale matrix, but not showing vacuolation, while the other three mitochondria appear normal. Scale bar = 0.2 μm.
F. Western blot analysis for protein carbonyls (Oxyblot) in the mitochondrial membrane-enriched fraction and the soluble fraction of spinal cord from wtSOD1 tg mice and G93Ahigh-mSOD1 tg mice at different stages of disease. Molecular weight standards are indicated at right.
G. Increased nitration of CyPD and ANT in G93Ahigh-mSOD1 mouse spinal cord. Nitrated proteins from soluble and mitochondrial membrane-enriched fractions from wtSOD1 and mSOD1 tg mice at different stages of disease were IP with monoclonal mouse 3-nitrotyrosine antibody followed by western blotting with monoclonal CyPD or polyclonal ANT antibodies.
To examine structurally a potential role for mitochondrial permeability transition in early pathological mechanisms of mSOD1 mice, contact sites between the outer and inner mitochondrial membranes were examined in electron micrographs (Fig. 4A,B, insets, arrows). Estimates revealed a greater than two-fold increase in the number of outer and inner mitochondrial membrane contact sites in mitochondrial profiles located in dendritic processes in the ventral horn (Fig. 4C). Other mitochondrial changes in G93Ahigh-mSOD1 mice at pre-symptomatic stages of disease were fragmentation of matrix-swollen mitochondria in ventral horn dendrites (Fig. 4D) and fission of mitochondria within the cell bodies of MNs (Fig. 4E). Mitochondria interpreted as undergoing fission had well-defined narrow connecting membranes (Fig. 4E, arrows).
Spinal cords of mSOD1 mice were examined for oxidative damage to proteins because oxidative stress can cause mitochondrial abnormalities, including permeability transition (McStay et al., 2002). The presence of protein carbonyl groups, a hallmark of the oxidative alteration of proteins, was identified by oxyblot analysis. Proteins in mitochondrial-enriched membrane fractions of G93Ahigh-mSOD1 mice at pre-symptomatic, symptomatic, and end-stage disease had enhanced carbonyl modification compared to control (Fig. 4F). The damage to proteins appeared in part to be an accentuation of baseline damage (Fig. 4F). As expected, the pattern of oxidative damage to soluble proteins in mSOD1 mouse spinal cord was different from that seen for the mitochondrial-enriched membrane fractions (Fig. 4F). Surprisingly, soluble proteins in pre-symptomatic and early symptomatic mice had protein carbonyl levels lower than baseline (Fig. 4F). A prominent band of carbonyl modified proteins at a low molecular mass was seen in both fractions (Fig. 4F).
mPTP proteins in spinal cords of mSOD1 mice were examined for nitration because peroxynitrite (ONOO-), a biologically significant oxidant formed by the reaction of nitric oxide (NO) with O2- (Beckman et al., 1993; Pacher et al., 2007) can induce permeability transition (Vieira et al., 2001). Immunoprecipitated CyPD in mitochondrial-enriched membrane and soluble fractions of spinal cord had basal levels of tyrosine nitration (Fig. 4G, top). CyPD in both spinal cord fractions of symptomatic G93Ahigh-mSOD1 mice had greater amounts of tyrosine nitration than control (Fig. 4G, top). The greatest change in CyPD nitration was seen in the soluble compartment of early symptomatic mice (Fig. 4G, top). The nitration of ANT in mouse spinal cord was very different from CyPD. ANT nitration was not detected in control mitochondrial-enriched membrane fractions of spinal cord (Fig. 4G, bottom). In G93Ahigh-mSOD1 mice, ANT nitration was found at highest amounts in pre-symptomatic and symptomatic mice, while it was barely detectable in mice at end-stage disease (Fig. 4G, bottom). Possibly two different isoforms of ANT showed nitration at pre-symptomatic disease (Fig. 4G, bottom).
CyPD deletion delays disease onset and prolongs survival of ALS mice in gender-preferential patterns
mSOD1 mice without CyPD were generated to determine if its absence alters the course of disease. Prominent gender and model severity effects were seen (Fig. 5). With G93Ahigh-expressers, the mean survival was extended ∼25% in male mSOD1 mice without CyPD and 79% in female mSOD1 mice without CyPD compared to gender-matched mSOD1 mice with both CyPD alleles (Fig. 5A). No significant effect on survival was seen with heterozygous male G93Ahigh mice, but heterozygous female G93Ahigh mice had modest, but significant, survival effect (Fig. 5A). A few (n=4) female G93Ahigh-expressing mSOD1-CyPD-/- mice, which were excluded from this analysis, had an unusually rapid death and subsequent autopsy revealed that these mice had either lymphoma or cardiomegaly (data not shown), with the former consistent with the finding that CyPD can function pro-apoptotically to delete malignant cells (Yasuda et al., 2006; Kang et al., 2007). With G93Alow-expressers, a slight (10%) improvement was seen with male mSOD1 mice without CyPD, but heterozygous and homozygous CyPD null female mouse groups showed a 20-22% increase in mean survival compared to gender-matched mSOD1 mice with both CyPD alleles (Fig. 5B). The time to disease onset was extended 60% in male G93Ahigh-expressers without CyPD and was over doubled in female G93Ahigh-expressers without CyPD (Fig. 5C). With G93Alow-expressers the time to disease onset was extended 30% and 52% in heterozygous and homozygous CyPD null males, respectively, and ∼81-85% in heterozygous and homozygous CyPD null females (Fig. 5D). The absence of CyPD in G93A-mSOD1 mice had no effect on body mass at end-stage disease (Fig. 5E,F).
