

Abstract
In the present study we tested the hypothesis that the cyclophilin D-dependent (CyD) mitochondrial permeability transition (CyD-mPT) plays an important role in glutamate-triggered delayed calcium deregulation (DCD) and excitotoxic neuronal death. We used cultured cortical neurons from wild-type C57BL/6 and cyclophilin D knockout mice (Ppif-/-). Induction of the mPT was identified by following the rapid secondary acidification of mitochondrial matrices monitored with mitochondrially targeted pH-sensitive yellow fluorescent protein. Suppression of the CyD-mPT due to genetic CyD ablation deferred DCD and mitochondrial depolarization, and increased the survival rate after exposure of neurons to 10μM glutamate, but not to 100μM glutamate. Ca2+ influx into Ppif-/- neurons was not diminished in comparison with WT neurons judging by 45Ca accumulation. In both types of neurons, 100μM glutamate produced greater Ca2+ influx than 10μM glutamate. We hypothesize that greater Ca2+ influx produced by higher glutamate rapidly triggered the CyD-independent mPT in both WT and Ppif-/- neurons equalizing their responses to supra-physiologic excitotoxic insults. In neurons exposed to moderate but pathophysiologically-relevant glutamate concentrations, an induction of the CyD-mPT appears to play an important role in mitochondrial injury contributing to DCD and cell death.
In various neurodegenerations, in traumatic brain injury and stroke, prolonged exposure of neurons to glutamate causes massive Ca2+ influx into the cytosol (Choi, 1988;Manev et al., 1989;Tymianski et al., 1993b). This produces a rapid jump in the cytosolic Ca2+ concentration ([Ca2+]c) followed by its transient decrease to a lower level (Nicholls and Budd, 2000). After some delay, this decrease in [Ca2+]c is followed by a secondary sustained elevation of [Ca2+]c or “delayed calcium deregulation” (DCD) (Tymianski et al., 1993a;Nicholls and Budd, 1998). Elevated [Ca2+]c activates Ca2+-dependent degradation enzymes and represents a serious danger to neurons by promoting neuronal death (Wu et al., 2004;Bano et al., 2005;Xu et al., 2007). Therefore, DCD is considered not only a hallmark of but also a potential mechanism leading to glutamate excitotoxicity (Manev et al., 1989;Randall and Thayer, 1992;Tymianski et al., 1993b). Despite obvious significance and extensive studies, the precise mechanisms leading to DCD still are not completely understood.
Mitochondria accumulate cytosolic Ca2+ and thus contribute to the clearance of elevated [Ca2+]c (Kiedrowski and Costa, 1995;Wang and Thayer, 1996;White and Reynolds, 1997). However, Ca2+ uptake can lead to an induction of the mitochondrial permeability transition (mPT) pore accompanied by depolarization of organelles, and therefore, inhibition of further Ca2+ accumulation (Bernardi, 1999). The molecular composition of the mPT pore is not yet clear, but it is well established that mitochondrial cyclophilin D (CyD) is a regulatory component of the pore (Baines et al., 2005;Basso et al., 2005;Schinzel et al., 2005). It is hypothesized that following excessive Ca2+ influx into mitochondria, CyD facilitates activation/assembly of the mPT pore (Rasola and Bernardi, 2007). In addition, CyD binds cyclosporin A (CsA), an inhibitor of the CyD-dependent mPT (CyD-mPT) (Crompton et al., 1988). However, under greater Ca2+ loading, pore activation/assembly may occur in a CyD-independent manner producing the CyD-independent mPT insensitive to CsA (Brustovetsky and Dubinsky, 2000).
In early studies, CsA and its non-immunosuppressive analog, N-methyl-valine-4-cyclosporin A (MetVal4-CsA), were found to be protective against DCD and/or neuronal death in experiments with oxygen-glucose deprivation (OGD) and in glutamate-treated neurons (Schinder et al., 1996;Nieminen et al., 1996;White and Reynolds, 1996;Khaspekov et al., 1999;Vergun et al., 1999;Almeida and Bolanos, 2001;Alano et al., 2002). CsA and its non-immunosuppressive derivative NIM811 appeared to be protective against neuronal death in traumatic brain and spinal cord injury emphasizing the key role of the mPT pore in these conditions (Sullivan et al., 1999;Scheff and Sullivan, 1999;Sullivan et al., 2000;Okonkwo et al., 1999;Sullivan et al., 2005;McEwen et al., 2007;Mbye et al., 2008). In addition, in a recent study, NIM811, protected against neuronal death in the model of transient focal cerebral ischemia, suggesting involvement of the CyD-mPT (Korde et al., 2007). These studies linked glutamate-induced DCD and neuronal death following ischemic insult to the CyD-mPT. However, other investigators have failed to confirm protective effects of CsA or MetVal4-CsA (Isaev et al., 1996;Castilho et al., 1998;Reynolds, 1999;Chinopoulos et al., 2004;Pivovarova et al., 2004). Recently, homozygous knockout mice lacking CyD (Ppif-/- mice) were generated, and it was shown that isolated mitochondria from these mice have increased resistance to Ca2+ (Baines et al., 2005;Forte et al., 2007;Nakagawa et al., 2005). Cultured cortical neurons from Ppif-/- mice appeared to be more resistant to oxidative stress than cells from wild-type animals (WT) (Forte et al., 2007). In addition, Ppif-/- mice revealed a reduction in brain infarct size after acute middle cerebral artery occlusion (Schinzel et al., 2005). All these effects were attributed to the suppression of the CyD-mPT.
In the present study, we investigated the role of the CyD-mPT in glutamate-triggered DCD and excitotoxic cell death in cultured cortical neurons derived from Ppif-/- mice and their genetic background C57BL/6 mice. Our experiments revealed that cultured neurons from Ppif-/-mice were more resistant to glutamate-triggered DCD and cell death than neurons from WT animals. This protection was limited to a moderate but pathophysiologically-relevant glutamate concentration. At higher concentrations, glutamate produced similar DCD and cell death in both Ppif-/- and WT neurons.
Experimental Procedures
Materials
Carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP), cyclosporin A (CsA), ADP, oligomycin, glutamate, and glycine were purchased from Sigma (St. Louis, MO). Fura-2FF AM was bought from Teflabs (Austin, TX) and Rhodamine-123 (FluoroPure™ grade) was purchased from Invitrogen (Carlsbad, CA).
Isolation and purification of brain mitochondria
Mitochondria from the brains of three cyclophilin D-knockout Ppif-/- mice (from Dr. Jeffery Molkentin, University of Cincinnati) and from the brains of three wild-type C57BL/6 mice (Harlan, Indianapolis, IN) were isolated in mannitol-sucrose medium, according to an IACUC approved protocol, and purified on a discontinuous Percoll gradient (Brustovetsky et al., 2002). Mitochondrial protein was determined by the Bradford method (Bradford, 1976) using BSA as a standard. Mitochondria isolated from C57/BL6 and Ppif-/- mice had a respiratory control index of 8.27±0.47, N=4, and 8.53±0.31, N=3, respectively. Respiratory control index is the ratio of the respiratory rate stimulated by addition of 200 μM ADP (State 3) to the respiratory rate after inhibition of ADP phosphorylation with 1μM oligomycin (State 4). Mitochondrial respiration was measured using a Clark-type oxygen electrode at 37°C in the standard incubation medium containing 125 mM KCl, 0.5 mM MgCl2, 3 mM KH2PO4, 3 mM glutamate plus 1 mM malate, 10μM EGTA, 0.1% BSA (free from fatty acids), 10 mM HEPES, pH 7.4.
Slow Ca2+ infusion
The experiments with slow Ca2+ infusion were performed as described previously (Chalmers and Nicholls, 2003;Shalbuyeva et al., 2007) with some modifications. Briefly, mitochondria were incubated in the standard incubation medium supplemented with 200μM ADP and 1μM oligomycin in a 0.3 ml chamber at 37°C under continuous stirring. Mitochondrial Ca2+ accumulation was followed by monitoring disappearance of Ca2+ from the incubation medium with a miniature Ca2+ selective electrode. A solution of CaCl2 was infused into the chamber at a constant rate of 330 nmol CaCl2/mg protein × min using a KDS 100 pump (KD Scientific, Holliston, MA) equipped with a Hamilton microsyringe. The Ca2+ uptake capacity was estimated as the amount of accumulated Ca2+ (μmol per mg of mitochondrial protein). The Ca2+ uptake capacity was assessed by linear fitting of the early fragment of the experimental trace and the final linear fragment of the trace and then finding the intersection point of these linear graphs (Fig. 1c).
