

Abstract
Transforming growth factor-beta (TGF-β) signaling is disrupted in many cancers, including cervical cancer, leading to TGF-β resistance. Although initially sensitive, human papillomavirus type 16 (HPV16) immortalized human keratinocytes (HKc/HPV16) become increasingly resistant to the growth inhibitory effects of TGF-β during in vitro progression to a differentiation resistant phenotype (HKc/DR). We have previously shown that loss of TGF-β sensitivity in HKc/DR is attributed to decreased expression of TGF-β receptor type I (TGF-β RI), while the levels of TGF-β receptor type II (TGF-β RII) remain unchanged. The present study explored molecular mechanisms leading to reduced TGF-β RI expression in HKc/DR. Using TGF-β RI and TGF-β RII promoter reporter constructs, we determined that acute expression of the HPV16 oncogenes E6 and E7 decreased the promoter activity of TGF-β RI and TGF-β RII by about 50%. However, promoter activity of TGF-β RI is decreased to a greater extent than TGF-β RII as HKc/HPV16 progress to HKc/DR. Reduced TGF-β RI expression in HKc/DR was found not to be linked to mutations within the TGF-β RI promoter or to promoter methylation. Electrophoretic mobility shift and supershift assays using probes encompassing Sp1 binding sites in the TGF-β RI promoter found no changes between HKc/HPV16 and HKc/DR in binding of the transcription factors Sp1 or Sp3 to the probes. Also, Western blots determined that protein levels of Sp1 and Sp3 remain relatively unchanged between HKc/HPV16 and HKc/DR. Overall, these results demonstrate that mutations in or hypermethylation of the TGF-β RI promoter, along with altered levels of Sp1 or Sp3, are not responsible for the reduced expression of TGF-β RI we observe in HKc/DR. Rather the HPV16 oncogenes E6 and E7 themselves exhibit an inhibitory effect on TGF-β receptor promoter activity.
Keywords: TGF-beta receptors, HPV-mediated transformation, Human keratinocytes
1. Introduction
The cytokine transforming growth factor-beta (TGF-β) plays important roles in tissue homeostasis, extracellular matrix production, angiogenesis, immune response and growth inhibition of epithelial cells [1-4]. The overall biological effects of TGF-β are dependent on the cell type and context [1-4]. TGF-β signals through receptors that are members of the serine/threonine receptor kinase superfamily [5-7]. Signaling is initiated with TGF-β binding to TGF-β receptor type II (TGF-β RII), whose serine/threonine kinase activity is constitutively active. The binding of TGF-β to TGF-β RII promotes the recruitment of a TGF-β receptor type I (TGF-β RI). TGF-β RI is activated via transphosphorylation by TGF-β RII on multiple serine residues. Activated TGF-β RI then phosphorylates one of the receptor regulated smads, smad2 or smad3. Upon phosphorylation the receptor regulated smads associate with smad4, a co-smad. The smad complex then translocates to the nucleus, interacts with a variety of transcription factors, including Sp1 and Sp3, binds to Smad binding elements in the promoter region of target genes, and modulates transcription [5-7].
A loss of sensitivity to the antiproliferative effects of TGF-β is a common feature of human tumors and is thought to play an important role in tumor progression and metastasis [8,9]. Many alterations/defects in TGF-β signaling, which lead to TGF-β resistance have been identified in a variety of human malignancies; including mutations in the genes that encode the TGF-β receptors and smad proteins and reduced or loss of TGF-β RI and TGF-β RII and smad expression [9].
We use a model system of human papillomavirus type 16 (HPV16)-mediated carcinogenesis to explore the cellular and molecular events that mark in vitro progression. In this model normal human keratinocytes (HKc) are immortalized by transfection with HPV16 DNA (HKc/HPV16) and progress towards malignancy through several phenotypically defined and reproducible stages that include growth factor independence (HKc/GFI), differentiation resistance (HKc/DR), and ultimately malignant conversion [10-12]. We previously reported that HKc/HPV16 are initially as sensitive as normal HKc to the growth inhibitory effects of TGF-β, but become increasingly resistant to TGF-β during in vitro progression [13]. Furthermore, we have linked the loss of sensitivity to TGF-β at the HKc/DR stage of our model system to decreased expression of TGF-β RI mRNA [14]. No change was found in the expression of TGF-β RII mRNA nor was any rearrangement of the TGF-β RI gene identified. Exogenous re-expression of TGF-β RI in HKc/DR restored TGF-β sensitivity, confirming that the loss of TGF-β RI is a key event leading to TGF-β resistance in HKc/DR [14].
The promoter for the human TGF-β RI gene has been partially characterized and some details concerning regulation of its transcription have been determined. The TGF-β RI promoter is GC rich and does not contain a distinct TATA box or a functional CAAT box [15]. It is common for such genes to be regulated by the Sp1 family of transcription factors. Consistent with this possibility is the presence of four consensus and several putative Sp1 binding sites in the human TGF-β RI promoter [15]. Furthermore, the importance of the Sp1 family of transcription factors in regulating TGF-β RI transcription is supported by the finding that decreased expression of Sp1 [16] or increased expression of Sp3 [17] can decrease the expression of TGF-β RI in GEO human colon carcinoma and MCF-7L breast cancer cells. Hypermethylation of CpG islands in the 5’ region of the TGF-β RI gene was found to decrease TGF-β RI expression and lead to TGF-β resistance in gastric cancer cells [18].
