

Abstract
In 2004 we proposed the mitochondrial cascade hypothesis of sporadic Alzheimer’s disease (AD). Our hypothesis assumed sporadic and autosomal dominant AD are not etiologically homogeneous, considered evidence that AD pathology is not brain-limited, and incorporated aging theory. The mitochondrial cascade hypothesis asserted: (1) inheritance determines mitochondrial baseline function and durability; (2) mitochondrial durability influences how mitochondria change with age; and (3) when mitochondrial change reaches a threshold, AD histopathology and symptoms ensue. We now review the reasoning used to formulate the hypothesis, discuss pertinent interim data, and update its tenants. Readers are invited to consider the conceptual strengths and weaknesses of this hypothesis.
Introduction
The amyloid cascade hypothesis has dominated the Alzheimer’s disease (AD) research field since 1992 (Hardy and Higgins, 1992). It followed the discovery that amyloid precursor protein (APP) gene mutation could produce dementia with neuritic plaques and neurofibrillary tangles (Goate et al., 1991). The amyloid cascade hypothesis was subsequently bolstered by work showing presenilin (PS) 1 and 2 mutations also cause AD, and that APP, PS1, and PS2 mutation all favor processing of APP to its 42 amino acid beta amyloid (Aβ) derivative (Levy-Lahad et al., 1995; Sherrington et al., 1995; Scheuner et al., 1996). The hypothesis has evolved over time, with the suspect Aβ42 bogey shifting from fibrils to protofibrils to oligomers to perhaps dimers (Haass and Steiner, 2001; Hardy and Selkoe, 2002; Lesne et al., 2006; Shankar et al., 2008). The core assumption, though, that Aβ42 induces tangles, neurodegeneration, and dementia has not changed. The amyloid cascade hypothesis argues forced Aβ production in transgenic animals should faithfully model the disease and Aβ reduction should arrest it. Much of the AD research field has invested heavily in AD transgenic modeling and Aβ-directed therapeutic development and consequently has a vested interest in the amyloid cascade hypothesis.
While the simplicity of the amyloid cascade hypothesis is in many ways appealing, it does oversimplify some controversial issues. First and foremost, it presumes brain aging and AD are uniformly divergent events. We believe, however, that sporadic AD and brain aging are convergent events (Swerdlow, 2007a; 2007b). To emphasize this we previously proposed the mitochondrial cascade hypothesis, which considers sporadic AD from the perspective of mitochondrial aging theory, our own cytoplasmic hybrid (cybrid) studies of AD mitochondria, basic cell biology principles, and the amyloid cascade hypothesis itself(Swerdlow and Khan, 2004). Since we first proposed this hypothesis in 2004 pertinent data have been published. We now review the conceptual basis of our hypothesis and discuss it from the context of recent AD research.
What is the Mitochondrial Cascade Hypothesis of Sporadic AD?
The mitochondrial cascade hypothesis asserts inheritance determines mitochondrial baseline function and durability, mitochondrial durability influences how mitochondria change with age, and AD histopathology and symptoms ensue when mitochondrial change reaches a threshold (Swerdlow and Khan, 2004). It presumes autosomal dominant and sporadic AD are not etiologically homogeneous. It is intend ed to apply only to sporadic AD, but makes predictions about autosomal dominant AD. Mitochondrial dysfunction is seen as a nexus between the sporadic and autosomal dominant forms. In the autosomal dominant forms excessive Aβ impairs mitochondrial function, and it is predicted Aβ-induced mitochondrial dysfunction initiates other AD-characteristic histopathologies. In the sporadic form, age-related mitochondrial changes drive mitochondrial function beyond a functional threshold that induces AD-characteristic histopathologies, including processing of APP to Aβ (Figure 1). Before discussing the specifics of the hypothesis it is necessary to briefly discuss mitochondrial function in AD.
Figure 1.
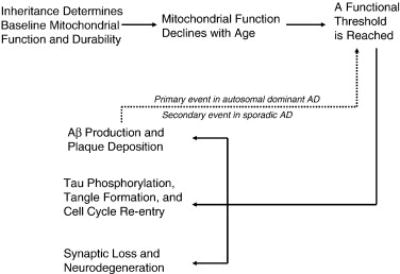
The mitochondrial cascade hypothesis.
Mitochondrial Function in AD
Several mitochondrial enzyme activities are altered in AD. Pyruvate dehydrogenase complex, ketoglutarate dehydrogenase complex, and cytochrome oxidase (CO) Vmax activities are all reduced (Swerdlow and Kish, 2002). The CO defect, in particular, is central to the mitochondrial cascade hypothesis. CO constitutes the terminal end of the mitochondrial electron transport chain (ETC). It accepts electrons from cytochrome c, which receives them from more up stream parts of the E TC. CO passes its acquired electrons to oxygen so that H20 rather than reactive oxygen species (ROS) is generated. CO is the single greatest site of cell oxygen consumption.
The AD CO Vmax activity reduction was first shown in platelets in 1990 and brain in 1992 (Parker et al., 1990; Kish et al., 1992). Anatomic distribution of the CO defect was initially debated; it is now accepted it occurs at least in platelets, fibroblasts, and large parts of the brain (Swerdlow and Kish, 2002). Reports using different experimental methods suggest CO activity in AD subjects is 10–50% less than in age-matched controls. In general, studies that isolate mitochondria report bigger reductions than studies that do not. CO reduction occurs at all stages of the disease, including mild cognitive impairment (MCI) (Valla et al., 2006).
Because CO activity is reduced outside the brain it presumably is not a neurodegeneration artifact. Whether the Vmax reduction reflects reduced CO enzyme or a structurally altered enzyme is still debated. The Vmax activity rate is reduced in tissue homogenates normalized to total cell protein, in highly purified mitochondrial fractions in which the activity rate is normalized to total mitochondrial protein, and when normalized to activity of the mitochondrial enzyme citrate synthase (Swerdlow and Kish, 2002). Spectral analysis of the enzyme finds it is kinetically altered in AD and lacks one of its two substrate binding sites (Parker and Parks, 1995). Studies report AD brains have increased CO immunochemical staining and CO gene expression (Hirai et al., 2001; Manczak et al. 2004). All this supports the view CO activity is reduced in AD because the enzyme is structurally different than it is in controls. Other studies, though, report CO protein levels and mRNA levels for several CO genes are reduced in AD and support the possibility CO activity is reduced because there is less CO (Chandrasekaran et al., 1994; Kish et al., 1999). Perhaps both scenarios occur. It is important to note as far as bioenergetics goes this debate does not stop with CO. Some studies suggest mitochondrial DNA (mtDNA) and mitochondrial number increase in AD, while others suggest mtDNA and mitochondrial number decline (de la Monte et al., 2000; Hirai et al., 2001; Baloyannis, 2006).