Figure 5.
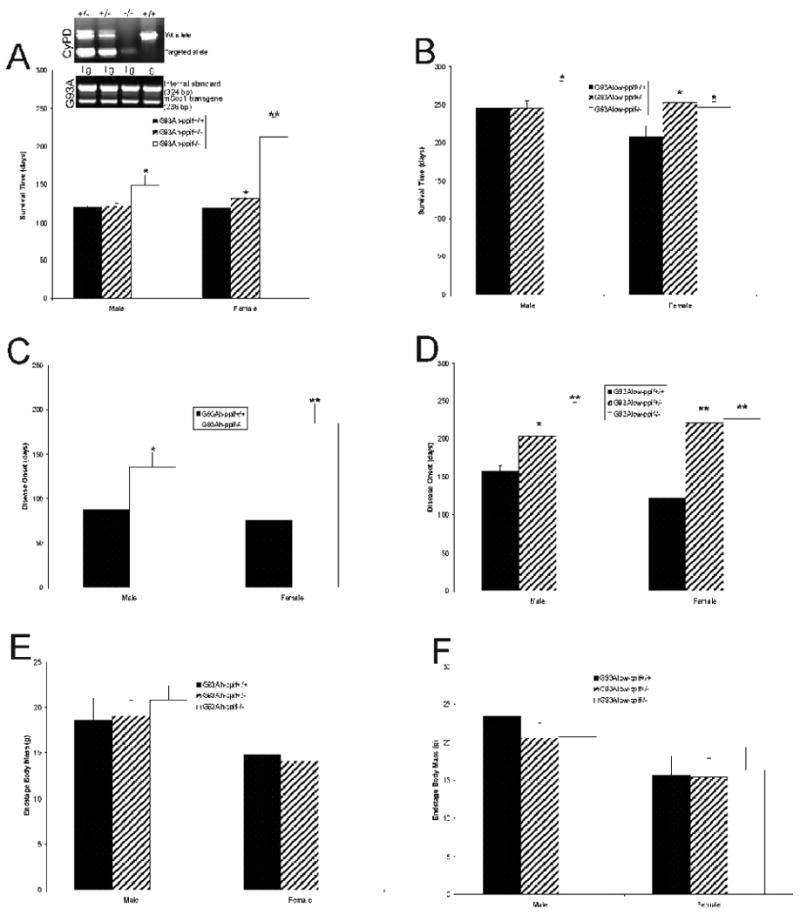
CyPD depletion improves survival of G93Ahigh- and G93Alow-mSOD1 mice and delayed the onset of disease. n=8-10 mice/genotype/gender. PCR genotyping of tail genomic DNA is shown for representative mice (A) above. Values are mean ± SD. Significant difference (*p < 0.05 or **p <0.01) from ppif+/+ control.
A histopathological assessment of spinal cords of mice at end-stage ALS was done to determine if the absence of CyPD altered the pathological degenerative process in MNs. Residual MNs in G93A-mSOD1 mice with CyPD showed the characteristic (Martin and Liu, 2004; Martin et al., 2007) cytoplasmic vacuolation (Fig. 6A) and mitochondrial swelling (Fig. 6C). In contrast, remaining MNs in G93A-CyPD null mice had Nissl-rich, darkly-stained cell bodies (Fig. 6B), and the cytoplasmic vacuolation and mitochondrial swelling was attenuated markedly or blocked (Fig. 6D). However, MNs in G93A-CyPD null mice accumulated numerous granule-like structures within their cell body cytoplasm (Fig. 6B, arrows).
Figure 6.
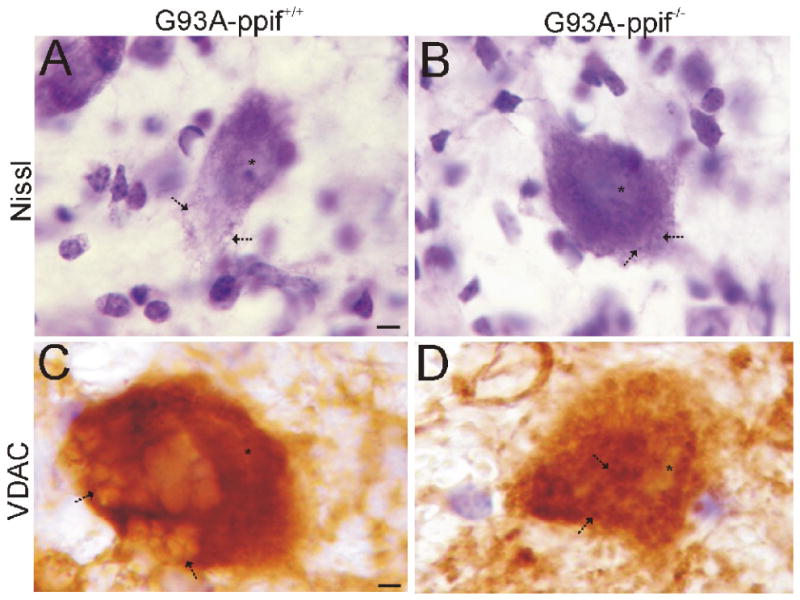
CyPD depletion changes the degenerative process in MNs of G93A-mSOD1 mice.