The focus of the present study was to investigate several potential cellular and molecular mechanisms that may be responsible for and ultimately lead to the decreased expression of TGF-β RI mRNA that is associated with TGF-β resistance during in vitro progression of HKc/HPV16. Overall, these studies determined that decreased expression of TGF-β RI in HKc/DR was not the result of mutations in or hypermethylation of the TGF-β RI promoter, nor to changes in the protein levels of the transcription factors Sp1 or Sp3. Using TGF-β RI and TGF-β RII promoter reporter constructs we found that TGF-β RI promoter activity decreased as HKc/HPV16 progressed to the HKc/DR stage. However, the precise cellular changes leading to reduced TGF-β RI promoter activity in HKc/DR remain to be identified.
2. Materials and methods
2.1. Cell culture
Normal human keratinocytes (HKc) were isolated from neonatal foreskins and immortalized by transfection with a plasmid containing a head-to-tail dimer of HPV16 DNA (HKc/HPV16) and cultured in serum-free MCDB153-LB basal medium supplemented with epidermal growth factor (5 ng/ml), bovine pituitary extract (35-50 μg protein/ml), hydrocortisone (0.2 μM), calcium chloride (0.1 mM), triiodothyronine (10 nM), transferrin (10 μg/ml), insulin (5 μg/ml), and gentamycin (50 μg/ml) as described in detail previously [13]. This medium will be referred to as complete medium. Growth factor-independent HKc (HKc/GFI) were selected by maintaining HKc/HPV16 in complete medium lacking epidermal growth factor and bovine pituitary extract. Differentiation resistant cells (HKc/DR) were selected from HKc/GFI in complete medium containing calcium chloride (1 mM) and fetal bovine serum (5%) [13]. All cells were maintained at 37° C in 5% CO2 in air. Cells were split 1:10 when confluent, and medium was replaced every 48 h.
2.2. Real time PCR
RNA was extracted from HKc/HPV16 or HKc/DR using the RNeasy mini kit (Qiagen, Valencia, CA) according to manufacturer’s instructions, except the DNase I treatment time was doubled to 30 min to ensure complete digestion. Total RNA was reverse transcribed using either the GeneAmp Core Kit (Applied Biosystems, Foster City, CA) (2 μg of total RNA) or the iScript cDNA Synthesis Kit (BioRad, Hercules, CA) (1 μg of total RNA). Real Time primers were designed to yield a product with an average size of 150 bp and the sequences were blasted in GenBank to ensure specificity. The following primers were used: TGF-β RI forward 5’ CTT AAT TCC TCG AGA TAG GC 3’, reverse 5’ GTG AGA TGC AGA CGA AGC 3’, TGF-β RII forward 5’ CAG AAA TCC TGC ATG AGC 3’, reverse 5’ GCA GCA TCT TCC AGA ATA AAG 3’, HPV16 E6 forward 5’ GAG ATG GGA ATC CAT ATG CTG 3’, reverse 5’ CGG TTT GTT GTA TTG CTG TTC 3’, HPV16 E7 forward 5’ ATG GTC CAG CTG GAC AAG CA 3’, reverse 5’ GGC ACA CAA TTC CTA GTG TGC 3’, RPLP0 forward 5’ TTA AAC CCC CTC GTG GCA ATC 3’, reverse 5’ CCA CAT TCC CCC GGA TAT GA 3’. Real Time PCR was performed using either the SYBR Green DNA PCR Core Reagents (Applied Biosystems, Foster City, CA) or the iQ SYBR Green Supermix (BioRad, Hercules, CA). RPLP0 was used as an internal control. The average threshold cycle of duplicate samples for the mRNA of interest was used in calculations to determine the fold change difference in that message normalized to the RPLPO control.
2.3. TGF-β RI and TGF-β RII promoter luciferase constructs
Construction of a TGF-β RI promoter luciferase construct (-1379 to +6), which will be referred to as TGF-β RI promoter-Luc, has been described in detail in a previous publication from our laboratory [14].
Approximately 1.3 Kb of the TGF–β RII promoter, including 50 base pairs of exon 1, was amplified by PCR from normal HKc genomic DNA (100 ng). The forward primer was designed to include a NheI restriction site and corresponded to nucleotides -1262 to -1240: 5’ TTG GGT GCT AGC TGG AGC ATC A 3’. The reverse primer contains a HindIII site and corresponds to nucleotides +50 to +28: 5’ ACT CAA CTT CAA GCT TGC GCT GC 3’. The restriction sites are shown in bold and underlined. The purified PCR product was subcloned into pGEMTeasy (Promega, Madison, WI) and the TGF-β RII promoter insert (nucleotides -1258 to +40) was then released by NheI and HindIII digestion and cloned into the pGL3-Basic luciferase vector (Promega, Madison, WI). The TGF-β RII promoter sequence was verified by direct sequencing of both DNA strands. We will refer to this construct as TGF-β RII promoter-Luc.