The CO enzyme complex contains 13 protein subunits. Ten are encoded by nuclear and three by mtDNA genes. Cybrid studies suggest mtDNA is at least partly responsible for reduced CO activity in AD (Swerdlow et al., 1997; Swerdlow, 2007c). Cybrids are cell lines created by placing mitochondria from individual human subjects into cultured cells (King and Attardi, 1989). Cybrids are also useful for assessing the integrity of an individual’s mitochondrial genes, because when the individual’s mitochondria are transferred the genes inside those mitochondria are also transferred. The mitochondria moved into the cultured cells are grown inside the cells until there are enough to study. Cybrid studies show mitochondria obtained from platelets of persons with AD have reduced CO activity (Swerdlow et al., 2007c). This specific biochemical defect persists over time in the cybrid lines, which supports the view mtDNA differs between AD and control subjects and that these differences can affect CO. Although some studies find specific sequence-level differences between AD and control subject brain and platelet mtDNA, it is not clear such differences also occur in cybrids (Corral-Debrinski et al., 1994; Mecocci et al., 1994; Coskun et al., 2004; Coon et al., 2006). It is still not known how exactly mtDNA from AD subjects differs from that of controls. Regardless, considerable data suggest maternally inherited genes could influence AD phenomena (discussed below).
Does Inheritance Influence AD Risk by Influencing Mitochondrial Function?
Data from epidemiologic, neuropsychological, biomarker, and cell biology studies suggest mitochondrial inheritance could influence AD risk and pathology. One study reported mtDNA haplogroups influence AD risk (van der Walt et al., 2004). Another study found for AD subjects with a demented parent, the demented parent was more often the mother (Edland et al., 1996). This relationship persisted after correcting for greater female longevity, and implies having an AD mother confers a greater risk of AD than having an AD father. A large analysis of Framingham study subject offspring (the Framingham Offspring Study) found neuropsychological test performance in non-demented, middle aged individuals with an AD- affected mother was deficient relative to those with an AD-affected father or no AD-affected parent (Wolf et al., 2005). This may or may not reflect a presymptomatic AD carriage state. Positron emission tomography (PET) studies are relevant to this question. PET quantifies brain glucose utilization and reports this as a “cerebral metabolic rate of glucose” (CMRglu). AD subjects have reduced CMRglu in particular brain regions as do cognitively intact persons with an APOE4 allele (Small et al., 1995; Reiman et al., 1996). Cognitively intact, middle aged individuals with AD mothers but not AD fathers also have AD-like patterns of CMRglu reduction (Mosconi et al., 2007).
It was also previously reported cybrid cell lines containing mtDNA from individuals with AD mothers had lower CO activity than cybrids containing mtDNA from individuals with AD fathers (Davis et al., 1997). Taken together, epidemiologic (maternal inheritance bias), neuropsychological (differences in cognitive testing between children of AD-affected mothers and fathers), biomarker (PET differences in CMRglu between children of AD mothers and fathers), and cell biology (perpetuation of AD pathology in cybrid cell lines, as well as a maternal inheritance effect on cybrid CO activity) data suggest mitochondrial inheritance could influence AD risk and pathology.
Nuclear DNA inheritance may also influence AD risk through effects on mitochondrial function. Under in vitro conditions different apolipoprotein E isoforms differentially affect mitochondrial function (Chang et al., 2005). Single nucleotide polymorphisms (SNP) in the gene for TFAM, a protein required for mtDNA replication and expression, and TOMM40, a translocase of the outer mitochondrial membrane protein, may associate with AD (Gunther et al., 2004; Belin et al., 2007; Alvarez et al., 2008; Bekris et al., 2008). We recently concluded a study in which SNP frequencies for the 13 CO genes was determined. Interim data are shown in Figure 2 (Swerdlow et al., 2006). We found non-synonymous nuclear CO gene SNPs in greater than 10% of the general population. CO gene nuclear SNPs occurred in combination with non-synonymous mtDNA CO gene SNPs, which were found in 20% of the general population. Nuclear CO gene untranslated region (UTR) polymorphisms were extremely common. The functional consequences of CO nuclear polymorphisms and CO nuclear DNA-mtDNA compound polymorphisms are currently under study in our laboratory.
Figure 2.
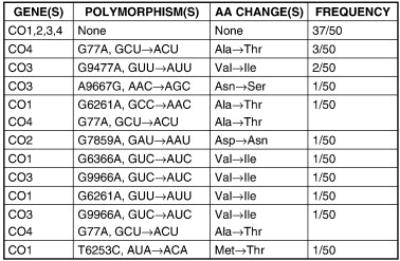
Non-synonymous open reading frame polymorphisms in 50 individuals who under went CO1, CO2, CO3, and CO4 gene sequencing. The listed polymorphisms from the four genes analyzed define 11 distinct CO enzyme complexes among the 50 individuals. Synonymous polymorphisms, untranslated region (UTR) polymorphisms, and variation in the other 9 CO genes are not listed. The nucleotide numbers for CO1, CO2, and CO3 are from the Cambridge mtDNA sequence. The CO4 polymorphism occurs at nucleotide 77 of the mRNA. AA=amino acid.
It is already known dramatic reductions in CO activity are functionally devastating (Prick et al., 1983; Flierl et al., 1997; Cooper and Brown, 2008). The question as far as AD goes is whether CO activity variation within the “normal” range has clinical relevance. We postulate if it does, inheritance-determined function of an individual’s CO could influence AD risk. Both maternal and paternal inheritance would play important roles, but maternal inheritance would play a greater role because of the mtDNA contribution. Interestingly, a secondary analysis of our AD cybrid data found the overall CO activity reduction we observed was mostly driven by cybrids containing mtDNA from younger AD subjects (Figure 3) (Swerdlow, 2007b). This suggests the lower an individual’s inherent CO activity is, the younger the age at which the individual reaches the dementia threshold and is diagnosed with AD. To summarize this section, several lines of evidence support the view inheritance influences mitochondrial function and mitochondria-relevant gene inheritance could influence AD risk.
Figure 3.
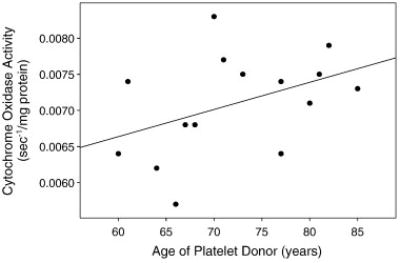
Cytochrome oxidase activities from15 cyb rid cell lines prepared by transferring AD subject platelet mitochondria to NT2 cells depleted of endogenous mtDNA. Enzyme activities are plotted as a function of the platelet donor’s age. The ages of the subjects roughly estimates the ages at which their AD developed, and suggests those with lower cytochrome oxidase activities had a younger age of onset.
Are Age-Related Mitochondrial Changes Relevant to AD?
Studies of aging brain mitochondria consistently report reductions of complex I activity, complex IV activity, and increased ROS production (Navarro and Boveris, 2007). Reduced numbers of mitochondria do not appear to account for these changes. Thus, a reasonable explanation is physical changes to the enzyme complexes are responsible. Other age-related mitochondrial changes include reduced membrane potential, increased size, and increased fragility.