Fig. 1. Ca2+ uptake capacity of isolated brain mitochondria from cyclophilin D-knockout mice (Ppif-/-) and their genetic background C57BL/6 mice (wild-type, WT). Cyclosporin A (CsA, 1μM) augments Ca2+ uptake capacity of mitochondria from C57BL/6 mice but fails to increase Ca2+ uptake capacity of mitochondria from Ppif-/- mice.
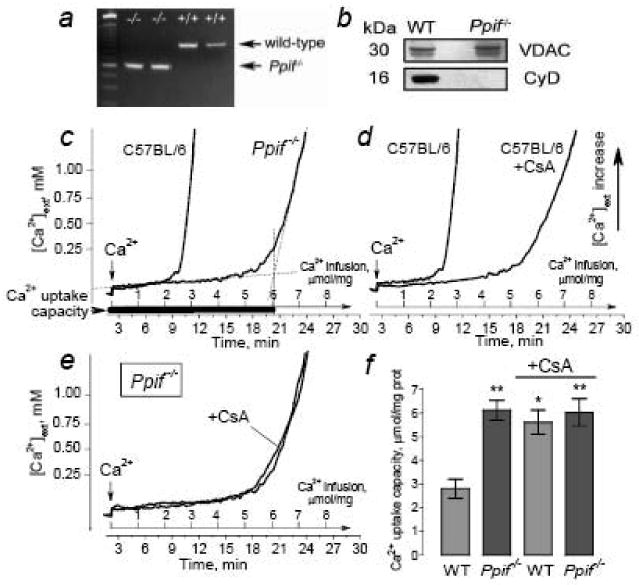
In a, genotyping CyD-knockout and wild-type mice: PCR products generated from genomic DNA obtained from cyclophilin D-knockout mice (Ppif-/-) and C57BL/6 mice (wild-type, WT) were separated by agarose gel electrophoresis and visualized with ethidium bromide. In b, western blot analysis of CyD expression in brain mitochondria isolated from Ppif-/- and wild-type (WT) mice. The protein loading for electrophoresis was 15μg protein per lane. VDAC was used as a loading control. In c, comparison of Ca2+ uptake capacity of brain mitochondria from C57BL/6 and Ppif-/- mice. In d, effect of CsA (1μM) on Ca2+ uptake capacity of mitochondria from C57BL/6 mice. In e, lack of CsA (1μM) effect on Ca2+ uptake capacity of mitochondria from Ppif-/-mice. In f, summary of data obtained with mitochondria from wild-type and Ppif-/- mice and incubated with and without 1μM CsA. Data are mean±SEM. *p<0.05, t=4.116, in comparison of Ca2+ uptake capacity in WT mitochondria incubated with or without CsA, N=3; **p<0.01 in comparison of Ca2+ uptake capacity in WT mitochondria versus Ca2+ uptake capacity in Ppif-/- mitochondria incubated with (t=4.742) or without CsA (t=4.860).
Western blot analysis of CyD
Isolated brain mitochondria pretreated with Protease Inhibitor Cocktail (Roche, Indianapolis, IN) were solubilized by incubation in a NuPAGE LDS sample buffer (Invitrogen, Carlsbad, CA) supplemented with a reducing agent at 70°C for 15 minutes. 4-12% of the Bis-Tris MOPS gels (Invitrogen) were used for electrophoresis (15μg protein/lane). After electrophoresis, the proteins were transferred to a Hybond™-ECL™ nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ). Blots were incubated for an hour at room temperature with mouse monoclonal anti-cyclophilin D antibody (1:1000 dilution) (EMD Chemicals, San Diego, CA) or goat polyclonal anti-VDAC1 antibody (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA). VDAC1 was used as a control for loading. Blots were developed using goat anti-mouse or donkey anti-goat IgG (1:20000) coupled with horseradish peroxidase (Jackson ImmunoResearch Labs, West Grove, PA) and Supersignal West Pico chemiluminescent reagents (Pierce, Rockford, IL). The molecular weight marker SeeBlue®Plus 2 Standard (5μl) (Invitrogen, Carlsbad, CA) was used to determine the molecular weights of the bands.
Genotyping
All Ppif-/- mice were genotyped to ensure that they were homozygous. After preparing the tail samples, the genomic DNA was re-suspended and hydrated in 75μl of Tris-EDTA buffer, pH 8.0. Then, the samples were diluted 1:20 and 1μl of this diluted solution was used for the PCR (50μl reaction volume). We used three primers in a single reaction: Exon3-F: CTC TTC TGG GCA AGA ATT GC; Neo-F: GGC TGC TAA AGC GCA TGC TCC; and Exon4-R: ATT GTG GTT GGT GAA GTC GCC. The reaction conditions were 95°C for 3 min, then 35 cycles at 95°C for 0.5 min, 56°C for 0.5 min, 72°C for 1 min, and then 72°C for 10 min. The wild-type allele was amplified as a band ∼850bp, and the null allele was amplified as a band ∼600bp.
Cell cultures
Primary cultures of cortical neurons were prepared from postnatal day 1 mouse pups according to IACUC approved protocols and procedures previously published for the hippocampus (Dubinsky, 1993), but without preplated glia and the use of antibiotics. For fluorescence measurements, neurons were plated on glass bottomed Petri dishes (Dubinsky, 1993). For all platings, 35 μg/ml uridine plus 15 μg/ml 5-fluoro-2′-deoxyuridine were added 24 hours after plating to inhibit proliferation of non-neuronal cells. Cultures were maintained in a 5% CO2 atmosphere at 37°C in Eagle's MEM supplemented with 10% NuSerum (BD Bioscience, Bedford, MA) and 27 mM glucose. Experiments were performed on neurons at 12-14 days in vitro.
Cyclophilin D immunocytochemistry and visualization of mitochondria with MitoTracker Red
To co-localize CyD immunostaining with mitochondria, neurons from C57BL/6 and Ppif-/- mice, respectively, were stained with MitoTracker Red (Molecular Probes, Eugene, OR). Prior to fixation for immunocytochemistry, neurons were incubated with 0.3μM MitoTracker Red in the growth medium for 15 minutes at 37°C. Then, cultured neurons were fixed with 0.05% glutaraldehyde for 20 minutes, followed by incubation with 4% paraformaldehyde for 15 minutes, and then washed with PBS. Next, cells were incubated with 0.1% glycine-PBS for 30 minutes and incubated with a blocking solution containing 2.5% IgG- and protease-free BSA (Jackson ImmunoResearch Laboratories, West Grove, PA), 2.5% goat serum, and 0.1% Triton X-100 in PBS for an hour at room temperature. Cells were incubated overnight at 4°C with the primary mouse monoclonal anti-cyclophilin D antibody (1:500 dilution) (EMD Chemicals, San Diego, CA). Then, cells were incubated with a secondary donkey anti-mouse antibody conjugated with AlexaFluor 488 (1:1000 dilution) (Invitrogen, Carlsbad, CA) for an hour at room temperature. Bright field and fluorescence images were acquired using a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon CFI Plan Apo 100× 1.4 NA objective and Photometrics cooled CCD camera CoolSNAPHQ (Roper Scientific, Tucson, AZ) controlled by MetaMorph 6.3 software (Molecular Devices, Downingtown, PA).