A and B. Cresyl violet (Nissl) staining shows that degenerating MNs in G93A mice with CyPD have prominent vacuolation of the cytoplasm (A, arrows), while MNs in G93A mice without CyPD have attenuated cytoplasmic vacuolation and the formation of numerous cytoplasmic granule-like structures (B, arrows). Asterisk marks nucleus. Scale bar in A (same for B) = 4 μm.
C and D. Immunoperoxidase staining for VDAC (brown signal) shows that degenerating MNs in G93A mice with CyPD have prominent mitochondrial swelling (C, arrows), while the mitochondrial swelling in MNs in G93A mice without CyPD is blocked (D, arrows). Asterisk marks nucleus. Scale bar in C (same for D) = 3.5 μm.
Discussion
The major findings of this study relate to mitochondria and the mPTP in MNs and mouse ALS. First, we demonstrate that the major components of the mPTP are present and seemingly enriched selectively, in mouse MNs. Second, early in the course of MN disease in ALS mice, mitochondria undergo profound trafficking abnormalities and dramatic remodeling of the cristae and matrix resulting in the formation of megamitochondria and coinciding with increased protein carbonyl formation and nitration of mPTP components. Third, the genetic deletion of a major mPTP component, CyPD, has robust effects in ALS mice by delaying disease onset and extending survival. These observations demonstrate that the mPTP participates in the causal mechanisms of MN degeneration in mouse ALS.
Mitochondrial permeability transition is a mitochondrial state in which the proton-motive force is disrupted (Crompton, 1999; van Gurp et al., 2003; Bernadri et al., 2006; Leung and Halestrap, 2008). This disruption involves the mPTP that functions as a voltage, thiol, and Ca2+ sensor. Conditions of mitochondrial Ca2+ overload, excessive oxidative stress, and decreased electrochemical gradient (ΔP), ADP, and ATP can favor mitochondrial permeability transition. The mPTP is believed to be a poly-protein transmembrane channel formed at the contact sites between the IMM and the OMM. The collective components of the mPTP are still controversial, but the VDAC in the OMM, the ANT in the IMM, and CyPD in the matrix are core components (Crompton, 1999; van Grup et al., 2003; Bernardi et al., 2006). The VDAC adopts an open conformation at low or zero membrane potential and a closed conformation at potentials above 30-40 mV making the OMM permeable to most small hydrophilic molecules up to 1.3 kDa for free exchange of respiratory chain substrates (Rostovtseva et al., 2005). The ANT is a multi-pass membrane protein, with odd-numbered transmembrane helices having kinks because of proline residues, which mediates the exchange of cytosolic ADP for mitochondrial ATP (Graham et al., 1997). During normal mitochondrial function the OMM and the IMM are separated by the intermembrane space, and the VDAC and the ANT do not interact (Crompton, 1999). Permeability transition is activated by the formation of the mPTP; the IMM looses its integrity and the ANT changes its conformation from its native state into a non-selective pore (Crompton, 2003). This process is catalyzed by CyPD which is a peptidylprolyl isomerase (PPIase) that functions in protein cis-trans isomerization and chaperoning (Waldmeier et al., 2003). The ANT and CyPD interact directly (Woodfield et al., 1998). The molar concentration of CyPD (in heart mitochondria) is much less (>5%) than ANT; thus, under normal conditions only a minor fraction of the ANT can be in a complex with CyPD (Johnson et al., 1999; Leung and Halestrap, 2008). When this occurs, small ions and metabolites permeate freely across the IMM and oxidation of metabolites by O2 proceeds with electron flux not coupled to proton pumping, resulting in collapse of ΔP, dissipation of ATP production, production of ROS, equilibration of ions between the matrix and cytosol, matrix volume increases, and mitochondrial swelling (van Grup et al., 2003; Rostovtseva et al., 2005).