Mitochondrial DNA mutations may contribute to age-related mitochondrial changes. In descriptive support of this, mtDNA deletions and oxidative damage accumulate with age in many tissues, especially brain (Corral-Debrinksi et al., 1992; Mecocci et al., 1993). It is easier to find low abundance, heteroplasmic point mutations (microheteroplasmy) in the brains of elderly individuals than in the brains of young individuals (Lin et al., 2002; Simon et al., 2004). To critically assess whether mtDNA mutation accumulation can promote aging, several groups have developed mice with deficient mtDNA polymerase γ(mtPOLG) proofreading capacity (Zhang et al., 2005; Trifunovic et al, 2004.; Kujoth et al., 2005). Characterizations of brain and brain mitochondria are scant, but immunostaining does reveal an increase in CO-negative fibers (Vermulst et al., 2008). Evaluations of multiple tissues, though, certainly show accelerated mtDNA deletion and point mutation accumulation, an age-dependent progressive reduction of ETC enzyme activities, and accelerated aging.
Similar to what is found across the mitochondrial genome, CO genes from aging individuals also accumulate sequence deviations. It was previously shown an age-related increase in CO1 gene microheteroplasmy inversely correlates with CO activity (Lin et al., 2002). If it is correct persons with lower inherent CO activities have an earlier AD onset age and by extension a greater risk of AD (Figure 3), this finding has direct implications for the mitochondrial cascade hypothesis. It is possible to postulate a scenario in which an individual’s CO activity at a given age (inherent CO activity minus the age-related CO activity decline at a particular age) either defines a threshold at which brain aging becomes brain disease, or provides a biomarker that defines when other mitochondrial parameters cross a line demarcating brain aging from brain disease (Figure 4). In this model, the distance between an individual’s inherent CO activity and the CO activity disease threshold, in conjunction with how rapidly an individual’s CO activity falls with aging, defines the age at which AD presents.
Figure 4.
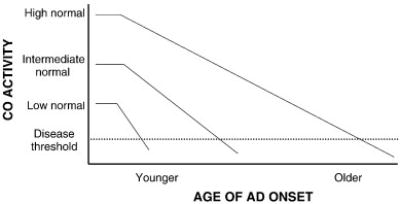
An individual’s cytochrome oxidase activity at a given age reflects their baseline activity minus an age-dependent activity decline. Activity baselines and rates of decline vary between individuals. For heuristic purposes we have shown a cytochrome oxidase activity threshold below which AD symptoms manifest. Those with high baseline activities and slow decline rates would remain above the disease threshold until a very advanced age, while those with lower baseline activities and more rapid decline rates would cross the threshold at a much younger age. CO=cytochrome oxidase.
Is Amyloidosis in Sporadic AD a Primary Event?
The amyloid cascade hypothesis assumes Aβ occupies the apex of a neurodegenerative cascade. Support for this comes from studies of autosomal dominant AD. Research on these variants collectively suggests Aβ more likely mediates disease than other APP derivatives or APP itself (Scheuner., 1996). Proponents extrapolate this paradigm to sporadic AD, which necessarily de-emphasizes the role aging may play by precluding it from the cascade apex.
The mitochondrial cascade hypothesis does not contest the view all pathology in autosomal dominant AD ultimately originates from aberrant Aβ production. We are unconvinced, though, that this is the case in sporadic AD. Unlike autosomal AD, sporadic AD is truly an age-dependent event, and amyloid protein formation is a recognized consequence of aging (Swerdlow, 2007a). In addition to the brain it occurs in the heart and pancreas, and the amyloid protein deposited in each organ is unique. This “senile amyloidosis” has in fact been recognized for decades, and generally is felt to represent a consequence of aging rather than a cause of disease (Ravid et al., 1967; Cornwell and Westermark, 1980). Indeed, aged individuals with a particular amyloid protein in one organ have an increased likelihood of having a different amyloid protein in a different organ (Schwartz, 1968; Storkel et al., 1983). This classic observation is supported by recent studies showing epidemiologic associations between AD and type II diabetes mellitus (Ott et al., 1999; Haan, 2006).
Cybrid modeling of AD mitochondrial function suggests AD subject mitochondria promote Aβ production. We found increased intra and extracellular Aβ42 and Aβ40 levels in AD cybrid cell lines with reduced CO activity (Khan et al., 2000). The extracellular Aβ formed adherent aggregates on flask surfaces. Living cells approached aggregate borders but did not grow on top of the aggregates. This is consistent with reports showing oxidative phosphorylation uncoupling, ATP synthase inhibition, and cytochrome oxidase inhibition converts APP processing towards amyloidogenic derivatives (Gabuzda et al., 1994; Gasparini et al., 1997; Webster et al., 1998).
Exposing cultured cells or isolated mitochondria to Aβ impairs the mitochondrial ETC in general and perhaps CO specifically (Pereira et al., 1998; Canaveri et al., 1999; Crouch et al., 2005). This does not impugn and is actually consistent with the mitochondrial cascade hypothesis. Our hypothesis asserts mitochondrial dysfunction activates a final common pathway that produces all aspects of AD histology. In autosomal dominant AD, Aβ overproduction is the primary cause of the mitochondrial dysfunction. In sporadic AD, mitochondrial function that declines beyond an aging-disease threshold is the primary cause. Aβ, as one downstream product of the mitochondrial functional decline, presumably acts as part of a positive feedback loop that exaggerates mitochondrial dysfunction. We originally proposed cells may have developed this loop to shut down perturbed mitochondria, or else help cells transition from aerobic to anaerobic metabolism (Swerdlow and Khan, 2004). In any case, we do not believe Aβ evolved solely as a toxic time bomb.
The existence of a mitochondria- Aβ nexus is supported by work showing APP, Aβ, PS proteins, and in fact the entire γ secretase complex is in fact found in mitochondria (Yamaguchi et al., 1992; Anandatheerthavarada et al., 2003; Lustbader et al., 2004; Hansson et al., 2004; Casperson et al., 2005; Teng and Tang, 2005; Manczak et al., 2006; Devi et al., 2006; Anandatheerthavarada and Devi, 2007; Hansson Petersen et al., 2008). The importance of the mitochondria- Aβ nexus is supported by experiments performed with mtDNA-depleted cells that lack ETC function. NT2 neuronal cells are killed by Aβ, while mtDNA-depleted NT2 cells are not (Cardoso et al., 2001). This implies Aβ toxicity is mediated through effects on mitochondria.
Are Tau Pathology and Cell Cycle Re-entry Related Events?