Calcium imaging and monitoring of mitochondrial membrane potential (Δψ) in cultured neurons
Cortical neurons were loaded at 37°C simultaneously with 2.6μM Fura-2FF-AM (Kd=5.5μM, λex 340, 380 nm/λem 512 nm, Molecular Probes, Eugene, OR), to follow changes in cytosolic Ca2+, and 1.7μM Rhodamine-123 (Rh123) (λex 507 nm/λem 529 nm, Molecular Probes, Eugene, OR) to monitor changes in mitochondrial membrane potential (Δψ) in the standard bath solution containing 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM NaHEPES, pH 7.4, 5 mM glucose, and 65 mM sucrose. The ion composition of the bath solution is similar or close to those used previously in studies of DCD and excitotoxicity (Wang and Thayer, 1996;White and Reynolds, 1996;Dubinsky et al., 1995;Kushnareva et al., 2005). Sucrose was used to maintain osmolarity similar to that in the growth medium (340 mosm). Osmolarity of the bath solution was measured with an osmometer, Osmette II™ (Precision Systems Inc., Natick, MA). Fluorescence imaging was performed with an inverted microscope, Nikon Eclipse TE2000-U, using a Nikon CFI Plan Fluor 20× 0.45 NA objective and a back-thinned EM-CCD camera Hamamatsu C9100-12 (Hamamatsu Photonic Systems, Bridgewater, NJ) controlled by Simple PCI software 6.1 (Compix Inc., Sewickley, PA). The excitation light was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA). The excitation light at 480 nm was attenuated by quartz neutral density filters to 10%. The excitation filters (340±5, 380±7, and 480±20) were controlled by a Lambda 10-2 optical filter changer (Sutter Instruments, Novato, CA). Fluorescence was recorded through a 505 nm dichroic mirror at 535±25 nm. To minimize photobleaching and phototoxicity, the images were taken every 15 seconds during the time-course of the experiment using the minimal exposure time that provided acceptable image quality. The changes in [Ca2+]c were monitored by following a ratio of F340/F380, calculated after subtracting the background from both channels. The changes in the Δψ were monitored by following changes in the fluorescence of Rh123 expressed as F/F0. The Rh123 fluorescence traces were also constructed after subtracting the background. The contribution of plasma membrane depolarization to the Rh123 signal appeared to be negligible as determined by applying Ca2+-free bath solution containing 50 mM KCl and a correspondingly decreased concentration of NaCl to Rh123-loaded neurons (not shown). After 3 minutes of fluorescence recording in the standard bath solution, various concentrations of glutamate (10 or 100μM) plus 10μM glycine were applied to the neurons. At the end of the experiment, the bath solution with glutamate and Ca2+ was replaced by a glutamate- and Ca2+-free solution, and then 1μM FCCP was applied to the neurons to depolarize neuronal mitochondria and to release Ca2+ accumulated in mitochondria.
Transfection of primary cortical neurons
To measure matrix pH (pHm) in mitochondria within live cells, hippocampal neurons were transfected in suspension during plating using an electroporator BTX 630 ECM (Harvard Apparatus, Holliston, MA) with a plasmid encoding mitochondrially targeted eYFP (enhanced Yellow Fluorescent Protein, generously provided by Dr. Roger Tsien, University of California, San Diego) as described previously (Shalbuyeva et al., 2006). Neurons were taken into the experiment 12-14 days after transfection.
Confocal microscopy
Laser-based spinning-disk confocal microscopy was used to visualize the mitochondrial network in neurons expressing mito-eYFP. The imaging was performed with an inverted microscope, Nikon Eclipse TE2000-U, equipped with spinning-disk confocal unit Yokogawa CSU-10 (Yokogawa Electric Corp. Tokyo, Japan) using a Nikon CFI Plan Apo 100× 1.3 NA objective and a back-thinned EM-CCD camera Andor iXon+ DU-897 (Andor, Morrisville, NC) controlled by Andor iQ (Andor, Morrisville, NC). A 2× extender was placed in front of the camera to increase spatial resolution. The serial images (z-stack) were acquired with a z-step 0.1μm using a piezo-electric positioning device PIFOC P-721 (Physik Instrumente, Karlsruhe, Germany). Image processing consisted of a blind 3D deconvolution using AutoQuant X 2.1.1 (MediaCybernetics, Bethesda, MD) and a 3D rendering using Imaris 5.7.2 (Bitplane, St. Paul, MN).
Matrix pH measurements
Matrix pH (pHm) measurements and pH calibration were performed as described previously (Bolshakov et al., 2008). In addition, neurons expressing mito-eYFP were loaded with Fura-2FF to follow pHm and changes in cytosolic Ca2+ concentration simultaneously. In these experiments we used a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon CFI SuperFluor 40× 1.3 NA objective and a Photometrics cooled CCD camera CoolSNAPHQ (Roper Scientific, Tucson, AZ) controlled by MetaMorph 6.3 software (Molecular Devices, Downingtown, PA).
45Ca accumulation
Experiments were performed as described previously (Hartley et al., 1993) with some modifications. Cortical neurons from WT or Ppif-/- mice were plated on a 12-well plate. The neurons were washed once with standard bath solution to remove growth medium. Then, neurons were incubated with a solution containing 10 or 100μM glutamate (plus 10μM glycine) and supplemented with 5μCi/ml 45Ca. The neurons were then washed three times in an ice-cold, Ca2+-free bath solution. Neurons were lysed with 0.5% SDS. An aliquot was added to the scintillation liquid Ecolite (MP Biomedicals, Santa Ana, CA) and counted in a Tri-Carb 2100TR liquid scintillation analyzer (Packard Instrument Co., Meriden, CT). Each condition was carried out three times.
Glutamate toxicity
After culturing mouse cortical neurons for 12-14 DIV, the medium was replaced with Eagle's MEM without serum supplemented with 27 mM glucose, 15 mM sucrose, 100μM glutamine, and 10μM glycine (Brustovetsky et al., 2004). The neurons were then exposed to various concentrations of glutamate (3-300μM) for 10 minutes. After that, glutamate was removed, and the cells were rinsed with Eagle's Balanced Salt Solution (EBSS) supplemented with 27 mM glucose and 15 mM sucrose, and left in the thermostat at 37°C for the next 24 hours. After 24 hours, cell death was assessed by nuclear staining with 4.5μM propidium iodide (PI) (Pivovarova et al., 2004). Nuclei staining with PI is associated with the loss of barrier properties of the plasma membrane and is considered an indication of necrosis (Orrenius et al., 2003). In addition, neuronal death was quantitatively evaluated with the Trypan Blue exclusion method (Dubinsky and Rothman, 1991). An induction of apoptosis was evaluated with Annexin V staining (Molecular Probes, Eugene, OR). Dying neurons were detected using a Nikon Eclipse TE2000-U inverted microscope equipped with a Nikon CFI SuperFluor 20× 0.75 NA objective and a Photometrics cooled CCD camera CoolSNAPHQ (Roper Scientific, Tucson, AZ) controlled by MetaMorph 6.3 software (Molecular Devices, Downingtown, PA). These toxicity experiments were also performed in triplicate on neurons from three separate platings.
Statistics
Every experiment was performed using at least three separate preparations of isolated mitochondria or three independent, separate neuronal platings. All data represent mean ± SEM of at least 3 separate and independent experiments. Statistical analysis of the experimental results consisted of a one-way ANOVA followed by Bonferroni's post hoc test (GraphPad Prism® 4.0, GraphPad Software Inc., San Diego, CA). t-scores calculated in GraphPad Prism® are shown in the legends to the Figures with statistical analysis.
Results
Calcium uptake capacity of isolated brain mitochondria from C57BL/6 and Ppif-/-mice
In our experiments, we used homozygous Ppif-/- mice lacking CyD (Baines et al., 2005) to test the hypothesis that the CyD-mPT contributes to mitochondrial injury in neurons exposed to glutamate, thus limiting the ability of the organelles to participate in the maintenance of [Ca2+]c. All mice used in our experiments were genotyped using PCR analysis of genomic DNA. Figure 1a shows the results of genotyping two C57BL/6 mice and two Ppif-/- mice. C57BL/6 mice are genetic background for Ppif-/- mice. Therefore, we used isolated mitochondria and cultured neurons derived form C57BL/6 mice as controls for the experiments with mitochondria and neurons derived from Ppif-/- mice. The wild-type allele amplifies as a band ∼850bp and the null allele amplifies as a band ∼600bp as shown. Western blotting analysis confirmed the lack of CyD in brain mitochondria isolated from Ppif-/- mice (Fig. 1b). Consistent with previous reports (Basso et al., 2005;Nakagawa et al., 2005;Schinzel et al., 2005;Forte et al., 2007;Naga et al., 2007), in the experiments with slow CaCl2 infusion, brain mitochondria isolated from Ppif-/- mice had greater Ca2+ uptake capacity than mitochondria from WT mice (Fig. 1c). In this study, we used non-synaptic mitochondria, which mostly represented a mixture of mitochondria from glial cells and neuronal somata. In our experiments with cultured neurons, we were focused on perturbations in cytosolic Ca2+ and mitochondrial membrane potential in the neuronal somata. Therefore, isolated non-synaptic mitochondria were well-suited to our purpose. CsA (1μM) significantly increased Ca2+ uptake capacity of mitochondria from WT mice (Fig. 1d) implicating the CyD-mPT as a mechanism that limits the ability of mitochondria to accumulate Ca2+. On the other hand, CsA was without effect in mitochondria from Ppif-/- mice where CyD, the target for CsA, was genetically ablated (Fig. 1e). Figure 1f shows the summary of data obtained with mitochondria from wild-type and Ppif-/- mice and incubated with or without 1μM CsA. Thus, isolated brain mitochondria from Ppif-/- mice with suppressed CyD-mPT demonstrated higher resistance to Ca2+ than mitochondria from WT animals. However, the increasing Ca2+ load eventually damaged mitochondria from both Ppif-/- and WT mice and precluded further Ca2+ accumulation probably due to the induction of the CyD-independent mPT.