2.4. Transfection and luciferase assays
HKc/HPV16 or HKc/DR were seeded into 60-mm dishes. Transfections were performed when cells reached approximately 50% confluency. TGF-β RI or TGF-β RII promoter-Luc constructs (2 μg DNA) were transfected in triplicate along with pRL-Null Renilla luciferase (20 ng DNA) (Promega, Madison, WI) as a control for transfection efficiency. Transfections were performed using TransFast Transfection Reagent (Promega, Madison, WI) according to the manufacturer’s instructions. All cells were maintained in complete media without gentamycin. Luciferase activity was measured 48 to 72 h after transfection using the Dual-Luciferase Assay System (Promega, Madison, WI). For each sample the firefly luciferase activity was normalized to Renilla luciferase activity and each triplicate was then averaged. The values are expressed as a percent of control. In the case of HKc/HPV16 comparisons, the HKc/DR is expressed as a percent of HKc/HPV16. In the case of HPV16 oncogene infected HKc, the oncogene infected cell value is expressed as a percent of the LXSN infected control.
2.5. Infection of normal HKc with retroviruses expressing the HPV16 oncogenes E6 and E7
PA317 retrovirus producing lines, containing LXSN retroviruses expressing the HPV16 oncogenes E6 and E7 were a gift from Dr. Denise Galloway. Normal HKc were seeded onto 60-mm dishes at passage 0. At 50% confluency, a single 60-mm dish was infected with LXSN, LXSN16E7, LXSH16E6, LXSN16E6/E7 retroviruses, or mock infected. Approximately 24 h after infection the cells were trypsinized and plated for selection into three to five 100-mm dishes, depending on confluency, along with one 100-mm dish of mock infected cells. At 24 h after plating, cells were fed complete media containing G418 (125 μg/ml for LXSN retroviruses) or hygromycin (10 μg/ml for the LXSH retrovirus) and allowed to grow in the selection media for 5 to 8 days. After selection cells were placed in complete media without antibiotics prior to plating for experiments.
2.6. Analysis for mutations in the TGF-β RI promoter
DNA was extracted from a single 100-mm dish of 10 different individual normal HKc strains and four independently-derived HKc/HPV16 and HKc/DR lines, each established from a different donor (designated HKc/HPV16d-1, -2, -4, and -5) using the GenElute Mammalian Genomic DNA Miniprep Kit (Sigma, St. Louis, MO), according to manufacturer’s instructions. The following two sets of primers were designed to amplify 1.3 Kb of the TGF-β RI promoter in two overlapping fragments, the nucleotides of the sequencing primer (Sp6 and T7 sites, respectively) are underlined; Fragment 1 forward primer (corresponding to TGF-β RI promoter nucleotides -1422 to -1403) GAT TTA GGT GAC ACT ATA GTC TCA GAT CCC AGC TCC AAT; Fragment 1 reverse primer (corresponding to TGF-β RI promoter nucleotides -644 to -663) CCG AAG ATC CCT CCT CAG TGC C; Fragment 2 forward primer (corresponding to TGF-β RI promoter nucleotides -710 to -693) TAA TAC GAC TCA CTA TAG GGG ACC GCT GGG AGC AGG AG; and Fragment 2 reverse primer (corresponding to TGF-β RI promoter nucleotides +14 to -5) ACC GCC GCC TCC ATG GTC C. PCR was performed using HotStar Taq DNA Polymerase along with 1x Solution Q (Qiagen, Valencia, CA). DNA sequencing was conducted by the DNA sequencing facility in the Department of Biology at the University of South Carolina and the sequences aligned against the DNA sequence published by Bloom et al. [15] in combination with GenBank accession number U51139.
2.7. TGF-β receptor type I promoter methylation analysis
The following four sets of primers were designed in order to determine the methylation status of both strands of the TGF-β RI promoter in the areas surrounding the two distal Sp1 sites and the two (three) proximal Sp1 sites; Sense strand 1 forward primer (corresponding to TGF-β RI promoter nucleotides -1143 to -1116) 5’ GAT TTA GGT GAC ACT ATA GGA TAG GAA GAA TTA TAA ATT TGG AGT TT 3’; Sense strand 1 reverse primer (corresponding to TGF-β RI promoter nucleotides -773 to -798) 5’ ACC CAA ATC CTA AAC CCA AAC ACA CC 3’; Sense strand 2 forward primer (corresponding to TGF-β RI promoter nucleotides -434 to -408) 5’ GAT TTA GGT GAC ACT ATA GGA AAG AGA TTT ATA TAG ATA TAT TTA T 3’; Sense strand 2 reverse primer (corresponding to TGF-β RI promoter nucleotides -44 to -65) 5’ CTA CCT CAC CCC AAC AAA CCT C 3’; Antisense strand 1 forward primer (corresponding to TGF-β RI promoter nucleotides -759 to -787) 5’ TAA TAC GAC TCA CTA TAG GGG ATT TTA GTG TTA GAT TTA GAT TTT GAG T 3’; Antisense strand 1 reverse primer (corresponding to TGF-β RI promoter nucleotides -1022 to -984) 5’ CAC CTC AAA ATT ATT ATA CAA CAA TAT CAA ACA CAA CTC 3’; Antisense strand 2 forward primer (corresponding to TGF-β RI promoter nucleotides -38 to -64) 5’ TAA TAC GAC TCA CTA TAG GGG TGT TGT TGT TTT ATT TTA GTA AAT TT 3’; and Antisense strand 2 reverse primer (corresponding to TGF-β RI promoter nucleotides -428 to -399) 5’ AAC TCA CAC AAA CAC ACC CAT CAC TCT ACC 3’. The underlined portion of the primer sequences represents either a Sp6 site (sense strand forward primer set 1 and 2) or a T7 site (antisense strand forward primer set 1 and 2) added to the primer for sequencing purposes. Note that the nucleotide numbering for the TGF-β RI promoter is based on that described by Bloom et al. [15].