Tangles consist of aggregated microtubule associated protein (MAP) tau, tangle tau is excessively phosphorylated at serine and threonine residues, and tau phosphorylation presumably facilitates aggregation (Billingsley and Kincaid, 1997; Lovestone and Reynolds, 1997; Johnson and Hartigan, 1999). Although MAP tau gene mutations drive neurodegeneration, the resultant phenotype is characteristic of frontotemporal dementia rather than AD and altered Aβ production is not characteristic of primary tauopathies (Cairns et al., 2007). Polymorphic variation of the tau gene defines two extended haplotypes, H1 and H2; H1 associates with progressive supranuclear palsy, corticobasal degeneration, Parkinson’s disease, but probably not AD (Baker et al., 1999; Pittman et al., 2006; Mukherjee et al., 2007). Data from autosomal dominant AD therefore indicate Aβ perturbations can drive tau phosphorylation and tangle formation, while data showing the converse are lacking. The amyloid cascade hypothesis assumes Aβ drives tangle formation in both autosomal dominant and sporadic AD but in both cases is mechanistically vague on this point.
The mitochondrial cascade hypothesis postulates reduced cell energy promotes tau phosphorylation in both sporadic and autosomal dominant AD (Swerdlow and Khan, 2004). Supporting data come from experiments showing prolonged fasting, complex I inhibition, CO inhibition, mitochondrial uncoupling, and hibernation (a state associated with mitochondrial uncoupling) induce tau phosphoryation (Blass et al., 1990; Yanagisawa et al., 1999; Arendt et al., 2003; Szabados et al., 2004; Escobar-Khondiker et al., 2007; Hartig et al., 2007). In addition to several neurodegenerative disorders, tau phosphorylation also occurs in normal embryogenesis and transformed cells. Repeated cell division in the latter two cases utilizes considerable energy and energy storage is not possible. In AD, we predict energy levels are low due to mitochondrial failure.
Cell growth and cycling are regulated by trophic stimulation and energy status. Trophins bind insulin, insulin-like growth factor 1 (IGF1), and other related receptors belonging to the tyrosine kinase receptor family. In neurons, ligand binding induces insulin receptor substrate 2 (IRS2) tyrosine phosphorylation, phospho-activated IRS2 phosphorylates phosphoinositide 3 kinase (PI3K), and this activating phosphorylation eventually drives phosphorylation and activation of the Akt proto-oncogene protein (Bondy and Cheng, 2004; Manning and Cantely, 2007). We believe cells use this trophin-stimulated pathway to sense whether their external environment will support growth or division. Cell energy status also regulates Akt activity. The mammalian target of rapamycin (mTOR) protein helps cells adapt to fluctuations in their energy supply (Schieke and Finkel, 2006). mTOR complexes with other proteins, and one of these complexes, mTORC2, also phosphorylates Akt (Hresko and Mueckler, 2005; Sarbassov et al., 2005a; Sarbassov et al., 2005b).
Cell growth sometimes occurs in conjunction with division and sometimes it does not. Growing to divide and growing not to divide (hypertrophy), though, are fundamentally different processes. Cytoskeletal maintenance is not advantageous in dividing cells and high energy demand precludes energy storage. Growth without division requires a cytoskeleton and energy storage is possible. It is not surprising a single Akt-regulated protein, glycogen synthase kinase 3β (GSK3β), coordinates cytoskeletal maintenance with energy storage. GSK3β phosphorylates tau and inhibits glycogen synthase (Cohen and Frame, 2001; Grimes and Jope, 2001; Bhat and Budd, 2002). Tau phosphorylation prevents cytoskeleton formation or promotes cytoskeleton disassembly, while glycogen synthase inhibition blocks glucose storage. GSK3β activation is therefore conducive to cell division.
Substantial evidence reveals AD neuron cell cycle re-entry is more common than it is in non-AD brain (Vincent et al., 1996; McShea et al., 1997; Nagy et al., 1997; Arendt et al., 2000; Yang et al., 2001; Herrup and Arendt, 2002; Yang et al., 2003; Mosch et al., 2007). This re-entry proceeds through several parts of the cycle but fails at the G2-M interface (Zhu et al., 2004). The mitochondrial cascade hypothesis proposes tau phosphorylation/tangle formation and cell cycle re-entry are related events, and experimental support for this prediction was recently published (McShea et al., 2007). We speculate preserved trophic stimulation in the face of low energy mimics a cycling profile and causes AD neurons to act as dividing cells. If correct, Akt must function differently under conditions of trophic stimulation/high energy than it does under conditions of trophic stimulation/low energy (Figure 5). Trophic stimulation/high energy would presumably cause Akt to inhibit GSK3β, thus facilitating growth without division. Trophic stimulation/low energy would have to activate the Akt proto-oncogene protein in such a way that GSK3β inhibition does not occur. We are currently exploring this possibility. Finally, it is necessary to postulate another factor, perhaps oxidative stress, causes phosphorylated tau to form tangle aggregations.
Figure 5.
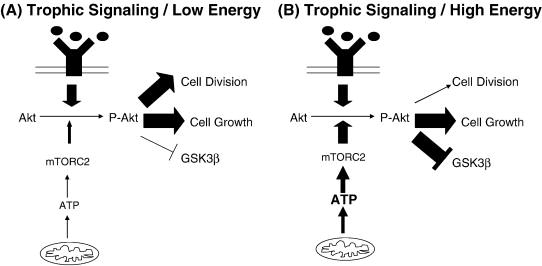
Hypothetical scheme in which Akt drives cell growth and division under conditions of trophic stimulation and low cell energy levels, and drives cell growth without division under conditions of trophic stimulation and higher energy levels. GSK3β=glycogen synthase kinase 3 beta; mTORC2=mammalian target of rapamycin complex 2; P=phospho.
Do Mitochondria Contribute Directly to AD Neuron Degeneration?
Published data suggest AD neurons die an apoptotic death (Su et al., 1994; Cotman, 1998; Rohn et al., 2002; Colurso et al., 2003; Blalock et al., 2004; Takuma et al., 2005). These data are sparse and inferential, since it is not possible to directly observe actual dying neurons in AD brain. Mitochondria presumably initiate neurodegeneration-related apoptosis by releasing proteins to the cytoplasm or nucleus that activate programmed cell death (PCD) pathways (Mattson, 2000). Accumulation of the mitochondrial proteins cytochrome c, apoptosis initiating factor (AIF), and Smac/DIABLO beyond their typical mitochondrial confines clearly induces PCD in laboratory models (Saelens et al., 2004). We previously reported cytoplasmic cytochrome c levels are elevated in AD cybrids, and caspase 3 activity is also increased (Khan et al., 2000). Mitochondrial depolarization is associated with cytochrome c egress, and AD cybrid mitochondria are relatively depolarized (Cassarino et al., 1998; Khan et al., 2000; Trimmer et al., 2000; Cardoso et al., 2004).
We believe apoptotic pathways are not maintained in absolute on or off states, but rather reflect mitochondrial integrity, number, or both. Cells with greater mitochondrial surface areas may have higher apoptotic set points, as might cells with failing mitochondria(Khan et al., 2000). One study of AD brain did show increased mitochondrial surface area as well as increased numbers of degrading mitochondria (Hirai et al., 2001). AD cybrids similarly show increased numbers of mitochondria and perturbed mitochondrial infrastructure (Trimmer et al., 2000; Trimmer et al., 2004).