Delayed calcium deregulation in cortical neurons from C57BL/6 and Ppif-/- mice
Next, we addressed the question of whether cultured cortical neurons derived from Ppif-/- mice have an increased resistance to glutamate-induced DCD in comparison with neurons from WT animals. To confirm the lack of CyD expression in mitochondria of cultured neurons, we performed an immunochemistry analysis using an antibody against CyD. To ensure mitochondrial localization of CyD, neurons were co-stained with a mitochondrial marker, MitoTracker Red, and then CyD and MitoTracker Red images were overlaid (Fig. 2). Immunostaining of cultured neurons derived from WT animals revealed an abundance of CyD in mitochondria, while in neurons from Ppif-/- mice CyD was below the detection limit. To evaluate the role of the CyD-mPT in glutamate-induced DCD, neurons were co-loaded with calcium-sensitive, low-affinity fluorescence dye Fura-2FF and mitochondrial membrane potential-sensitive dye Rhodamine-123 (Rh123). Figure 3 shows Fura-2FF and Rh123 fluorescence traces from the representative experiments with cultured neurons derived from WT (a,c) and Ppif-/-mice (b,d). The individual traces (thin grey lines) were obtained from different individual neurons and overlapped by average traces (thick black lines for Fura-2FF and for Rh123, mean±SEM). Following a few minutes of incubation in the standard bath solution, neurons were exposed to 10μM glutamate plus 10μM glycine for 20 minutes. This concentration of glutamate was chosen based on early data obtained after cerebral ischemia in rats (Benveniste et al., 1984;Van Hemelrijck et al., 2005) and on clinical reports indicating an increase of glutamate concentration in the cerebrospinal fluid of stroke patients up to 7-8μM (Castillo et al., 1997;Castillo et al., 1996). After 20 minutes of incubation, the glutamate and Ca2+-containing bath solution was replaced with a glutamate- and Ca2+-free solution to assess the ability of neurons to recover [Ca2+]c. Finally, 1μM FCCP was applied to neurons to completely depolarize the entire mitochondrial population and release accumulated Ca2+.
Fig. 2. Immunocytochemical detection of cyclophilin D (CyD) in cultured cortical neurons derived from wild-type C57BL/6 mice (a-d) and cyclophilin D-knockout (Ppif-/-) mice (e-h).

In a and e, phase contrast bright field images of cortical neurons from C57BL/6 and Ppif-/- mice, respectively. In b and f, neurons from wild-type and Ppif -/- mice, respectively, were exposed to mouse anti-CyD antibody and donkey anti-mouse antibody conjugated with Alexa Fluor 488 (Invitrogen). In c and g, mitochondrial staining with MitoTracker Red in neurons from C57BL/6 and Ppif-/- mice, respectively. In d and h, co-localization of CyD and MitoTracker staining in neurons from wild-type and Ppif-/- mice.
Fig. 3. The changes in cytosolic Ca2+ ([Ca2+]c) and mitochondrial membrane potential (Δψ) in response to elevated glutamate (Glu) in WT (a,c) and Ppif-/- (b,d) neurons.

In a-d, the original (thin grey traces) and the averaged fluorescence traces (thick black traces, mean±SEM) from the representative experiments are shown. Cytosolic Ca2+ was followed by monitoring Fura-2FF fluorescence and Δψ was followed by monitoring Rhodamine-123 (Rh123) fluorescence. In these experiments, neurons were exposed to 10μM or to 100μM glutamate plus 10μM glycine as indicated. At the end of the experiments, glutamate and Ca2+ were removed by replacing the bath solution with glutamate- and Ca2+-free solution containing 1mM EGTA. 1μM FCCP was added to neurons as indicated to completely depolarize mitochondria and release Ca2+ accumulated in mitochondria. In a, tDCD is the time from the beginning of glutamate exposure to the completion of the DCD.
In these experiments, both WT and Ppif-/- neurons responded to glutamate with a fast initial jump in [Ca2+]c followed by a decrease to a new, somewhat elevated level (Fig. 3). Soon after that, a secondary slower but greater increase in [Ca2+]c took place in WT neurons indicating the onset of DCD (Fig. 3a). The recovery of [Ca2+]c after the removal of glutamate and Ca2+ was slow in WT neurons, and the release of Ca2+ from mitochondria in response to FCCP-induced mitochondrial depolarization was negligible (Fig. 3a). Simultaneous to the increases in [Ca2+]c we observed two waves of mitochondrial depolarization manifested in the increase of Rh123 fluorescence. Interestingly, at the end of the experiments, FCCP increased the Rh123 signal presumably by depolarizing uninjured mitochondria. This hypothesis is supported by the observations that mitochondria are functionally heterogeneous within the cell (Collins et al., 2002) and that glutamate-treated neurons may contain both damaged and normal, uninjured mitochondria at the same time (Pivovarova et al., 2004). Alternatively, it could be an artifact due to pH change in the cytosol and in the mitochondrial matrix. However, FCCP did not produce significant changes in cytosolic or matrix pH following DCD in cultured neurons exposed to glutamate (Bolshakov et al., 2008), therefore this scenario seems unlikely.
Mitochondria significantly contribute to maintenance of calcium homeostasis in neuronal cells (Herrington et al., 1996;Pivovarova et al., 2004). Mitochondrial damage due to induction of the CyD-mPT might cause depolarization of the organelles precluding further Ca2+ accumulation by mitochondria. Therefore, we hypothesized that protection of mitochondria from the CyD-mPT by genetic ablation of CyD could protect mitochondria, enhance their ability to accumulate Ca2+ and thereby increase resistance of neurons to DCD and secondary mitochondrial depolarization. Indeed, in Ppif-/- neurons lacking CyD, the induction of DCD and secondary mitochondrial depolarization were significantly deferred (Fig. 3b). However, even Ppif-/- neurons eventually experienced DCD and secondary mitochondrial depolarization. This is consistent with the facts that CyD ablation does not provide an absolute protection against Ca2+-induced mitochondrial damage (Fig. 1c) and mPT may occur in a CyD-independent manner (Basso et al., 2005).
In some pathological conditions, the concentration of glutamate in the extracellular milieu might jump up to 100μM within a few minutes of global ischemia, leading to severe neuronal injury and cell death (Benveniste et al., 1984;Nakayama et al., 2002). In our next experiments, we investigated whether the protective effects of CyD ablation could be extended to a greater glutamate challenge. It appeared that both WT and Ppif-/- neurons responded similarly to 100μM glutamate (Fig. 3c,d). Thus, the protective role of CyD ablation was evident only with a moderate glutamate concentration.
In the experiments with calcium and Δψ imaging, different neurons in the same dish responded to glutamate somewhat differently (Fig. 3). This is a typical behavior of individual cultured neurons (Schinder et al., 1996;Nieminen et al., 1996;White and Reynolds, 1996;Khaspekov et al., 1999;Vergun et al., 1999;Almeida and Bolanos, 2001;Alano et al., 2002;Isaev et al., 1996;Castilho et al., 1998;Reynolds, 1999;Chinopoulos et al., 2004;Pivovarova et al., 2004;Manev et al., 1989;Tymianski et al., 1993b;Thayer and Miller, 1990;Budd and Nicholls, 1996). In addition, in our experiments we observed some variations in responses of neurons from different platings. To provide a statistical analysis of the data, we introduced a parameter: the time from the beginning of glutamate exposure to the completion of the DCD (t DCD) (Fig. 3a,b). A similar approach was used previously to analyze secondary mitochondrial depolarization in cultured neurons exposed to glutamate (Vergun et al., 2003). Our statistical analysis confirmed that glutamate triggered DCD more rapidly in WT neurons than in Ppif-/- neurons (Fig. 4). At the same time, tDCD was similar both in WT and in Ppif-/- neurons exposed to 100μM glutamate. There were 8 separate, independent experiments in each group with neurons from five different platings. N indicates total number of cells in each group. Thus, the statistical analysis of data obtained with calcium imaging showed that the protection against DCD evoked by CyD ablation was restricted to a moderate glutamate concentration and was not evident with a greater glutamate challenge.