DNA was extracted from 10 different normal HKc strains as well from our HKc/HPV16 and HKc/DR lines (HKc/HPV16d-1, -2, -4, and -5) using the Genelute mammalian genomic DNA extraction kit (Sigma, St. Louis, MO) according to the manufacturer’s instructions. Methylation analysis of the TGF-β receptor type I promoter was conducted on DNA (1 μg) from normal HKc and the HKc/HPV16 cell lines, along with a DNA methylation positive control (Intergen, Purchase, NY) and the TGF-β RI promoter luciferase plasmid. The DNA was bisulfite treated using the CpGenome DNA Modification kit (Intergen, Purchase, NY) for 17 to 19 h. The bisulfite treated DNA was then subjected to PCR amplification using the primer sets described above and purified PCR product was then sequenced. Primers yielded products of approximately 300-400 bases. The products were sequenced and then aligned using VectorNTI Suite 6 software (Invitrogen Corporation, Carlsbad, CA) using default settings.
2.8. Immunoblots for Sp1 and Sp3 in HKc/HPV16 and HKc/DR
Nuclear extracts were prepared from our HKc/HPV16 and HKc/DR lines (HKc/HPV16d-1, -2, -4, -5), cultured in complete media for 48 h, as described by Baldwin et al. [19]. Western blots for Sp1 (10 μg total nuclear protein) or Sp3 (20 μg total nuclear protein) were conducted using a 1:1000 dilution of either an anti-Sp1 or an anti-Sp3 polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) along with a 1:1000 dilution of an anti-actin antibody (Sigma, St. Louis, MO). Membranes were incubated with the primary antibodies for 1 h at room temperature with shaking, rinsed, and then incubated with a 1:500 dilution of a goat anti-rabbit polyclonal HRP-conjugated secondary antibody (Transduction Laboratories, Lexington, KY). Proteins were detected using SuperSignal Chemiluminescence substrate (Pierce, Rockford, IL).
2.9. Electrophoretic mobility shift assays (EMSA) of the TGF-β RI promoter
EMSAs were performed using nuclear extracts from the HKc/HPV16 and HKc/DR lines. Oligonucleotides, approximately 20 base pairs in length, were chosen to include the two proximal Sp1 binding sites in the TGF-β RI promoter. Double stranded complementary oligonucleotides with the following sequences were used in the EMSAs. Each oligonucleotide will be referred to based on the position of the 5’ most nucleotide in the TGF-β RI promoter:, oligonucleotide -174, CGG GGA GGC GGG GCC GGC GGG, -363 GCT GGC AGA CCC CGC CCC CAC G; Random, ACT GAA TCA CTA TGT ACA TTG TGT CAT A. The underlined nucleotides represent potential Sp1 binding sites. Each reaction contained 12 μg of nuclear extract. All competing unlabeled oligonucleotides were added to the reaction along with the nuclear extracts at a concentration 125x that of the labeled probe. In the case of supershift analysis, antibody (1 μg protein) against Sp1 (Santa Cruz, Pep2) or Sp3 (Santa Cruz, D-20) was added after probe addition. All samples were run on a nondenaturing polyacrylamide gel (4%) at room temperature in 1x Tris/glycine buffer.
2.10. RNA turnover studies for TGF-β receptor type I and TGF-β receptor type II in HKc/HPV16 and HKc/DR
HKc/HPV16d-1 and HKc/DRd-1 were seeded into 100-mm dishes and grown in complete media. Treatment with 5,6-dichloro-1-β-D-ribofuranosyl-benzimidazole (DRB), (50 ug/ml, Sigma, St. Louis, MO) was began when the cells were approximately 75% confluent. RNA was extracted from one 100-mm dish at various times (0, 4, 8, 12, and 16, 24 h) following DRB treatment using the RNeasy mini kit (Qiagen, Valencia, CA). To compare RNA turnover rates of the TGF-β receptors in HKc/HPV16 to HKc/DR, RNA collected at different times after DRB treatment was reverse transcribed and TGF-β RI and TGF-β RII mRNA levels determined by Real Time PCR. Samples were assayed in duplicate from two separate experiments using specific primers for TGF-β RI and the TGF-β RII (see section 2.2 above for primer sequences).