Considerations and Implications
We believe aging and sporadic AD are fundamentally convergent, not divergent, entities. This reflects clinical and biomarker experience showing cognitive change and Aβ plaque accumulation are common (although not obligatory) consequences of aging and can precede dementia by years if not decades (Swerdlow, 2007a; Aizenstein et al., 2008). The mitochondrial cascade hypothesis asserts declining mitochondrial function initiates these clinical and bio marker changes and therefore represents the primary underlying problem in sporadic AD. We do not claim, though, that AD pathology simply exaggerates age-related physiologic changes. Whether mitochondrial biogenesis increases or decreases in sporadic AD is pertinent to this point. Studies of brain aging suggest mtDNA copy number and ETC protein levels increase with age; we assume this represents a compensatory response to falling ETC efficiency (Barrientos et al., 1997; Hirai et al., 2001). Data addressing these parameters in AD brain are not uniform, and some investigators conclude mitochondrial biogenesis increases further in AD while others conclude it decreases (de la Monte et al., 2000; Hirai et al., 2001). We await this issue’s definitive resolution. If mitochondrial biogenesis increases, it would suggest a physiologic divide between aging and AD arises at the point compensation is no longer possible. If mitochondrial biogenesis decreases, the point at which the cell abandons compensatory efforts could define this divide.
We have not critically questioned whether Aβ is more relevant to AD than APP or other APP derivatives. Most proponents of the amyloid cascade hypothesis believe Aβ is central although this assumption has been challenged (Galvan et al., 2007). While our hypothesis states mitochondrial dysfunction increases Aβ levels and increased Aβ affects mitochondrial function, our Aβ emphasis is partly for heuristic purposes and APP or other APP derivatives could ultimately prove more relevant. A better understanding of how APP, Aβ, or secretase enzymes access mitochondria and whether APP processing occurs in or on mitochondria could help address this.
There are data that argue Aβ is not toxic under physiologic in vivo conditions and may even mitigate other sporadic AD-associated pathologies (Nunomura et al., 2001; Aizenstein et al., 2008). Aβ and oxidative stress marker levels inversely correlate in particular brain regions, suggesting Aβ reduces ROS production (Nunomura et al., 2001). Limited Aβ exposure has also been associated with mitochondrial biogenesis in animal and cell models (Reddy et al., 2004; Diana et al., 2008). Our hypothesis is comfortable with both sides of this debate. As we stated in 2004, we believe Aβ production (or other changes in APP physiology) in sporadic AD reflects an adaptive but failed attempt to disable impaired mitochondria, or else an adaptive but failed attempt by aerobically marginal cells to acquire an anaerobic phenotype (Swerdlow and Khan, 2004).
The mitochondrial cascade hypothesis predicts effective autosomal dominant AD treatments could prove partly or wholly ineffective in sporadic AD. Even if Aβ exacerbates mitochondrial failure in sporadic AD, its removal may not adequately restore mitochondrial function to tolerable levels. There have been enormous efforts in recent years to treat sporadic AD by removing fibrillar Aβ, blocking Aβ oligomerization, and preventing formation of Aβ42. Testing these approaches in small numbers of autosomal dominant AD subjects before attempting them in large numbers of sporadic AD subjects might prove useful. Our hypothesis may also help explain why interventions that benefit APP transgenic mice have thus far not benefited sporadic AD patients (Swerdlow, 2007a; Holmes et al., 2008).
When we first proposed the mitochondrial cascade hypothesis it was acknowledged to at least a small degree by the AD research field and an independent group subsequently advocated a mitochondria-centric AD hypothesis (Swerdlowand Khan, 2005; Roses et al., 2007; Mancuso et al., 2007). A mitochondrial cascade hypothesis has been formulated for sporadic Parkinson’s disease (PD), and it would seem reasonable to also formulate a mitochondrial cascade hypothesis for sporadic amyotrophic lateral sclerosis (ALS) (Domingues et al., 2008). Like AD, PD and ALS have rare autosomal forms in which mitochondrial dysfunction may play an intermediary role in neurodegeneration, and common sporadic forms that are associated with mitochondrial dysfunction (Swerdlow, 2007d).
In formulating the mitochondrial cascade hypothesis we considered epidemiologic, clinical, biochemical, and molecular studies of human sporadic AD (Swerdlow and Khan, 2004). We used cybrid cell lines to model sporadic AD mitochondrial function, and data from this model are critical to this hypothesis. Animal data from ETC inhibition models, mtDNA manipulation models, and Mendelian manipulation models also contributed. For example, APP transgenic mice with increased mitochondrial oxidative stress as a consequence of reduced manganese superoxide dismutase show increased plaque deposition, while APP transgenic mice with decreased mitochondrial oxidative stress as a consequence of reduced CO holoenzyme show decreased plaque deposition (Li et al., 2004; Fukui et al., 2007). These studies support our view that mitochondrial physiology influences amyloidosis. How best to model a sporadic disease, though, is a critical question that will impact the popularity of our hypothesis. Without accessible models, interest will likely remain limited regardless of how conceptually reasonable our hypothesis seems.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Russell H. Swerdlow, Departments of Neurology and Molecular & Integrative Physiology, University of Kansas School of Medicine, Kansas City, KS.
Shaharyar M. Khan, Gencia Corporation, Charlottesville, VA.