Fig. 4. Statistical analysis of tDCD obtained with WT and Ppif-/- neurons exposed to moderate (10μM) and high (100μM) glutamate concentrations.

Data are mean±SEM. tDCD was determined by finding the time between the beginning of glutamate exposure and the intersection point of two linear graphs approximating the uprising fragment of the averaged Fura-2FF fluorescence trace and the fragment corresponding to the elevated [Ca2+]c plateau as shown in Fig. 3a. Data are mean±SEM. *p<0.01, t=3.525, in a comparison between WT neurons exposed to 10 and 100μM glutamate; **p<0.001, t=14.35, in a comparison between WT and Ppif-/- neurons exposed to 10μM glutamate; #p<0.001, t=18.25, in a comparison between Ppif neurons exposed to 10 and 100μM glutamate. There were 8 independent experiments. N in the Figure shows the total number of cells examined in these experiments. Statistical analysis of the experimental results consisted of one-way ANOVA followed by Bonferroni's post hoc test (GraphPad Prism® 4.0, GraphPad Software Inc., San Diego, CA).
The lack of protection due to CyD ablation in neurons exposed to high glutamate could be because of a greater Ca2+ influx and faster mitochondrial damage. In our experiments with isolated mitochondria, increasing Ca2+ loading ultimately overcame protection conferred by CsA or by CyD ablation (Fig. 1c-f). To assess the kinetics and the amount of Ca2+ influx into neurons, we measured 45Ca accumulation in the cells. Every experiment was performed in triplicate with neurons from three different platings. Glutamate at a higher concentration (100μM) caused faster 45Ca influx into neurons than a lower concentration of glutamate (10μM) (Fig. 5). Therefore, with higher glutamate mitochondrial damage might occur earlier and the protection against DCD in Ppif-/- neurons could be significantly diminished or completely vanished. In addition, Ppif-/-neurons accumulated 45Ca more rapidly in comparison with WT neurons indicating the lack of decrease in Ca2+ influx into Ppif-/- neurons. Thus, it seemed very unlikely that the deferment in DCD and mitochondrial depolarization in Ppif-/- neurons exposed to moderate glutamate was due to attenuation of the Ca2+ influx into the cells.
Fig. 5. 45Ca accumulation in glutamate-treated WT (thick lines) and Ppif-/- neurons (thin lines).

Neurons were incubated in the standard bath solution supplemented with 5μCi of 45Ca. 45Ca accumulation was measured at 1, 3, and 5 minutes after application of 10μM (open symbols) or 100μM glutamate (filled symbols) as indicated. Non-specific 45Ca binding, measured in the presence of 10μM MK801 (an inhibitor of NMDA receptors), 10μM CNQX (an inhibitor of AMPA receptors), and 5μM nifedipine (a blocker of voltage-gated Ca2+ channels) was subtracted from 45Ca accumulation in the absence of inhibitors. Data are mean±SEM, N=3.
Recently, a complex dynamics of mitochondrial matrix acidification in neurons exposed to glutamate was revealed with pH-sensitive enhanced yellow fluorescent protein targeted to mitochondria (mito-eYFP) (Bolshakov et al., 2008). In these experiments, the second phase of matrix acidification following glutamate application reflected an induction of the mPT pore leading to a drastic increase in ion permeability of the inner mitochondrial membrane and pH equilibration between the cytosol (more acidic) and the mitochondrial matrix (initially more alkaline). In the current study, we used this approach in the experiments with WT and Ppif-/-neurons to demonstrate the relationship between the onset of DCD and the induction of the CyD-mPT. We examined 39-48 individual neurons in each group in 12 separate, independent experiments with cells from six different platings. Figure 6a shows representative bright field (right) and fluorescent (left) images of neurons expressing mito-eYFP. The expression rate was in the range of 10-15%. Figure 6b demonstrates 3D reconstruction of the mitochondrial network in the neuron expressing mito-eYFP. Figures 6c-e illustrate results from the experiments with WT or Ppif-/- neurons exposed to 10μM glutamate (plus 10μM glycine). Statistical analysis of these experiments is shown in Figure 6f. The onset of secondary matrix acidification (SMac) was quantitatively assessed in all tested WT neurons and in those Ppif-/- neurons that experienced SMac within the time course of the experiment. The onset of DCD in WT and Ppif-/- neurons coincided with SMac. (Fig. 6c,d). In the case with WT neurons, the addition of FCCP at the end of the experiment did not produce a significant change in pHm. In contrast to WT neurons, in Ppif-/- neurons SMac and DCD were significantly deferred in 38 (79.2%) or absent in 10 (20.8%) out of 48 neurons examined in 12 separate experiments (Fig. 6d,e). With Ppif-/- neurons that did not experience SMac and DCD, FCCP produced significant matrix acidification due to equilibration of pHm and cytosolic pH accompanied by a strong increase in [Ca2+]c, suggesting a massive release of accumulated Ca2+ from mitochondria given that external Ca2+ was removed. Such a strong FCCP-induced increase in [Ca2+]c was not observed after DCD in WT neurons. In both WT and Ppif-/- neurons, SMac was not observed when neurons were exposed to glutamate in Ca2+-free bath solution (not shown). When neurons were exposed to 100μM glutamate, the onset of SMac occurred simultaneously in WT and Ppif-/- neurons (Fig. 6f). Thus, in neurons exposed to moderate glutamate, mitochondrial damage due to induction of the CyD-mPT substantially contributed to DCD. Suppression of the CyD-mPT induction, and hence preservation of mitochondrial Ca2+ uptake, at least temporarily protected neurons from the collapse of calcium homeostasis leading to postponement of DCD. With a high level of glutamate, mitochondrial damage occurred in a CyD-independent manner presumably due to induction of the CyD-independent mPT.
Fig. 6. The relationship between delayed calcium deregulation and induction of the mitochondrial permeability transition in WT and Ppif-/- neurons.

In a, representative bright field (right) and fluorescent (left) images of cultured hippocampal neurons expressing mito-eYFP. In b, a 3D reconstruction of the mitochondrial network in the neuron expressing mito-eYFP. In c-e, simultaneous recordings of cytosolic Ca2+ with Fura-2FF (thick traces) and matrix pH (pHm) with mito-eYFP (traces with symbols). Glutamate (Glu, 10μM, plus glycine, 10μM) was added as indicated. In f, statistical analysis of tSMac measured with WT and Ppif-/- neurons exposed to moderate (10μM) and high (100μM) glutamate concentrations. tSMac is the time from the beginning of glutamate exposure to the onset of the secondary matrix acidification. Data are mean±SEM. *p<0.01, t=3.575, in a comparison between WT neurons exposed to 10 and 100μM glutamate; **p<0.001, t=37.01, in a comparison between WT and Ppif-/- neurons exposed to 10μM glutamate; #p<0.001, t=38.83, in a comparison between Ppif-/- neurons exposed to 10 and 100μM glutamate. N in the Figure shows the total number of cells examined in these experiments. Statistical analysis of the experimental results consisted of one-way ANOVA followed by Bonferroni's post hoc test (GraphPad Prism® 4.0, GraphPad Software Inc., San Diego, CA).
Excitotoxic cell death in cortical neurons from C57BL/6 and Ppif-/- mice
There is a substantial body of evidence suggesting a causal link between sustained elevation of cytosolic Ca2+ and excitotoxic neuronal death (Tymianski et al., 1993c;Tymianski et al., 1994;Limbrick, Jr. et al., 1995;Brustovetsky et al., 2004). In our next experiments, we addressed the question of whether suppression of the glutamate-triggered, Ca2+-dependent CyD-mPT due to genetic CyD ablation leads to an increase in the survival rate of neurons exposed to glutamate. Glutamate-induced cell death was evaluated following propidium iodide (PI) nuclear staining (Pivovarova et al., 2004). This method has been chosen because it detects necrosis, which significantly contributes to neuronal death following prolonged glutamate exposure (Ankarcrona et al., 1995). However, there is a possibility that in Ppif-/- neurons necrosis might be substituted for another type of cell death, apoptosis. To examine this possibility, we double stained neurons with PI, a marker of necrosis, and Annexin V, a marker of the early stage of apoptosis. 10 minute exposure of neurons from WT or Ppif-/- mice to 10μM glutamate (plus 10μM glycine) 24 hours later caused significant cell death in WT neurons but not in Ppif-/- neurons (Fig. 7). Thus, CyD ablation resulted in remarkable protection against glutamate toxicity. With both WT and Ppif-/-neurons, Annexin V staining was negligible. As a positive control for Annexin V staining, we used Ppif-/- neurons treated for 24 hours with 30μM cisplatin (Cis), an agent that induces apoptosis. Treatment of neurons with cisplatin caused strong Annexin V staining, indicating an induction of apoptosis (Fig. 7). Thus, protection of Ppif-/- neurons from glutamate-triggered necrotic cell death was not accompanied by increased apoptosis.