Our study demonstrates for the first time the localization of the core components of the mPTP directly in MNs. Antibodies to CyPD, ANT, and VDAC all yielded strong signals in mouse MNs compared to other regions and cells in spinal cord. We used antibodies that are not isoform specific, thus we were unable to distinguish the predominant forms of ANT and VDAC in MNs. There are only two other reports of CyPD localization in mammalian CNS which found CyPD enriched in subsets of neurons with relative low levels in astrocytes (Naga et al., 2007; Hazelton et al., 2008). Moreover, we found that CyPD, ANT, and VDAC have mitochondrial and non-mitochondrial localizations in MN. They are all nuclear-encoded mitochondrial-targeted proteins, thus a possible explanation for their non-mitochondrial localizations is that they are pre-mitochondrial forms. Other cyclophilin isoforms are located in the cytoplasm (Bose and Freedman, 1994), but this immunoreactivity was abolished in ppif-/- mice, demonstrating that it is CyPD. Work on ANT localization in nervous tissue is very scarce. ANT appears to be expressed in reactive astrocytes (Buck et al., 2003). Previous studies of nervous tissue have found VDAC in neurons and glial cells (McEnery et al., 1993) and associated with mitochondria, the endoplasmic reticulum, and the plasma membrane (Shoshan-Barmatz et al., 2004; Akanda et al., 2008), although the non-mitochondrial localizations of VDAC have been disputed (Yu et al., 1996). We observed that spinal cord, brainstem, and forebrain had similar levels of CyPD as well as similar levels of ANT and VDAC. Thus, differences in the levels of individual mPTP components fails to explain the intrinsic differences in the sensitivity to Ca2+-induced mitochondrial permeability transition seen in spinal cord versus brain (Sullivan et al., 2004; Morota et al., 2007). We also observed that not all mitochondria within MNs contained CyPD, ANT, and VDAC. This observation supports that idea of that mitochondria are heterogeneous in shape (Collins and Bootman, 2003; Jensen, 2005) and biochemical composition, notably metabolism (Hamburger et al., 1970), CyPD content (Naga et al., 2007), and genetics (Wallace, 2005).
Mitochondrial pathology has been implicated in the mechanisms of ALS (Martin, 2006), but the evidence was largely circumstantial. In mSOD1 mouse models of ALS, mitochondria in spinal cord exhibit pathology (Wong et al., 1995; Kong and Xu, 1998; Bendotti et al., 2001) and some of the mitochondrial degeneration occurs very early in the course of the disease (Bendotti et al., 2001; Martin et al., 2007). Wildtype SOD1 and human mSOD1 associate with mitochondria (Okado-Matsumoto and Fridovich, 2001; Higgins et al., 2002). One interpretation is that the mSOD1 causes mitochondrial degeneration by inducing OMM extension and leakage and intermembrane space expansion (Higgins et al, 2003). Our data reported here suggest that the OMM remains relatively intact to permit formation of mega-mitochondria in MNs cell bodies in G93Ahigh mice. Our observations here also demonstrate that mitochondria in dendrites in spinal cord ventral horn undergo extensive cristae and matrix remodeling. Mechanisms for this damage could be related to mSOD1 gaining access to the matrix (Vijayvergiya et al., 2005) and inducing disturbances in oxidative phosphorylation (Mattiazzi et al., 2002). This mitochondrial conformation might favor the formation of the mPTP; indeed, we found evidence for increased contact sites between the OMM and IMM in G93Ahigh mice. Some mitochondria with vesiculated matrix form bud-like structures continuous with the matrix and appear to undergo fragmentation. Another feature of mitochondria in MNs of young mice without symptoms was apparent fission of ultrastructurally normal mitochondria. Our interpretation of electron micrographs as mitochondrial fission is consistent with those of others (Ju et al., 2008; Han et al., 2008), but corroboration is required using alternative methods. It is not clear if the cristae and matrix remodeling and the apparent fragmentation and fission mitochondria are related or independent events; nevertheless, our morphological observations enforce the idea that mitochondria are critical to the pathobiology of mSOD1 toxicity to MNs.
The possibility of changes in mitochondrial trafficking in MNs of mSOD1 mice is mostly unexplored. We show here that MNs in mSOD1 mice at pre-symptomatic disease accumulate mitochondria from their distal axons/terminals. We demonstrated previously that MNs in mSOD1 mice at pre-symptomatic disease generate higher levels of superoxide, NO, and ONOO- than MNs in tg mice expressing human wtSOD1 (Martin et al., 2007). This mitochondrial accumulation occurs at a time when cell body volume is increasing (Martin et al., 2007). G93A-mSOD1 perturbs anterograde axonal transport of mitochondria in cultured primary MNs (de Vos et al., 2007) making it possible that retrogradely transported mitochondria with toxic properties from the neuromuscular junction fail to be returned to distal processes. It is noteworthy is this regard that mitochondria within synapses are more susceptible to Ca2+ overload than non-synaptic mitochondria (Brown et al., 2006). Mitochondria with enhanced toxic potential from distal axons and terminals could therefore have a “Trojan horse” messenger role in the degeneration of MNs in ALS via retrograde transport from skeletal muscle.
Aberrant oxidative chemistry could contribute to the mechanisms of mitochondriopathy in G93Ahigh mice (Beckman et al., 2001; Liochev and Fridovich, 2003). G93A-mSOD1 has enhanced free radical-generating capacity compared to wildtype enzyme (Yim et al., 1996) and can catalyze protein oxidation by hydroxyl-like intermediates and carbonate radical (Pacher et al., 2007). We found that protein carbonyl formation in the mitochondrial membrane-enriched fraction of spinal cord is a robust signature of pre- and incipient-disease, while, surprisingly, in the same pre-symptomatic mice, oxidative damage to soluble proteins appeared lower than control. In human sporadic ALS, protein carbonyls are elevated in motor cortex (Ferrante et al., 1997). A pervious study of G93Ahigh mice showed increased protein carbonyl formation in total spinal cord extracts at pre-symptomatic disease, and one of these oxidatively damaged proteins was SOD1 (Andrus et al., 1998), but it was not identified as human or mouse SOD1. We observed a similar low molecular weight band of oxidatively damaged protein that could be SOD1, and we also found oxidative damage to higher molecular mass proteins not seen earlier (Andrus et al., 1998). A more recent mass spectroscopy study of G93Ahigh mice identified proteins in total spinal cord extracts with greater than baseline carbonyl modification, including SOD1, translationally controlled tumor protein, and ubiquitin carboxyl-terminal hydrolase-L1 in G93Ahigh mice (Poon et al., 2005).