References
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez V, Corao AI, Alonso-Montes C, Sanchez-Ferrero E, De Mena L, Morales B, Garcia-Castro M, Coto E. Mitochondrial transcription factor A (TFAM) gene variation and risk of late-onset Alzheimer’s disease. J Alzheimers Dis. 2008;13:275–280. doi: 10.3233/jad-2008-13305. [DOI] [PubMed] [Google Scholar]
- Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anandatheerthavarada HK, Devi L. Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist. 2007;13:626–638. doi: 10.1177/1073858407303536. [DOI] [PubMed] [Google Scholar]
- Arendt T, Holzer M, Stobe A, Gartner U, Luth HJ, Bruckner MK, Ueberham U. Activated mitogenic signaling induces a process of dedifferentiation in Alzheimer’s disease that eventually results in cell death. Ann N Y Acad Sci. 2000;920:249–255. doi: 10.1111/j.1749-6632.2000.tb06931.x. [DOI] [PubMed] [Google Scholar]
- Arendt T, Stieler J, Strijkstra AM, Hut RA, Rudiger J, Van der Zee EA, Harkany T, Holzer M, Hartig W. Reversible paired helical filament-like phosphorylation of tau is an adaptive process associated with neuronal plasticity in hibernating animals. J Neurosci. 2003;23:6972–6981. doi: 10.1523/JNEUROSCI.23-18-06972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M. Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999;8:711–715. doi: 10.1093/hmg/8.4.711. [DOI] [PubMed] [Google Scholar]
- Baloyannis SJ. Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis. 2006;9:119–126. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Casademont J, Cardellach F, Estivill X, Urbano-Marquez A, Nunes V. Reduced steady-state levels of mitochondrial RNA and increased mitochondrial DNA amount in human brain with aging. Brain Res Mol Brain Res. 1997;52:284–289. doi: 10.1016/s0169-328x(97)00278-7. [DOI] [PubMed] [Google Scholar]
- Bekris LM, Millard SP, Galloway NM, Vuletic S, Albers JJ, Li G, Galasko DR, DeCarli C, Farlow MR, Clark CM, Quinn JF, Kaye JA, Schellenberg GD, Tsuang D, Peskind ER, Yu CE. Multiple SNPs within and surrounding the apolipoprotein E gene influence cerebrospinal fluid apolipoprotein E protein levels. J Alzheimers Dis. 2008;13:255–266. doi: 10.3233/jad-2008-13303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin AC, Bjork BF, Westerlund M, Galter D, Sydow O, Lind C, Pernold K, Rosvall L, Hakansson A, Winblad B, Nissbrandt H, Graff C, Olson L. Association study of two genetic variants in mitochondrial transcription factor A (TFAM) in Alzheimer’s and Parkinson’s disease. Neurosci Lett. 2007;420:257–262. doi: 10.1016/j.neulet.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Budd SL. GSK3beta signalling: casting a wide net in Alzheimer’s disease. Neurosignals. 2002;11:251–261. doi: 10.1159/000067423. [DOI] [PubMed] [Google Scholar]
- Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323 (Pt 3):577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer’s disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass JP, Baker AC, Ko L, Black RS. Induction of Alzheimer antigens by an uncoupler of oxidative phosphorylation. Arch Neurol. 1990;47:864–869. doi: 10.1001/archneur.1990.00530080046009. [DOI] [PubMed] [Google Scholar]
- Bondy CA, Cheng CM. Signaling by insulin-like growth factor 1 in brain. Eur J Pharmacol. 2004;490:25–31. doi: 10.1016/j.ejphar.2004.02.042. [DOI] [PubMed] [Google Scholar]
- Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canevari L, Clark JB, Bates TE. beta-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999;457:131–134. doi: 10.1016/s0014-5793(99)01028-5. [DOI] [PubMed] [Google Scholar]
- Cardoso SM, Santos S, Swerdlow RH, Oliveira CR. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001;15:1439–1441. doi: 10.1096/fj.00-0561fje. [DOI] [PubMed] [Google Scholar]
- Cardoso SM, Santana I, Swerdlow RH, Oliveira CR. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. J Neurochem. 2004;89:1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- Cassarino DS, Swerdlow RH, Parks JK, Parker WD, Jr, Bennett JP., Jr Cyclosporin A increases resting mitochondrial membrane potential in SY5Y cells and reverses the depressed mitochondrial membrane potential of Alzheimer’s disease cybrids. Biochem Biophys Res Commun. 1998;248:168–173. doi: 10.1006/bbrc.1998.8866. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran K, Giordano T, Brady DR, Stoll J, Martin LJ, Rapoport SI. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res Mol Brain Res. 1994;24:336–340. doi: 10.1016/0169-328x(94)90147-3. [DOI] [PubMed] [Google Scholar]
- Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Colurso GJ, Nilson JE, Vervoort LG. Quantitative assessment of DNA fragmentation and beta-amyloid deposition in insular cortex and midfrontal gyrus from patients with Alzheimer’s disease. Life Sci. 2003;73:1795–1803. doi: 10.1016/s0024-3205(03)00512-5. [DOI] [PubMed] [Google Scholar]
- Coon KD, Valla J, Szelinger S, Schneider LE, Niedzielko TL, Brown KM, Pearson JV, Halperin R, Dunckley T, Papassotiropoulos A, Caselli RJ, Reiman EM, Stephan DA. Quantitation of heteroplasmy of mtDNA sequence variants identified in a population of AD patients and controls by array-based resequencing. Mitochondrion. 2006;6:194–210. doi: 10.1016/j.mito.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008 doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- Cornwell GG, 3rd, Westermark P. Senile amyloidosis: a protean manifestation of the aging process. J Clin Pathol. 1980;33:1146–1152. doi: 10.1136/jcp.33.12.1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, Graham BH, Wallace DC. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics. 1994;23:471–476. doi: 10.1006/geno.1994.1525. [DOI] [PubMed] [Google Scholar]
- Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW. Apoptosis decision cascades and neuronal degeneration in Alzheimer’s disease. Neurobiol Aging. 1998;19:S29–32. doi: 10.1016/s0197-4580(98)00042-6. [DOI] [PubMed] [Google Scholar]
- Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper- dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid- beta1–42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE, Miller S, Herrnstadt C, Ghosh SS, Fahy E, Shinobu LA, Galasko D, Thal LJ, Beal MF, Howell N, Parker WD., Jr Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- de la Monte SM, Luong T, Neely TR, Robinson D, Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab Invest. 2000;80:1323–1335. doi: 10.1038/labinvest.3780140. [DOI] [PubMed] [Google Scholar]
- Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana A, Simic G, Sinforiani E, Orru N, Pichiri G, Bono G. Mitochondria morphology and DNA content upon sublethal exposure to beta-amyloid(1–42) peptide. Coll Antropol. 2008;32(Suppl 1):51–58. [PubMed] [Google Scholar]
- Domingues AF, Arduino DM, Esteves AR, Swerdlow RH, Oliveira CR, Cardoso SM. Mitochondria and ubiquitin-proteasomal system interplay: relevance to Parkinson’s disease. Free Radic Biol Med. 2008;45:820–825. doi: 10.1016/j.freeradbiomed.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology. 1996;47:254–256. doi: 10.1212/wnl.47.1.254. [DOI] [PubMed] [Google Scholar]