Fig. 7. An evaluation of necrotic cell death and the induction of apoptosis with propidium iodide (PI) and Annexin V staining after exposure of WT and Ppif-/- neurons to glutamate or cisplatin.
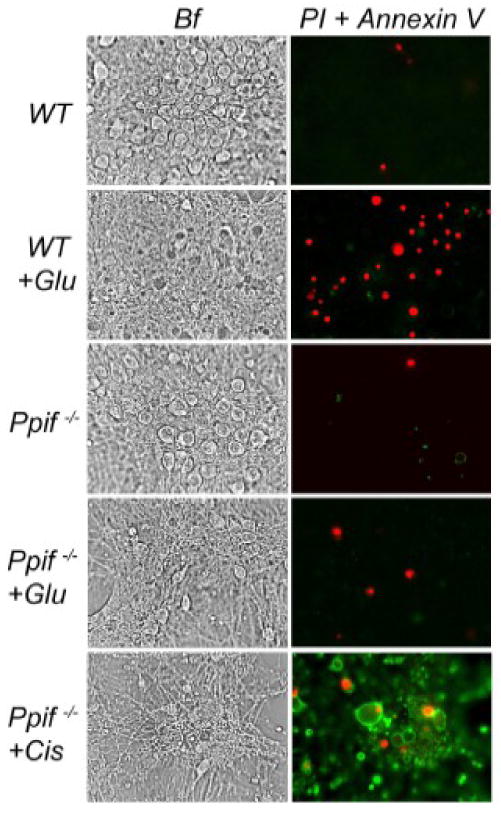
Neurons were exposed for 10 minutes to 10μM glutamate plus 10μM glycine (+Glu) as indicated. Then, glutamate was removed and necrotic cell death or induction of apoptosis was evaluated 24 hours later following the appearance of staining with PI or Annexin V. Ppif-/- neurons treated with cisplatin (+Cis, 30μM, 24 hours) were used as a positive control for Annexin V staining. Bf, phase contrast bright field. These experiments with similar results were performed in triplicate with neurons from three different platings.
In addition, neuronal death was quantitatively assessed across the range of glutamate concentrations (3-300μM) by the Trypan Blue exclusion method (Dubinsky and Rothman, 1991). There were 9 independent experiments with neurons from nine different platings. In each experiment, cells were counted three times in randomly chosen fields and the averages we used for statistical analysis. In these experiments, neurons were exposed to various concentrations of glutamate for 10 minutes. At this time, all WT neurons treated with 10μM glutamate experienced DCD and reached the final elevated [Ca2+]c plateau, whereas Ppif-/- neurons still had a relatively low level of [Ca2+]c (Fig. 3a,b). On the other hand, after 10 minutes of exposure to 100μM glutamate, both Ppif-/- and WT neurons had completed DCD (Fig. 3c,d). Accordingly, the glutamate toxicity experiments revealed greater survival of Ppif-/- neurons than WT neurons in the range of 10μM glutamate, while at higher glutamate concentrations the survival rate of Ppif-/- and WT neurons appeared to be similar (Fig. 8). Thus, the protective effect of CyD ablation against excitotoxic neuronal death was found to be significant at moderate but pathophysiologically-relevant concentrations of glutamate, while at higher glutamate concentrations the protection was not evident.
Fig. 8. Dose-dependence of glutamate-induced neuronal death.

Trypan Blue exclusion method was used to evaluate neuronal death (Dubinsky and Rothman, 1991). Neurons derived from wild-type (WT) C57BL/6 and Ppif-/- mice were exposed to various glutamate concentrations (3-300μM) for 10 minutes, then glutamate was removed and cell death was evaluated after 24 hours by counting Trypan Blue stained neurons in a blind manner. Data are mean±SEM. *p<0.001, t=12.12, in a comparison between WT and Ppif-/- neurons exposed to 10μM glutamate. N=9.
Discussion
In this study we demonstrated for the first time the key role of CyD-mPT in DCD and excitotoxic neuronal death using cultured neurons derived from CyD-knockout Ppif-/- mice. In contrast to pharmacological inhibitors which may have off-target effects, genetic ablation of CyD is free of such confounding complications, and therefore represents a more advantageous model for studying the role of the CyD-mPT in glutamate excitotoxicity and other pathologies.
The main finding of our study is that genetic ablation of CyD, which leads to suppression of the CyD-mPT, improves handling of calcium homeostasis and preserves mitochondrial membrane potential in cultured cortical neurons exposed to pathophysiologically-relevant glutamate concentration. Most importantly, CyD ablation increases survival rate of neurons exposed to moderate glutamate, and thus unequivocally demonstrates the important role of the CyD-mPT in glutamate excitotoxicity. However, while suppression of the CyD-mPT due to CyD ablation defers the collapse of calcium homeostasis and improves survival rate of neurons, this genetic manipulation fails to completely protect neurons against glutamate, emphasizing the potential multiplicity of mechanisms contributing to neuronal injury.
Early studies suggested an important role for the mPT in the disturbance of calcium homeostasis and neuronal death in the experimental model of traumatic brain injury (Sullivan et al., 1999;Scheff and Sullivan, 1999;Sullivan et al., 2000;Okonkwo et al., 1999;Sullivan et al., 2005;McEwen et al., 2007;Mbye et al., 2008), in oxygen/glucose deprivation (OGD) and in neurons exposed to excitotoxic glutamate (Schinder et al., 1996;Nieminen et al., 1996;White and Reynolds, 1996;Almeida and Bolanos, 2001;Alano et al., 2002;Khaspekov et al., 1999;Vergun et al., 1999). In these studies, CsA appeared to be neuroprotective, linking DCD and excitotoxic neuronal injury to induction of the mPT. However, in other studies, pharmacological inhibition of CyD appeared to be futile (Isaev et al., 1996;Castilho et al., 1998;Reynolds, 1999;Chinopoulos et al., 2004;Pivovarova et al., 2004). Since then, the lack of CsA protection against glutamate was used as the main argument against mPT involvement in DCD and excitotoxic neuronal death. The contradictory results obtained in early studies with CsA could be explained by the fact that CsA binds to both CyD and cytosolic cyclophilin A, and the latter leads to inhibition of calcineurin, which has numerous targets in the cell (Liu et al., 1991;Yakel, 1997). N-methyl-valine-4-cyclosporin A (MetVal4-cyclosporin), a CsA derivative that does not inhibit calcineurin (Zenke et al., 1993) but potently suppresses the mPT (Friberg and Wieloch, 2002), has been used in several studies to distinguish between inhibition of calcineurin and suppression of the mPT (Vergun et al., 1999;Alano et al., 2002;Khodorov, 2004;Castilho et al., 1998). Yet, even with MetVal4-cyclosporin, the results appeared to be rather controversial. Some investigators reported that MetVal4-cyclosporin is neuroprotective (Vergun et al., 1999;Alano et al., 2002;Khodorov, 2004) while others did not observe protection with this pharmacological agent (Castilho et al., 1998). The reason for this discrepancy is not quite clear. A possible explanation lies in the fact that CsA protection against the mPT is highly variable and strongly depends on experimental conditions. It significantly diminishes or completely vanishes following an increase in the magnitude of Ca2+ loading, mitochondrial depolarization, increased duration of the insult, or interaction of mitochondria with some agents such as free fatty acids (FFA) (Bernardi et al., 1993;Bernardi et al., 1992;Broekemeier and Pfeiffer, 1989;Broekemeier and Pfeiffer, 1995;Brustovetsky and Dubinsky, 2000).