Beckman, Estevez, and colleagues have proposed that mSOD1 and wtSOD1 can catalyze the formation of ONOO- that mediates tyrosine nitration in proteins causing alteration in function (Beckman et al., 1993; Crow et al., 1997; Estevez et al., 1999). Studies have found increased protein nitration and free 3-nitrotyrosine in CNS tissues from human ALS patients (Abe et al., 1995; Beal et al., 1997) and animal models of ALS (Martin et al., 1999; Martin et al., 2005; Martin et al., 2007) to support of this theory. Very few studies have emerged identifying these nitrated proteins (Casoni et al., 2005). We found cytochrome c oxidase subunit I and SOD2 with elevated nitration in G93Ahigh mouse spinal cord at early disease (Martin et al., 2007). Here, we identify two mPTP proteins as targets of nitration in ALS mice. The nitration of CyPD is very interesting in several aspects. First, CyPD in both mitochondrial membrane-enriched and soluble compartments is nitrated in wtSOD1 tg mouse spinal cord. Second, in G93Ahigh mice, CyPD nitration is elevated in early- to mid-symptomatic stages, but declines to baseline at end-stage disease. Third, CyPD nitration is greater in the soluble protein compartment compared to proteins in the mitochondrial membrane-enriched fraction. We also found ANT to be nitrated in G93Ahigh mouse spinal cord. This finding is notable particularly because nitrated ANT is found in pre-symptomatic and symptomatic stages but not at end-stage disease or in wtSOD1 tg mice. In vitro protein and cell experiments have shown that NO and ONOO- can act directly on the ANT to induce mitochondrial permeabilization in a cyclosporine A-sensitive manner (Vieira et al., 2001). Oxidative stress can enhance CyPD binding to the ANT (McStay et al., 2002). Interestingly, copper interactions with ANT and thiol modification of ANT can cause mPTP opening (Constantini et al 1998; Costantini et al., 2000; Garcia et al., 2007). Together these data and future work could reveal that oxidative and nitrative damage to proteins, some of which are core components of the mPTP, in G93Ahigh mice is targeted rather than stochastic and could impinge on the functioning of the mPTP.
CyPD is the best known regulator of the mPTP (Baines et al., 2005; Bernardi et al., 2006). We therefore examined the role of CyPD in the process of MN disease in mice through gene ablation. There is precedence for this logic in mouse models of stroke (Schinzel et al., 2005), multiple sclerosis (Forte et al., 2007), myocardial infarction (Nakayma et al., 2007), muscular dystrophy (Millay et al 2008), Alzheimer's disease (Du et al., 2008), and cell models of necrosis (Baines et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005) where mitochondrial abnormalities have been interpreted in the causal mechanisms of cell death. G93Ahigh-mSOD1 mice without CyPD had a marked delay in the onset of disease and lived significantly longer than tg mice with CyPD. The effect of CyPD deletion was much more prominent in females than in males. Female mice showed positive effects with haplo-deletion of CyPD. It has been argued that mitochondrial damage in G93Ahigh-mSOD1 mice is related to supra-normal levels of SOD1 and might not be related causally to the disease process (Jaarsma, 2006), but mitochondrial abnormalities have been found in G93Alow-mSOD1 mice (Sasaki et al., 2004). We therefore repeated the experiment using a tg mouse line with much lower levels of human mSOD1 expression and a slower disease progression. G93Alow-mSOD1 mice without CyPD also had a significant delay in the onset of disease and lived significantly longer than tg mice with CyPD. Thus, some form of mitochondrial pathobiology is occurring regardless of whether transgene expression of G93A is high or low. Three other reports have implicated the mPTP in ALS using pharmacological approaches. Cyclopsorine A treatment, delivered intracerebroventricularly or systemically in combination with a multiple drug resistance type 1a/b mouse background, modestly improved outcome in G93Ahigh mice (Keep et al., 2001; Karlsson et al., 2004; Kirkinezos et al., 2004), but these studies are confounded by the immunosuppressant actions of cyclopsorine A through calcineurin inhibition. CyPD inhibitors devoid of effects on calcineurin need to be tested in ALS mice. Nevertheless, G93A-mSOD1 mice without CyPD develop eventually MN disease. The effects of CyPD deficiency on MN cell mechanisms need to be examined in more detail, but the cell death phenotype appears to be switched or converted to another form with the attenuation of mitochondrial swelling. A switch in the cell death morphology and molecular mechanisms in MNs of mSOD1 mice without CyPD is an outcome consistent with the cell death continuum (Portera-Cailliau et al., 1997; Martin et al., 1998).