- Escobar-Khondiker M, Hollerhage M, Muriel MP, Champy P, Bach A, Depienne C, Respondek G, Yamada ES, Lannuzel A, Yagi T, Hirsch EC, Oertel WH, Jacob R, Michel PP, Ruberg M, Hoglinger GU. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007;27:7827–7837. doi: 10.1523/JNEUROSCI.1644-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flierl A, Reichmann H, Seibel P. Pathophysiology of the MELAS 3243 transition mutation. J Biol Chem. 1997;272:27189–27196. doi: 10.1074/jbc.272.43.27189. [DOI] [PubMed] [Google Scholar]
- Fukui H, Diaz F, Garcia S, Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabuzda D, Busciglio J, Chen LB, Matsudaira P, Yankner BA. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem. 1994;269:13623–13628. [PubMed] [Google Scholar]
- Galvan V, Gorostiza OF, Banwait S, Ataie M, Logvinova AV, Sitaraman S, Carlson E, Sagi SA, Chevallier N, Jin K, Greenberg DA, Bredesen DE. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc Natl Acad Sci U S A. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L, Racchi M, Benussi L, Curti D, Binetti G, Bianchetti A, Trabucchi M, Govoni S. Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci Lett. 1997;231:113–117. doi: 10.1016/s0304-3940(97)00536-3. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Gunther C, von Hadeln K, Muller-Thomsen T, Alberici A, Binetti G, Hock C, Nitsch RM, Stoppe G, Reiss J, Gal A, Finckh U. Possible association of mitochondrial transcription factor A (TFAM)genotype with sporadic Alzheimer disease. Neurosci Lett. 2004;369:219–223. doi: 10.1016/j.neulet.2004.07.070. [DOI] [PubMed] [Google Scholar]
- Haan MN. Therapy Insight: type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat Clin Pract Neurol. 2006;2:159–166. doi: 10.1038/ncpneuro0124. [DOI] [PubMed] [Google Scholar]
- Haass C, Steiner H. Protofibrils, the unifying toxic molecule of neurodegenerative disorders? Nat Neurosci. 2001;4:859–860. doi: 10.1038/nn0901-859. [DOI] [PubMed] [Google Scholar]
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, Ito A, Winblad B, Cowburn RF, Thyberg J, Ankarcrona M. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J Biol Chem. 2004;279:51654–51660. doi: 10.1074/jbc.M404500200. [DOI] [PubMed] [Google Scholar]
- Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartig W, Stieler J, Boerema AS, Wolf J, Schmidt U, Weissfuss J, Bullmann T, Strijkstra AM, Arendt T. Hibernation model of tau phosphorylation in hamsters: selective vulnerability of cholinergic basal forebrain neurons- implications for Alzheimer’s disease. Eur J Neurosci. 2007;25:69–80. doi: 10.1111/j.1460-9568.2006.05250.x. [DOI] [PubMed] [Google Scholar]
- Herrup K, Arendt T. Re-expression of cell cycle proteins induces neuronal cell death during Alzheimer’s disease. J Alzheimers Dis. 2002;4:243–247. doi: 10.3233/jad-2002-4315. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo- controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Hresko RC, Mueckler M. mTOR. RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J Biol Chem. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- Johnson GV, Hartigan JA. Tau protein in normal and Alzheimer’s disease brain: an update. J Alzheimers Dis. 1999;1:329–351. doi: 10.3233/jad-1999-14-512. [DOI] [PubMed] [Google Scholar]
- Khan SM, Cassarino DS, Abramova NN, Keeney PM, Borland MK, Trimmer PA, Krebs CT, Bennett JC, Parks JK, Swerdlow RH, Parker WD, Jr, Bennett JP., Jr Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol. 2000;48:148–155. [PubMed] [Google Scholar]
- King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, Wilson JM, DiStefano LM, Nobrega JN. Brain cytochrome oxidase in Alzheimer’s disease. J Neurochem. 1992;59:776–779. doi: 10.1111/j.1471-4159.1992.tb09439.x. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Mastrogiacomo F, Guttman M, Furukawa Y, Taanman JW, Dozic S, Pandolfo M, Lamarche J, DiStefano L, Chang LJ. Decreased brain protein levels of cytochrome oxidase subunits in Alzheimer’s disease and in hereditary spinocerebellar ataxia disorders: a nonspecific change? J Neurochem. 1999;72:700–707. doi: 10.1046/j.1471-4159.1999.0720700.x. [DOI] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Flint Beal M, Lin MT, Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- Lin MT, Simon DK, Ahn CH, Kim LM, Beal MF. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet. 2002;11:133–145. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Reynolds CH. The phosphorylation of tau: a critical stage in neurodevelopment and neurodegenerative processes. Neuroscience. 1997;78:309–324. doi: 10.1016/s0306-4522(96)00577-5. [DOI] [PubMed] [Google Scholar]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Coppede F, Murri L, Siciliano G. Mitochondrial cascade hypothesis of Alzheimer’s disease: myth or reality? Antioxid Redox Signal. 2007;9:1631–1646. doi: 10.1089/ars.2007.1761. [DOI] [PubMed] [Google Scholar]
- Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004;5:147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16and CDK4 in Alzheimer’s disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- McShea A, Lee HG, Petersen RB, Casadesus G, Vincent I, Linford NJ, Funk JO, Shapiro RA, Smith MA. Neuronal cell cycle re-entry mediates Alzheimer disease-type changes. Biochim Biophys Acta. 2007;1772:467–472. doi: 10.1016/j.bbadis.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age- dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, Arendt T. Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J Neurosci. 2007;27:6859–6867. doi: 10.1523/JNEUROSCI.0379-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, Tsui W, De Santi S, de Leon MJ. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci U S A. 2007;104:19067–19072. doi: 10.1073/pnas.0705036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee O, Kauwe JS, Mayo K, Morris JC, Goate AM. Haplotype-based association analysis of the MAPT locus in late onset Alzheimer’s disease. BMC Genet. 2007;8:3. doi: 10.1186/1471-2156-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy Z, Esiri MM, Cato AM, Smith AD. Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol. 1997;94:6–15. doi: 10.1007/s004010050665. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- Parker WD, Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- Parker WD, Jr, Parks JK. Cytochrome c oxidase in Alzheimer’s disease brain: purification and characterization. Neurology. 1995;45:482–486. doi: 10.1212/wnl.45.3.482. [DOI] [PubMed] [Google Scholar]
- Pereira C, Santos MS, Oliveira C. Mitochondrial function impairment induced by amyloid beta-peptide on PC12 cells. Neuroreport. 1998;9:1749–1755. doi: 10.1097/00001756-199806010-00015. [DOI] [PubMed] [Google Scholar]
- Pittman AM, Fung HC, de Silva R. Untangling the tau gene association with neurodegenerative disorders. Hum Mol Genet. 2006;15(Spec No 2):R188–195. doi: 10.1093/hmg/ddl190. [DOI] [PubMed] [Google Scholar]
- Prick MJ, Gabreels FJ, Trijbels JM, Janssen AJ, le Coultre R, van Dam K, Jaspar HH, Ebels EJ, Op de Coul AA. Progressive poliodystrophy (Alpers’ disease) with a defect in cytochrome aa3 in muscle: a report of two unrelated patients. Clin Neurol Neurosurg. 1983;85:57–70. doi: 10.1016/0303-8467(83)90024-0. [DOI] [PubMed] [Google Scholar]