Consistent with this notion, the protection of neurons against glutamate due to the inhibition of the CyD-mPT achieved by genetic CyD ablation depended on the severity of the glutamate insult. This fact was not established, and therefore was not appreciated in early studies with pharmacological inhibitors of CyD. We found in our experiments that excessive Ca2+ influx into neurons via activated glutamate receptors readily overrode protection imposed by inhibition of the CyD-mPT achieved by genetic CyD ablation. While neurons from Ppif-/-mice were more resistant to DCD following exposure to 10μM glutamate, with 100μM glutamate the difference between Ppif-/- and WT neurons became negligible. Importantly, cultured neurons exposed to 100μM glutamate accumulated 45Ca faster than neurons exposed to 10μM glutamate. It is conceivable that the faster delivery of Ca2+ to mitochondria and, correspondingly faster Ca2+ loading into mitochondria, might more rapidly trigger an induction of the mPT. The greater Ca2+ influx into neurons exposed to higher glutamate could also rapidly induce the CyD-independent mPT, which damages mitochondria due to dramatic permeabilization of the IMM regardless of the level of CyD expression. This might be accountable for the similar responses of WT and Ppif-/- neurons to high glutamate. The rapid and excessive Ca2+ influx into neurons could also be one of the reasons why CsA or its derivatives were not effective in the early experiments with high glutamate concentrations (Isaev et al., 1996;Castilho et al., 1998;Reynolds, 1999;Chinopoulos et al., 2004;Pivovarova et al., 2004).
It has been well established in early studies that an induction of the mPT pore in isolated mitochondria can cause a loss of pyridine nucleotides (Vinogradov et al., 1972;Di et al., 2001). This, in turn, can cause an inhibition of mitochondrial respiration that could be restored either by addition of NAD+ (Fontaine et al., 1998) or by addition of succinate (Brustovetsky et al., 2002;Brustovetsky et al., 2003). However, the inhibition of mitochondrial respiration due to loss of pyridine nucleotides primarily happens if mitochondria are fueled by Complex I-linked substrates exclusively. This seems quite unlikely in in situ conditions when in addition to Complex I-linked substrates mitochondria are also fueled by succinate. Nevertheless, the loss of pyridine nucleotides via the mPT pore might decrease reserve respiratory capacity of mitochondria thus contributing to mitochondrial dysfunction.
In contrast to the experiments with isolated mitochondria, identifying and assessing the role of the CyD-mPT in the experiments with cultured neurons appeared to be a daunting task. In addition to the use of pharmacological inhibitors of the CyD-mPT, some investigators employed alternative approaches in attempt to better understand the role of the CyD-mPT in disturbances of calcium homeostasis and excitotoxicity. Recently, Kushnareva et al. found increased amounts of Ca2+ accumulated in mitochondria isolated from cultured cortical neurons exposed to excitotoxic glutamate (Kushnareva et al., 2005). This was interpreted as strong evidence against involvement of the mPT in glutamate-triggered DCD, since the opening of the mPT pore should cause a release of previously accumulated Ca2+ (Bernardi and Petronilli, 1996). However, in another elegant study, Pivovarova et al., using electron probe X-ray microanalysis, observed significant calcium precipitates in swollen mitochondria within glutamate-treated cultured hippocampal neurons (Pivovarova et al., 2004). The swelling of mitochondria was attributed to the induction of the mPT, and it was proposed that, probably due to poor solubility (Chalmers and Nicholls, 2003), calcium precipitates remain in mitochondria even after the opening of the mPT pore (Pivovarova et al., 2004). Recently, this point of view received additional experimental support in experiments with isolated brain mitochondria (Kristian et al., 2007). Thus, an increased amount of Ca2+ in mitochondria from glutamate-treated neurons does not necessarily indicate a lack of the mPT, and therefore obviously cannot serve as evidence against mPT induction in neurons exposed to excitotoxic glutamate.
The advent of CyD-knockout Ppif-/- mice, which were used in the present study, permitted us to address the role of the CyD-mPT in DCD, secondary mitochondrial depolarization, and glutamate excitotoxicity without the use of pharmacological agents. The total elimination of CyD in neuronal mitochondria of Ppif-/- mice ensured the maximal suppression of the CyD-dependent mPT, which cannot be exceeded by any existing or future pharmacological inhibitors of CyD. The experiments performed in this study provided a deeper insight into the mechanisms of glutamate-triggered DCD and excitotoxicity and demonstrated that the use of neurons from Ppif-/- mice represents a new, valuable approach in examining the role of the CyD-mPT in pathophysiological processes including DCD and glutamate excitotoxicity.
Acknowledgments
We are thankful to Dr. Jeffery D. Molkentin (University of Cincinnati) and Cincinnati Children's Research Foundation for providing us with cyclophilin D-knockout mice. Dr. Natalia Shalbuyeva is gratefully acknowledged for help with mitochondrial experiments. This work was supported by the NIH/NINDS R01 NS 050131 to NB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Alano CC, Beutner G, Dirksen RT, Gross RA, Sheu SS. Mitochondrial permeability transition and calcium dynamics in striatal neurons upon intense NMDA receptor activation. J Neurochem. 2002;80:531–538. doi: 10.1046/j.0022-3042.2001.00738.x. [DOI] [PubMed] [Google Scholar]
- Almeida A, Bolanos JP. A transient inhibition of mitochondrial ATP synthesis by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J Neurochem. 2001;77:676–690. doi: 10.1046/j.1471-4159.2001.00276.x. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A, Diemer NH. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem. 1984;43:1369–1374. doi: 10.1111/j.1471-4159.1984.tb05396.x. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabo I, Zoratti M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem. 1992;267:2934–2939. [PubMed] [Google Scholar]
- Bernardi P, Veronese P, Petronilli V. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. I. Evidence for two separate Me2+ binding sites with opposing effects on the pore open probability. J Biol Chem. 1993;268:1005–1010. [PubMed] [Google Scholar]
- Bolshakov AP, Mikhailova MM, Szabadkai G, Pinelis VG, Brustovetsky N, Rizzuto R, Khodorov BI. Measurements of mitochondrial pH in cultured cortical neurons clarify contribution of mitochondrial pore to the mechanism of glutamate-induced delayed Ca(2+) deregulation. Cell Calcium. 2008;43:602–614. doi: 10.1016/j.ceca.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Broekemeier KM, Pfeiffer DR. Cyclosporin A-sensitive and insensitive mechanisms produce the permeability transition in mitochondria. Biochem Biophys Res Commun. 1989;163:561–566. doi: 10.1016/0006-291x(89)92174-8. [DOI] [PubMed] [Google Scholar]
- Broekemeier KM, Pfeiffer DR. Inhibition of the mitochondrial permeability transition by cyclosporin A during long time frame experiments: relationship between pore opening and the activity of mitochondrial phospholipases. Biochemistry. 1995;34:16440–16449. doi: 10.1021/bi00050a027. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem. 2002;80:207–218. doi: 10.1046/j.0022-3042.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Purl KJ, Capano M, Crompton M, Dubinsky JM. Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J Neurosci. 2003;23:4858–4867. doi: 10.1523/JNEUROSCI.23-12-04858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, Dubinsky JM. Limitations of cyclosporin A inhibition of the permeability transition in CNS mitochondria. J Neurosci. 2000;20:8229–8237. doi: 10.1523/JNEUROSCI.20-22-08229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky T, Purl K, Young A, Shimizu K, Dubinsky JM. Dearth of glutamate transporters contributes to striatal excitotoxicity. Exp Neurol. 2004;189:222–230. doi: 10.1016/j.expneurol.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Castilho RF, Hansson O, Ward MW, Budd SL, Nicholls DG. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 1998;18:10277–10286. doi: 10.1523/JNEUROSCI.18-24-10277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo J, Davalos A, Naveiro J, Noya M. Neuroexcitatory amino acids and their relation to infarct size and neurological deficit in ischemic stroke. Stroke. 1996;27:1060–1065. doi: 10.1161/01.str.27.6.1060. [DOI] [PubMed] [Google Scholar]
- Castillo J, Davalos A, Noya M. Progression of ischaemic stroke and excitotoxic aminoacids. Lancet. 1997;349:79–83. doi: 10.1016/S0140-6736(96)04453-4. [DOI] [PubMed] [Google Scholar]