3. Results
3.1. mRNA levels of TGF-β RI, but not TGF-β RII, decrease during in vitro progression of HPV16-immortalized HKc
Using ribonuclease protection assays, we previously reported that resistance to growth inhibition by TGF-β in HKc/DR was linked to reduced steady-state levels of mRNA for TGF-β RI, but not TGF-β RII, when compared to TGF-β sensitive HKc/HPV16 [14]. Using Real Time PCR, we have now compared TGF-β RI and TGF-β RII mRNA levels at the HKc/HPV16 and HKc/DR stages from individually established HPV16-immortalized cell lines (designated HKc/HPV16d-1, -2, -4, and -5). We found that the steady-state levels of TGF-β RI mRNA decreased 18-72% from the HKc/HPV16 stage to HKc/DR, while the mRNA levels of TGF-β RII remained relatively unchanged (data not shown).
3.2. Acute expression of the HPV16 oncogenes in normal HKc decreases both TGF-β RI and TGF-β RII promoter activity
To investigate what role the HPV16 oncogenes E6 and E7 may play in TGF-β receptor mRNA expression, we examined the effect of acute expression of E6 and E7 in normal HKc on TGF-β RI and RII promoter activity. We infected primary normal HKc with retroviruses expressing either HPV16 E6 or HPV16 E7 alone or HPV16 E6 and E7 together. After selection with the appropriate antibiotics, the cells were plated and transfected with the TGF-β RI promoter-Luc or the TGF-β RII promoter-Luc reporter constructs, along with pRL-Null Renilla luciferase as a control for transfection efficiency. Luciferase activity was measured 72 h after transfection. We observed a 40-60% decrease in the promoter activity of both TGF-β RI and TGF-β RII with acute expression of HPV16 E6 or HPV16 E7, with no synergism when the HPV16 E6 and E7 oncogenes were expressed together (Fig. 1). These results demonstrate that the acute expression of the HPV oncogenes E6 and E7 decreases TGF-β receptor type I and type II promoter activity to approximately the same extent and thus do not explain our finding that mRNA levels for only the TGF-β receptor type I (but not TGF-β receptor type II) are reduced in HKc/DR compared to HKc/HPV16.
Fig. 1.
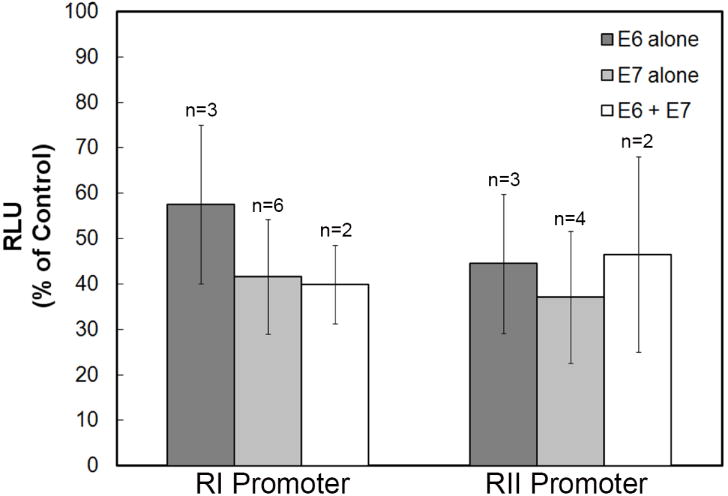
Effect of acute expression of the HPV16 oncogenes in normal HKc on TGF-β RI and RII promoter activity. Normal HKc were infected with retroviruses containing either HPV16 E6 or HPV16 E7 alone or HPV16 E6 and E7 together or with control LXSN retrovirus, selected, and transfected with 2 μg of the TGF-β RI promoter-Luc construct or a TGF-β RII promoter-Luc reporter construct, along with pRL-Null Renilla luciferase as a control for transfection efficiency. Luciferase activity was measure using the Dual Luciferase Assay Kit 72 h after transfection. The data are presented as a percent of the vector infected control (LXSN). n=the number of different normal HKc strains used for each condition and the error bars represent SD.
3.3. TGF-β RI promoter activity is decreased to a greater extent than that of the TGF-β RII promoter as HKc/HPV16 progress in vitro
Since HKc/DR express less TGF-β RI mRNA than HKc/HPV16, we compared the promoter activity of TGF-β RI and TGF-β RII in HKc/HPV16d-1 and HKc/DRd-1 using the TGF-β receptor promoter reporter constructs. The TGF-β RI promoter-Luc or the TGF-β RII promoter-Luc construct was transfected into HKc/HPV16 or HKc/DR along with the Renilla luciferase control plasmid (pRL-null). Luciferase activity was measured 48 h after transfection and values were normalized to Renilla. As shown in Fig. 2, the promoter activity for TGF-β RI is consistently decreased to a greater extent than the promoter activity for TGF-β RII in HKc/DR compared to HKc/HPV16 (approximately 65% for TGF-β RI versus 35% for TGF-β RII).
Fig. 2.