- Ravid M, Gafni J, Sohar E, Missmahl HP. Incidence and origin of non-systemic microdeposits of amyloid. J Clin Pathol. 1967;20:15–20. doi: 10.1136/jcp.20.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer’s disease brain. Neurobiol Dis. 2002;11:341–354. doi: 10.1006/nbdi.2002.0549. [DOI] [PubMed] [Google Scholar]
- Roses AD, Saunders AM, Huang Y, Strum J, Weisgraber KH, Mahley RW. Complex disease-associated pharmacogenetics: drug efficacy, drug safety, and confirmation of a pathogenetic hypothesis (Alzheimer’s disease) Pharmacogenomics J. 2007;7:10–28. doi: 10.1038/sj.tpj.6500397. [DOI] [PubMed] [Google Scholar]
- Saelens X, Festjens N, Vande Walle L, van Gurp M, van Loo G, Vandenabeele P. Toxic proteins released from mitochondria in cell death. Oncogene. 2004;23:2861–2874. doi: 10.1038/sj.onc.1207523. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005a;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005b;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Schieke SM, Finkel T. Mitochondrial signaling, TOR, and life span. Biol Chem. 2006;387:1357–1361. doi: 10.1515/BC.2006.170. [DOI] [PubMed] [Google Scholar]
- Schwartz P. New patho-anatomic observations on amyloidosis in the aged. Fluorescence microscopic investigations. In: Mandema E, Ruinen L, Scholten JH, Cohen AS, editors. Amyloidosis. Amsterdam: Excerpta Medica Foundation; 1968. pp. 400–417. [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Simon DK, Lin MT, Zheng L, Liu GJ, Ahn CH, Kim LM, Mauck WM, Twu F, Beal MF, Johns DR. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson’s disease. Neurobiol Aging. 2004;25:71–81. doi: 10.1016/s0197-4580(03)00037-x. [DOI] [PubMed] [Google Scholar]
- Small GW, Mazziotta JC, Collins MT, Baxter LR, Phelps ME, Mandelkern MA, Kaplan A, La Rue A, Adamson CF, Chang L, et al. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA. 1995;273:942–947. [PubMed] [Google Scholar]
- Storkel S, Bohl J, Schneider HM. Senile amyloidosis: principles of localization in a heterogeneous form of amyloidosis. Virchows Arch B Cell Pathol Incl Mol Pathol. 1983;44:145–161. doi: 10.1007/BF02890166. [DOI] [PubMed] [Google Scholar]
- Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Parks JK, Cassarino DS, Maguire DJ, Maguire RS, Bennett JP, Jr, Davis RE, Parker WD., Jr Cybrids in Alzheimer’s disease: a cellular model of the disease? Neurology. 1997;49:918–925. doi: 10.1212/wnl.49.4.918. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Kish SJ. Mitochondria in Alzheimer’s disease. Int Rev Neurobiol. 2002;53:341–385. doi: 10.1016/s0074-7742(02)53013-0. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63:8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH, Khan S. Alzheimer research forum live discussion: A “Mitochondrial Cascade Hypothesis” for sporadic Alzheimer’s disease. Journal of Alzheimer’s Disease. 2005;8:311–315. [Google Scholar]
- Swerdlow RH, Ghosh S, Wang KX. Polymorphism-determined variation of the cytochrome oxidase enzyme. Soc Neurosci Abstr. 2006;32:79.1. [Google Scholar]
- Swerdlow RH. Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiol Aging. 2007a;28:1465–1480. doi: 10.1016/j.neurobiolaging.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH. Pathogenesis of Alzheimer’s disease. Clin Interv Aging. 2007b;2:347–359. [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res. 2007c;85:3416–3428. doi: 10.1002/jnr.21167. [DOI] [PubMed] [Google Scholar]
- Swerdlow RH. Treating neurodegeneration by modifying mitochondria: potential solutions to a “complex” problem. Antioxid Redox Signal. 2007d;9:1591–1603. doi: 10.1089/ars.2007.1676. [DOI] [PubMed] [Google Scholar]
- Szabados T, Dul C, Majtenyi K, Hargitai J, Penzes Z, Urbanics R. A chronic Alzheimer’s model evoked by mitochondrial poison sodium azide for pharmacological investigations. Behav Brain Res. 2004;154:31–40. doi: 10.1016/j.bbr.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Takuma K, Yan SS, Stern DM, Yamada K. Mitochondrial dysfunction, endoplasmic reticulum stress, and apoptosis in Alzheimer’s disease. J Pharmacol Sci. 2005;97:312–316. doi: 10.1254/jphs.cpj04006x. [DOI] [PubMed] [Google Scholar]
- Teng FY, Tang BL. Widespread gamma-secretase activity in the cell, but do we need it at the mitochondria? Biochem Biophys Res Commun. 2005;328:1–5. doi: 10.1016/j.bbrc.2004.12.131. [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Jr, Miller SW, Davis RE, Parker WD., Jr Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- Trimmer PA, Keeney PM, Borland MK, Simon FA, Almeida J, Swerdlow RH, Parks JP, Parker WD, Jr, Bennett JP., Jr Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer’s disease worsen with passage in culture. Neurobiol Dis. 2004;15:29–39. doi: 10.1016/j.nbd.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Valla J, Schneider L, Niedzielko T, Coon KD, Caselli R, Sabbagh MN, Ahern GL, Baxter L, Alexander G, Walker DG, Reiman EM. Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion. 2006;6:323–330. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, Kroner CC, Welsh-Bohmer KA, Saunders AM, Roses AD, Small GW, Schmechel DE, Murali Doraiswamy P, Gilbert JR, Haines JL, Vance JM, Pericak-Vance MA. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett. 2004;365:28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer’s disease? J Cell Biol. 1996;132:413–425. doi: 10.1083/jcb.132.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MT, Pearce BR, Bowen DM, Francis PT. The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J Neural Transm. 1998;105:839–853. doi: 10.1007/s007020050098. [DOI] [PubMed] [Google Scholar]
- Wolf PA, Beiser A, Au R, Auerbach S, DeCarli C. Parental Occurrence of Dementia Linked to Lower Cognitive Function in the Framingham Offspring Study. Neurology. 2005;64(Suppl 1):A267–A268. [Google Scholar]
- Yamaguchi H, Yamazaki T, Ishiguro K, Shoji M, Nakazato Y, Hirai S. Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol. 1992;85:15–22. doi: 10.1007/BF00304629. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Planel E, Ishiguro K, Fujita SC. Starvation induces tau hyperphosphorylation in mouse brain: implications for Alzheimer’s disease. FEBS Lett. 1999;461:329–333. doi: 10.1016/s0014-5793(99)01480-5. [DOI] [PubMed] [Google Scholar]
- Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21:2661–2668. doi: 10.1523/JNEUROSCI.21-08-02661.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Mott JL, Chang SW, Stevens M, Mikolajczak P, Zassenhaus HP. Mitochondrial DNA mutations activate programmed cell survival in the mouse heart. Am J Physiol Heart Circ Physiol. 2005;288:H2476–2483. doi: 10.1152/ajpheart.00670.2004. [DOI] [PubMed] [Google Scholar]
- Zhu X, McShea A, Harris PL, Raina AK, Castellani RJ, Funk JO, Shah S, Atwood C, Bowen R, Bowser R, Morelli L, Perry G, Smith MA. Elevated expression of a regulator of the G2/M phase of the cell cycle, neuronal CIP-1-associated regulator of cyclin B, in Alzheimer’s disease. J Neurosci Res. 2004;75:698–703. doi: 10.1002/jnr.20028. [DOI] [PubMed] [Google Scholar]