We conclude that the mPTP actively participates in the mechanisms of MN death in ALS mice. mPTP activation is a possible triggering event for MN degeneration and MN selective vulnerability in ALS could be related to amount, composition, and trafficking of mitochondria in MNs. This work defines new mitochondrial mechanisms for MN degeneration in ALS and can lead to the identification of molecular mechanism-based therapies for treating this fatal disease.
Acknowledgments
This work was supported by grant NS034100 and NS052098 from the NIH-NINDS and AG016282 from the NIH-NIA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe K, Pan LH, Watanabe M, Kato T, Itoyama Y. Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci Lett. 1995;199:152–4. doi: 10.1016/0304-3940(95)12039-7. [DOI] [PubMed] [Google Scholar]
- Akanda N, Tofight R, Brask J, Tamm C, Elinder F, Ceccatelli S. Voltage-dependent anion channels (VDAC) in the plasma membrane play a critical role in apoptosis in differentiated hippocampal neurons but not in neural stem cells. Cell Cycle. 2008;7:3225–3234. doi: 10.4161/cc.7.20.6831. [DOI] [PubMed] [Google Scholar]
- Andrus PK, Fleck TJ, Gurney ME, Hall ED. Protein oxidative damage in a transgenic mouse medel of familial amyotrophic lateral sclerosis. J Neurochem. 1998;71:2041–2048. doi: 10.1046/j.1471-4159.1998.71052041.x. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair HS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, II, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH., Jr Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann Neurol. 1997;42:644–54. doi: 10.1002/ana.410420416. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993;364:548. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Estévez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–S20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Calvaresi N, Chiveri L, Prelle A, Moggio M, Braga M, Silani V, De Biasi S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J Neurol Sci. 2001;191:25–33. doi: 10.1016/s0022-510x(01)00627-x. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Carrì MT. Lessons from models of SOD1-linked familial ALS. Trends Mol Med. 2004;10:393–400. doi: 10.1016/j.molmed.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blalchy-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Lee MK, Slunt HH, Guarnieri M, Xu ZS, Wong PC, Brown RH, Jr, Price DL, Sisodia SS, Cleveland DW. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. PNAS USA. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose S, Freedman RB. Peptidyl prolyl cis-trans-isomerase activity associated with the lumen of the endoplasmic reticulum. Biochem J. 1994;300:865–870. doi: 10.1042/bj3000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Geddes JW. Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- Buck CR, Jurynec MJ, Upta DE, Law AKT, Bilger J, Wallace DC, McKeon RJ. Increased adenine nucleotide translocator 1 in reactive astrocytes facilitates glutamate transport. Exp Neurol. 2003;181:149–158. doi: 10.1016/s0014-4886(03)00043-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casoni F, Basso M, Massignam T, Gianazza E, Cheroni C, Salmona M, Bendotti C, Bonetto V. Protein nitration in a mouse model of familial amyotrophic lateral sclerosis. J Biol Chem. 2005;280:16295–16304. doi: 10.1074/jbc.M413111200. [DOI] [PubMed] [Google Scholar]
- Collins TJ, Bootman MD. Mitochondria are morphologically heterogeneous within cells. J Exp Biol. 2003;206:1993–2000. doi: 10.1242/jeb.00244. [DOI] [PubMed] [Google Scholar]
- Costantini P, Colonna R, Bernardi P. Induction of the mitochondrial permeability transition by N-ethylmaleimide depends on secondary oxidation of critical thiol groups. Potentiation by copper-ortho-phenthroline without dimerization of the adenine nucleotide translocase. Biochim Biophys Acta. 1998;1365:385–392. doi: 10.1016/s0005-2728(98)00090-5. [DOI] [PubMed] [Google Scholar]
- Costantini P, Belzacq AS, Vieira HLA, Larochette N, de Pablo MA, Zamzami N, Susin SA, Brenner C, Kroemer G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene. 2000;19:307–314. doi: 10.1038/sj.onc.1203299. [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Ferri A, Carri MT. Amyotrophic lateral sclerosis: From current developments in the laboratory to clinical implications. Antioxid Redox Signal. 2008;10:405–443. doi: 10.1089/ars.2007.1760. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Crompton M. Mitochondria and aging: a role for the permeability transition? Aging Cell. 2004;3:3–6. doi: 10.1046/j.1474-9728.2003.00073.x. [DOI] [PubMed] [Google Scholar]
- Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–9. [PMC free article] [PubMed] [Google Scholar]
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericak-Vance MA, Siddique T. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Browlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CCJ, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondrial content. Hum Mol Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Aranico O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estévez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH, Jr, Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, Rotilio G, Valentine JS, Carri MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. PNAS USA. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu Z, Fowlkes J, Rahder M, Stern K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. PNAS USA. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García N, Martínez-Abundis E, Pavón N, Correa F, Chávez E. Copper induces permeability transition through its interaction with the adenine nucleotide translocase. Cell Biol Int. 2007;31:893–899. doi: 10.1016/j.cellbi.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Golden WC, Brambrink AM, Traystman RJ, Shaffner DH, Martin LJ. Nitration of the striatal NA,K-ATPase α3 isoform occurs in normal brain development but is not increased during hypoxia-ischemia in newborn piglets. Neurochem Res. 2003;28:1883–1889. doi: 10.1023/a:1026132110850. [DOI] [PubMed] [Google Scholar]