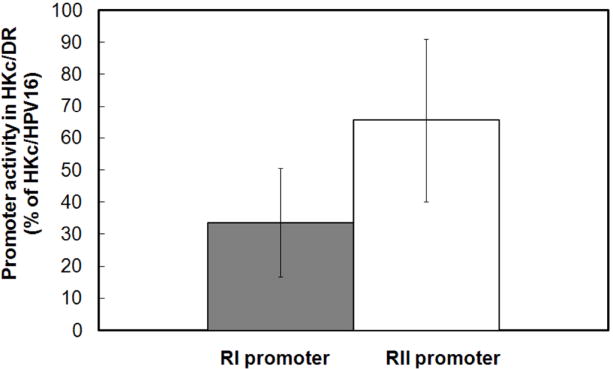
TGF-β RI and RII promoter activity in HKc/DR compared to HKc/HPV16. HKc/HPV16d-1 and HKc/DRd-1 were transfected with either 2 μg of the TGF-β RI promoter-Luc or the TGF-β RII promoter-Luc reporter constructs along with pRL-Null Renilla luciferase as a control for transfection efficiency. Luciferase activity was measured using the Dual Luciferase Assay Kit 48 h after transfection. After normalization for Renilla, the luciferase activity measured in HKc/DR is expressed as a percent of the luciferase activity measured in HKc/HPV16. The data shown are the average of seven experiments. The error bars represent SD.
3.4. Mutations in the TGF-β RI promoter are not responsible for decreased expression of TGF-β RI as HKc/HPV16 progress in vitro
We next examined the promoter region of the TGF-β RI gene in the HKc/HPV16 lines for mutations that may be responsible for decreased TGF-β RI expression in HKc/DR. We designed two sets of primers that would allow us to amplify approximately 1400 base pairs 5’ of the translation start site in two overlapping fragments (-1395 to -652 and -683 to +5). DNA was extracted from the HKc/HPV16 and HKc/DR stages from each of our individually derived HPV16-immortalized lines (HKc/HPV16d-1, -2, -4, and -5), along with ten different normal HKc strains. The TGF-β RI promoter was amplified by PCR and the DNA sequence of the products determined. We aligned our samples against the sequence published by Bloom et al. [15] and found inserts at nucleotides −194 (a C), -96 (a C), and −23 to −21 (CGG). However, our sequence was identical to the human genome sequence using BLAST, indicating that the Bloom et al. [15] sequence was likely incorrect. Thus we did not identify any mutations in TGF-β RI promoter in the HPV16 transformed cell lines, nor did we observe any polymorphisms in the TGF-β RI promoter in the 14 individuals examined (ten normal HKc strains and four independently-derived cell lines).
3.5. Methylation analysis of the TGF-β RI promoter
Methylation of DNA is a common epigenetic event that usually results in decreased rates of gene transcription. We thus explored whether methylation of the TGF-β RI promoter may be playing a role in the decreased expression of TGF-β RI we observe in HKc/DR. We used the CpGenome DNA modification kit (Intergen) to bisulfite treat DNA isolated from ten normal HKc strains and also DNA isolated from the four HPV16-immortalized cell lines (HKc/HPV16d-1, -2, -4, and -5) at both the HKc/HPV16 and HKc/DR stages. Universal methylated DNA (Intergen) served as a positive control, while bisulfite treated TGF-β RI promoter-Luc plasmid was used as a negative control. Four different primer sets were designed, two against the sense and two against the antisense strand which allowed us to explore for methylation in areas surrounding the two proximal Sp1 sites and the two distal Sp1 sites. We found no evidence for methylation in any of the amplified products. However, as expected, in the methylation positive control we did observe methylation of cytosine bases that were followed by guanines. Since we failed to find methylation of the TGF-β RI promoter in the normal HKc strains or the HPV16-immortalized cell lines at either the HKc/HPV16 or HKc/DR stages, promoter methylation is unlikely to be responsible for the decreased expression of the TGF-β RI in HKc/DR.
3.6. Sp1 and Sp3 nuclear protein levels do not change dramatically during in vitro progression of HKc/HPV16
Using nuclear extracts isolated from HKc/HPV16 and HKc/DR from each of our four independently derived cell lines (HKc/HPV16d-1, -2, -4, and -5) we performed Western blots to determine whether or not the protein levels of the transcription factors Sp1 and Sp3 change during in vitro progression. The anti-Sp1 antibody used recognizes isoforms of Sp1, of approximately 95 and 105 kDa; the 105 kDa isoform is suggested to be due to phosphorylation [20,21]. Actin was used as a loading control for each of the blots. We found no dramatic differences in the protein levels of Sp1 between the HKc/HPV16 and HKc/DR stages (Fig. 3A). These results indicate altered protein levels of Sp1 at the HKc/DR stage are unlikely to be responsible for the changes in TGF-β RI expression that we observe during in vitro progression. Overall, the protein level of Sp1 among the cell lines is relatively uniform.
Fig. 3.