- Chalmers S, Nicholls DG. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem. 2003;278:19062–19070. doi: 10.1074/jbc.M212661200. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Gerencser AA, Doczi J, Fiskum G, Adam-Vizi V. Inhibition of glutamate-induced delayed calcium deregulation by 2-APB and La3+ in cultured cortical neurones. J Neurochem. 2004;91:471–483. doi: 10.1111/j.1471-4159.2004.02732.x. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- Di LF, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. Journal of Neuroscience. 1993;13:623–631. doi: 10.1523/JNEUROSCI.13-02-00623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky JM, Kristal BS, Elizondo-Fournier M. An obligate role for oxygen in the early stages of glutamate-induced, delayed neuronal death. J Neurosci. 1995;15:7071–7078. doi: 10.1523/JNEUROSCI.15-11-07071.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky JM, Rothman SM. Intracellular calcium concentrations during “chemical hypoxia” and excitotoxic neuronal injury. Journal of Neuroscience. 1991;11:2545–2551. doi: 10.1523/JNEUROSCI.11-08-02545.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine E, Eriksson O, Ichas F, Bernardi P. Regulation of the permeability transition pore in skeletal muscle mitochondria. Modulation By electron flow through the respiratory chain complex i. J Biol Chem. 1998;273:12662–12668. doi: 10.1074/jbc.273.20.12662. [DOI] [PubMed] [Google Scholar]
- Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, Yu X, Fowlkes J, Rahder M, Stem K, Bernardi P, Bourdette D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc Natl Acad Sci U S A. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friberg H, Wieloch T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie. 2002;84:241–250. doi: 10.1016/s0300-9084(02)01381-0. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Isaev NK, Zorov DB, Stelmashook EV, Uzbekov RE, Kozhemyakin MB, Victorov IV. Neurotoxic glutamate treatment of cultured cerebellar granule cells induces Ca2+ -dependent collapse of mitochondrial membrane potential and ultrastructural alterations of mitochondria. FEBS Lett. 1996;392:143–147. doi: 10.1016/0014-5793(96)00804-6. [DOI] [PubMed] [Google Scholar]
- Khaspekov L, Friberg H, Halestrap A, Viktorov I, Wieloch T. Cyclosporin A and its nonimmunosuppressive analogue N-Me-Val-4-cyclosporin A mitigate glucose/oxygen deprivation-induced damage to rat cultured hippocampal neurons. Eur J Neurosci. 1999;11:3194–3198. doi: 10.1046/j.1460-9568.1999.00743.x. [DOI] [PubMed] [Google Scholar]
- Khodorov B. Glutamate-induced deregulation of calcium homeostasis and mitochondrial dysfunction in mammalian central neurones. Prog Biophys Mol Biol. 2004;86:279–351. doi: 10.1016/j.pbiomolbio.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L, Costa E. Glutamate-induced destabilization of intracellular calcium concentration homeostasis in cultured cerebellar granule cells: role of mitochondria in calcium buffering. Mol Pharmacol. 1995;47:140–147. [PubMed] [Google Scholar]
- Korde AS, Pettigrew LC, Craddock SD, Pocernich CB, Waldmeier PC, Maragos WF. Protective effects of NIM811 in transient focal cerebral ischemia suggest involvement of the mitochondrial permeability transition. J Neurotrauma. 2007;24:895–908. doi: 10.1089/neu.2006.0122. [DOI] [PubMed] [Google Scholar]
- Kristian T, Pivovarova NB, Fiskum G, Andrews SB. Calcium-induced precipitate formation in brain mitochondria: composition, calcium capacity, and retention. J Neurochem. 2007;102:1346–1356. doi: 10.1111/j.1471-4159.2007.04626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnareva YE, Wiley SE, Ward MW, Andreyev AY, Murphy AN. Excitotoxic injury to mitochondria isolated from cultured neurons. J Biol Chem. 2005;280:28894–28902. doi: 10.1074/jbc.M503090200. [DOI] [PubMed] [Google Scholar]
- Limbrick DD, Jr, Churn SB, Sombati S, DeLorenzo RJ. Inability to restore resting intracellular calcium levels as an early indicator of delayed neuronal cell death. Brain Res. 1995;690:145–156. doi: 10.1016/0006-8993(95)00552-2. [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Manev H, Favaron M, Guidotti A, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989;36:106–112. [PubMed] [Google Scholar]
- Mbye LH, Singh IN, Sullivan PG, Springer JE, Hall ED. Attenuation of acute mitochondrial dysfunction after traumatic brain injury in mice by NIM811, a non-immunosuppressive cyclosporin A analog. Exp Neurol. 2008;209:243–253. doi: 10.1016/j.expneurol.2007.09.025. [DOI] [PubMed] [Google Scholar]
- McEwen ML, Sullivan PG, Springer JE. Pretreatment with the cyclosporin derivative, NIM811, improves the function of synaptic mitochondria following spinal cord contusion in rats. J Neurotrauma. 2007;24:613–624. doi: 10.1089/neu.2006.9969. [DOI] [PubMed] [Google Scholar]
- Naga KK, Sullivan PG, Geddes JW. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J Neurosci. 2007;27:7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Nakayama R, Yano T, Ushijima K, Abe E, Terasaki H. Effects of dantrolene on extracellular glutamate concentration and neuronal death in the rat hippocampal CA1 region subjected to transient ischemia. Anesthesiology. 2002;96:705–710. doi: 10.1097/00000542-200203000-00029. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal glutamate excitotoxicity. Biochim Biophys Acta. 1998;1366:97–112. doi: 10.1016/s0005-2728(98)00123-6. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-D-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- Okonkwo DO, Buki A, Siman R, Povlishock JT. Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport. 1999;10:353–358. doi: 10.1097/00001756-199902050-00026. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci. 2004;24:5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Bernardi P. The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis. 2007;12:815–833. doi: 10.1007/s10495-007-0723-y. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ. Mitochondrial membrane potential and the permeability transition in excitotoxicity. Ann N Y Acad Sci. 1999;893:33–41. doi: 10.1111/j.1749-6632.1999.tb07816.x. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. J Neurotrauma. 1999;16:783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalbuyeva N, Brustovetsky T, Bolshakov A, Brustovetsky N. Calcium-dependent spontaneously reversible remodeling of brain mitochondria. J Biol Chem. 2006;281:37547–37558. doi: 10.1074/jbc.M607263200. [DOI] [PubMed] [Google Scholar]
- Shalbuyeva N, Brustovetsky T, Brustovetsky N. Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J Biol Chem. 2007;282:18057–18068. doi: 10.1074/jbc.M702134200. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? J Neurosci Res. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson M, Scheff SW. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Exp Neurol. 2000;161:631–637. doi: 10.1006/exnr.1999.7282. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J Physiol. 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993a;607:319–323. doi: 10.1016/0006-8993(93)91523-u. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993b;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Properties of neuroprotective cell-permeant Ca2+ chelators: effects on [Ca2+]i and glutamate neurotoxicity in vitro. J Neurophysiol. 1994;72:1973–1992. doi: 10.1152/jn.1994.72.4.1973. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Wallace MC, Spigelman I, Uno M, Carlen PL, Tator CH, Charlton MP. Cell-permeant Ca2+ chelators reduce early excitotoxic and ischemic neuronal injury in vitro and in vivo. Neuron. 1993c;11:221–235. doi: 10.1016/0896-6273(93)90180-y. [DOI] [PubMed] [Google Scholar]
- Van Hemelrijck A, Sarre S, Smolders I, Michotte Y. Determination of amino acids associated with cerebral ischaemia in rat brain microdialysates using narrowbore liquid chromatography and fluorescence detection. J Neurosci Methods. 2005;144:63–71. doi: 10.1016/j.jneumeth.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Vergun O, Han YY, Reynolds IJ. Glucose deprivation produces a prolonged increase in sensitivity to glutamate in cultured rat cortical neurons. Exp Neurol. 2003;183:682–694. doi: 10.1016/s0014-4886(03)00243-7. [DOI] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol. 1999;519(Pt 2):451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradov A, Scarpa A, Chance B. Calcium and pyridine nucleotide interaction in mitochondrial membranes. Arch Biochem Biophys. 1972;152:646–654. doi: 10.1016/0003-9861(72)90261-5. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Thayer SA. Sequestration of glutamate-induced Ca2+ loads by mitochondria in cultured rat hippocampal neurons. J Neurophysiol. 1996;76:1611–1621. doi: 10.1152/jn.1996.76.3.1611. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. J Physiol. 1997;498(Pt 1):31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Tomizawa K, Oda Y, Wei FY, Lu YF, Matsushita M, Li ST, Moriwaki A, Matsui H. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–4940. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. [DOI] [PubMed] [Google Scholar]
- Yakel JL. Calcineurin regulation of synaptic function: from ion channels to transmitter release and gene transcription. Trends Pharmacol Sci. 1997;18:124–134. doi: 10.1016/s0165-6147(97)01046-8. [DOI] [PubMed] [Google Scholar]
- Zenke G, Baumann G, Wenger R, Hiestand P, Quesniaux V, Andersen E, Schreier MH. Molecular mechanisms of immunosuppression by cyclosporins. Ann N Y Acad Sci. 1993;685:330–335. doi: 10.1111/j.1749-6632.1993.tb35882.x. [DOI] [PubMed] [Google Scholar]