- Graham BH, Waymire KG, Cottrell B, Trounce IA, Macgregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet. 1997;16:226–234. doi: 10.1038/ng0797-226. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hamberger A, Blomstrand C, Lehninger AL. Comparative studies of mitochondria isolated from neuron-enriched and glia-enriched fractions of rabbit and beef brain. J Cell Biol. 1970;45:221–234. doi: 10.1083/jcb.45.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Narin AC, Takei K, Matsui H, Matsushita M. CaM kinase Iα-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelton JL, Petrasheuskaya M, Fiskum G, Kristian T. Cyslophilin D is expressed predominantly in mitochondria of γ-aminobutyric acidergic interneurons. J Neurosci Res. 2008 doi: 10.1002/jnr.21921. 10.1002.jnr.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CMJ, Jung C, Ding H, Xu Z. Mutant Cu, Zn Superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22(RC215):1–6. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Xu Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003;4:16. doi: 10.1186/1471-2202-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- Jaarsma D. Swelling and vacuolation or mitochondria in transgenic SOD1-ALS mice: a consequence of supranormal SOD1 expression? Mitochondrion. 2006;6:48–49. doi: 10.1016/j.mito.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Jensen RE. Control of mitochondrial shape. Cur Opin Cell Biol. 2005;17:384–388. doi: 10.1016/j.ceb.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Johnson N, Khan A, Virji S, Ward JM, Crompton M. Import and processing of heart mitochondrial cyclophilin D. Eur J Biochem. 1999;263:353–359. doi: 10.1046/j.1432-1327.1999.00490.x. [DOI] [PubMed] [Google Scholar]
- Ju WK, Kim KY, Lindsey JD, Angert M, Duong-Polk KX, Scott RT, Kim JJ, Kukhmazov I, Ellisman MH, Perkins GA, Weinreb RN. Intraocular pressure elevation induces mitochondrial fision and triggers OPA1 release in glaucomatous optic nerve. Invest Opthalmol Vis Sci. 2008;49:4903–4911. doi: 10.1167/iovs.07-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmains PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Dohi T, Risa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific HSp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Karlsson J, Fong KS, Hansson MJ, Elmer E, Csiszar K, Keep MF. Life span extension and reduced neuronal death after weekly intraventricular cyclosporine injections in the G93A transgenic mouse model of amyotrophic lateral sclerosis. J Neurosurg. 2004;101:128–137. doi: 10.3171/jns.2004.101.1.0128. [DOI] [PubMed] [Google Scholar]
- Keep M, Elmér E, Fong KSK, Csiszar K. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res. 2001;894:27–331. doi: 10.1016/s0006-8993(01)02012-1. [DOI] [PubMed] [Google Scholar]
- Kirkinezos IG, Hernandez D, Bradley WG, Moraes CT. An ALS mouse model with a permeable blood-brain barrier benefits from systemic cyclosporine A treatment. J Neurochem. 2004;88:821–826. doi: 10.1046/j.1471-4159.2003.02181.x. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–50. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung AWC, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim Biophys Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Liochev SI, Fridovich I. Mutant Cu,Zn superoxide dismutases and familial amyotrophic lateral sclerosis: evaluation of oxidative hypotheses. Free Radic Biol Med. 2003;34:1383–9. doi: 10.1016/s0891-5849(03)00153-9. [DOI] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson A, Vande Velde C, Ward CW, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Anderson PM, Brannstrom TM, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–15. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Martin LJ. Mitochondriopathy in Parkinson disease and amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2006;65:1103–1110. doi: 10.1097/01.jnen.0000248541.05552.c4. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z. Opportunities for neuroprotection in ALS using cell death mechanism rationales. Drug Discov Today: Disease Mod. 2004;1:135–143. [Google Scholar]
- Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Kaiser A, Price AC. Motor neuron degeneration after sciatic nerve avulsion in adult rat evolves with oxidative stress and is apoptosis. J Neurobiol. 1999;40:185–201. [PubMed] [Google Scholar]
- Martin LJ, Liu Z. Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and bax-dependent. J Neurobiol. 2002;5:181–197. doi: 10.1002/neu.10026. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Price AC, McClendon KB, Al-Abdulla NA, Subramaniam JR, Wong PC, Liu Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J Neurochem. 2003;85:234–247. doi: 10.1046/j.1471-4159.2003.01659.x. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Chen K, Liu Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J Neurosci. 2005;25:6449–59. doi: 10.1523/JNEUROSCI.0911-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Liu Z, Chen K, Price AC, Pan Y, Swaby JA, Golden WC. Motor neuron degeneration in amyotrophoc lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500:20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- McCord JM, Fridovich I. Superoxide dismutase, an enzymic function for erythrocuprein (hemocuprein) J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- McEnery MW, Dawson TM, Verma A, Gurley D, Colombini M, Snyder SH. Mitochondrial voltage-dependent anion channel. J Biol Chem. 1993;268:23289–23296. [PubMed] [Google Scholar]