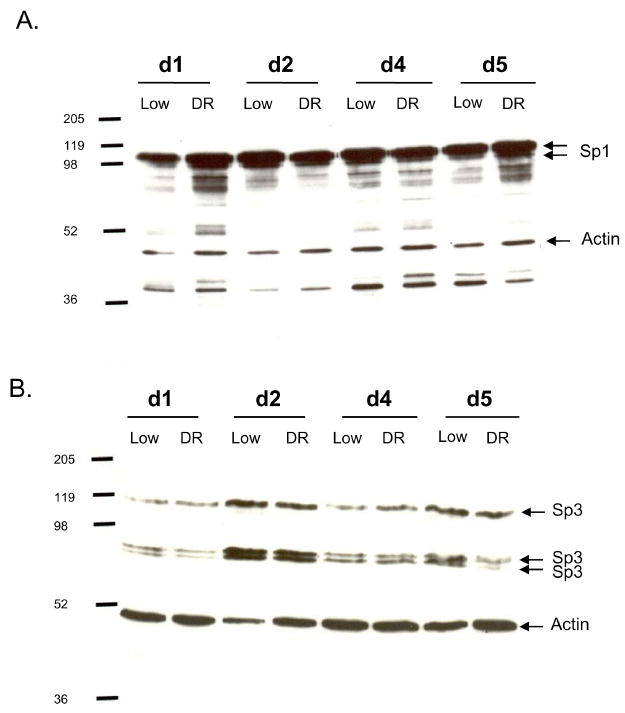
Western blots for Sp1 and Sp3 in HKc/HPV16 and HKc/DR lines. A. Western blot analysis for Sp1 and actin (loading control) was performed using nuclear extracts (10 μg protein) from each of four HPV16-immortalized cell lines (HKc/HPV16d-1, -2, -4, and -5) at both the HKc/HPV16 (Low) and the HKc/DR (DR) stages of in vitro progression. Molecular weight (kDa) markers are labeled to the left of the gel and arrows point to Sp1 and actin. B. Western blot analysis for Sp3 and actin was performed using nuclear extracts (20 μg protein) from each of the HKc/HPV16 cell lines as described above. Molecular weight markers (kDa) are labeled to the left of the gel and arrows point to Sp3 and actin bands.
The Sp3 antibody that we used in our Western blots recognizes three isoforms of Sp3: 78, 80, (arising from differential internal translational initiation) [17] and 115 kDa, the presumably biologically active form. Like Sp1, the nuclear levels of Sp3 did not show dramatic differences between the HKc/HPV16 and the HKc/DR stages, although we do see some differences among the cell lines (Fig. 3B). For example, the greatest level of Sp3 protein was observed in HKc/HPV16d-2. Since the Sp1/Sp3 ratios are known to be important for transcriptional activity, we would have expected the Sp1 levels to decrease or the Sp3 levels to increase as HKc/HPV16 progress to the HKc/DR stage.
3.7. EMSA analysis of Sp1 and Sp3 binding sites within the TGF-β RI promoter
Since Western blots showed no major differences in the nuclear protein levels of Sp1 and Sp3 we decided to investigate whether or not there was any differences between HKc/HPV16 and HKc/DR in the DNA binding activity of these transcription factors to their potential binding sites within the TGF-β RI promoter. We performed electrophoretic mobility shift assays (EMSAs) using labeled oligonucleotides (-363 and −174 probes) that encompassed the two proximal Sp1 binding sites within the TGF-β RI promoter, and nuclear extracts isolated from HKc/HPV16d-4 and its corresponding HKc/DR line. As shown in Fig. 4, we observed three separate bands of protein binding to each of the probes, but did not find any major differences in total binding when comparing HKc/HPV16 to HKc/DR (compare lanes 1 and 2 for probe -174 and lanes 5 and 6 for probe -363). To confirm the protein binding to the −174 and −363 probes was specific, we performed competition with unlabeled oligonucleotides at 125-fold excess of labeled probe, which completely inhibited binding to the labeled probes (Fig. 4, lanes 3 and 4 for probe -174 and lanes 7 and 8 for probe -363). Also binding to the -363 probe could in great part be competed by excess unlabeled -174 probe (Fig. 4, lanes 9 and 10). Binding levels of the fastest migrating band are inconsistent and competed by excess unlabeled oligonucleotides lacking a Sp1/Sp3 site (not shown); therefore this band is considered to be nonspecific (NS). Similar EMSA results using both the -174 and -363 probes were obtained using nuclear extracts isolated from the other three independently derived HKc/HPV16 lines and their HKc/DR counterparts (data not shown). Overall we conclude that nuclear proteins bind with specificity to both the −174 and −363 probes from the TGF-β RI promoter which encompass the two proximal Sp1 binding sites. However, the extent of binding to these probes is similar between HKc/HPV16 and HKc/DR.
Fig. 4.
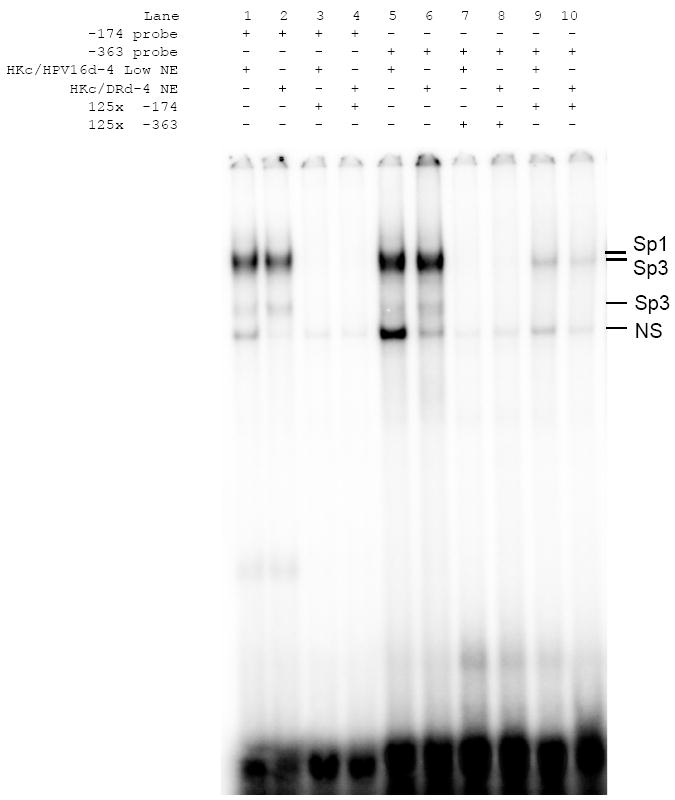
EMSA analysis of an Sp1 binding sites in the TGF-β RI promoter. EMSAs were performed on 4% polyacrylamide gels using nuclear extracts (12 μg protein) from HKc/HPV16d-4 and HKc/DRd-4. Double stranded oligonucleotides matching the sequence surrounding the Sp1 site at −363 in the TGF-β RI promoter were end labeled with 32P. Competition assays, using unlabeled oligonucleotides, were performed to demonstrate binding specificity. All competitors were added at 125-fold excess over the respective labeled probe. The probe/protein complexes are labeled to the right of the gel (NS=nonspecific binding).