- McStay GP, Clarke SJ, Halestrap AP. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem J. 2007;367:541–548. doi: 10.1042/BJ20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, Sweeney HL, Robbins J, Molkentin JD. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med. 2008;14:442–447. doi: 10.1038/nm1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morota S, Hansson MJ, Ishii N, Kudo Y, Elmer E, Uchino H. Spinal cord mitochondria display lower retention capacity compared with brain mitochochondria without inherent differences in sensitivity to cyclophilin D inhibition. J Neurochem. 2007;103:2066–2076. doi: 10.1111/j.1471-4159.2007.04912.x. [DOI] [PubMed] [Google Scholar]
- Naga KK, Sullivan PG, Geddes JW. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci. 2007;27:7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulated some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Chen X, Baines CO, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BHL, Hewett TE, Robbins J, Houser SR, Molkentin JD. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide (SOD) in rat liver. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown RH., Jr Amyotrophic lateral sclerosis-associated SOD1 mutant protein bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron. 2004;43:19–30. doi: 10.1016/j.neuron.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Poon HF, Hensley K, Thongboonkerd V, Merchant ML, Lynn BC, Pierce WM, Klein JB, Calabrese V, Butterfield DA. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice- a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;39:435–462. doi: 10.1016/j.freeradbiomed.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau CP, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuroal death in adult brain are morphologically distinct: further evidence for an apoptosis necrosis continuum. J Comp Neurol. 1997;378:88–104. [PubMed] [Google Scholar]
- Price DL, Ackerley S, Martin LJ, Koliatsos V, Wong PC. Motor neuron diseases. In: Siegel GJ, Albers RW, Brady ST, Price DL, editors. Basic neurochemistry: molecular, cellular, and medical aspects. New York: Elsevier; 2006. pp. 731–743. [Google Scholar]
- Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A. Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic sclerosis. J Biol Chem. 2004;279:15499–15504. doi: 10.1074/jbc.M313295200. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figiewicz DA, Sapp P, Hentati A, Donalsson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston AM, Berger R, Tanzi RE, Halperin JJ, Harzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Tan W, Colombini M. On the role of VDAC in apoptosis: fact and fiction. J Bioenerget Biomembr. 2005;37:129–142. doi: 10.1007/s10863-005-6566-8. [DOI] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Warita H, Murakami T, Abe K, Iwata M. Ultrastructural study of mitochondria in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2004;107:461–474. doi: 10.1007/s00401-004-0837-z. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hertz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. PNAS USA. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymick JC, Talbot K, Traynor GJ. Genetics of amyotrophic lateral sclerosis. Hum Mol Genet. 2007;16:R233–R242. doi: 10.1093/hmg/ddm215. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V, Zalk R, Gincel D, Vardi N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum, Biochim, Biophys. Acta. 2004;1657:105–114. doi: 10.1016/j.bbabio.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Keller JN, Lovell M, Sodhi A, Hart RP, Scheff SW. Intrinsic differences in brain and spinal cord mitochondria: implications for therapeutic interventions. J Comp Neurol. 2004;474:524–534. doi: 10.1002/cne.20130. [DOI] [PubMed] [Google Scholar]
- Turner BJ, Talbot K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog Neurobiol. 2008;85:94–134. doi: 10.1016/j.pneurobio.2008.01.001. [DOI] [PubMed] [Google Scholar]
- van Gurp M, Festjens N, van Loo G, Saeleus X, Vandenabeele P. Mitochondrial intermembrane proteins in cell death. Biochem Biophys Res Comm. 2003;304:487–497. doi: 10.1016/s0006-291x(03)00621-1. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. PNAS USA. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira HLA, Belzacq AS, Haouzu D, Bernassola F, Cohen I, Jacotot E, Ferri KF, Hamel CE, Bartle LM, Melino G, Brenner C, Goldmacher V, Kroemer G. The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxxynonenal. Oncogene. 2001;20:4305–4316. doi: 10.1038/sj.onc.1204575. [DOI] [PubMed] [Google Scholar]
- Vijayverguya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmeier PC, Zimmermann K, Qian T, Tintelnot-Blomley M, Lemasters JJ. Cyclophilin D as a drug target. Curr Med Chem. 2003;10:1485–1506. doi: 10.2174/0929867033457160. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn of evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Woodfield K, Rück A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336:287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda O, Fukuo K, Sun X, Nishitani M, Yotsui T, Higuchi M, Suzuki T, Rakugi H, Smithies O, Maeda N, Ogihara T. Apop-1, a novel protein inducing cyclophilin D-dependent but Bax/Bak-related channel-independent apoptosis. J Biol Chem. 2006;281:23899–23907. doi: 10.1074/jbc.M512610200. [DOI] [PubMed] [Google Scholar]
- Yim MB, Kang JH, Yim HS, Kwak HS, Chock PB, Stadtman ER. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: an enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. PNAS USA. 1996;93:5709–5714. doi: 10.1073/pnas.93.12.5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, Wolfgang W, Forte M. Subcellular localization of human voltage-dependent anion channel isoforms. J Biol Chem. 1995;270:13998–14006. doi: 10.1074/jbc.270.23.13998. [DOI] [PubMed] [Google Scholar]