To confirm that the nuclear proteins binding to the −363 probe were the transcription factors Sp1 and Sp3 we performed supershift assays (Fig. 5). As expected, similar amounts of protein bound to the probes when nuclear extracts from HKc/HPV16d-4 and HKc/DRd-4 were compared (Fig. 5, lanes 1 and 2). The addition of an anti-Sp1 antibody to the EMSA resulted in a supershifting of the uppermost protein band (Fig. 5, lanes 3 and 4). Sp3 has been shown to migrate as two bands in EMSA [22]. We were able to partially shift the two presumptive Sp3 bands with anti-Sp3 antibodies (Figure 5, lanes 5 and 6). There was no apparent difference in the amount of Sp1 or Sp3 shifted between the HKc/HPV16d-4 and HKc/DRd-4 stages. Similar supershift results were obtained using the -174 probe and nuclear extracts from the HKc/HPV16d-4 and HKc/DRd-4 lines as well as the -363 and -174 probes and nuclear extracts from the HKc/HPV16d-1 and HKc/DRd-1 lines (data not shown).
Fig. 5.
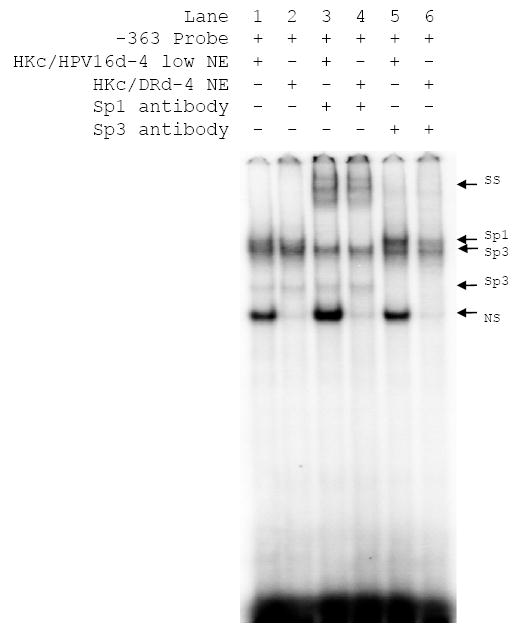
Supershift analysis for Sp1/Sp3 binding to an Sp1 binding site in the TGF-β RI promoter. Double stranded oligonucleotides matching the sequence surrounding the Sp1 site at −363 in the TGF-β RI promoter were end labeled with 32P. Nuclear extracts (12 μg protein) from HKc/HPV16d-4 or HKc/DRd-4 were used in the EMSAs. Supershifts analysis were performed as indicated using anti-Sp1 or anti-Sp3 antibodies (1 μg protein) which were added after the labeled probe was incubated with the nuclear extracts. The probe/protein complexes are labeled to the right of the gel (SS=supershift, NS=nonspecific binding).
We conclude from the EMSA/supershift analysis with the probes encompassing the two proximal Sp1 binding sites of the TGF-β RI promoter that the transcription factors Sp1 and Sp3 bind to these sites. However, there is no difference in the level of Sp1 or Sp3 binding between the HKc/HPV16 and HKc/DR stages.
3.8. The mRNA turnover rate of both TGF-β receptors type I and type II are increased in HKc/DR
The turnover rate for TGF-β RI and the TGF-β RII mRNA was examined in HKc/HPV16d-1 and HKc/DRd-1. The cells were treated with DRB (0, 4, 8, 12, and 16 h), the RNA was extracted at the various times and the mRNA levels for TGF-β RI and the TGF-β RII were determined using Real Time PCR. The half-life of TGF-β RI mRNA was 10.5 h in HKc/HPV16d-1 compared to 7.0 h for HKc/DRd-1, an increase in the turnover rate of TGF-β RI in HKc/DR of 3.5 h (Fig. 6A). We also observed a 4.25 h increase in the mRNA turnover rate of the TGF-β RII from (5.5 h to 9.75 h) in HKc/DR compared to HKc/HPV16 (Fig. 6B).
Fig. 6.
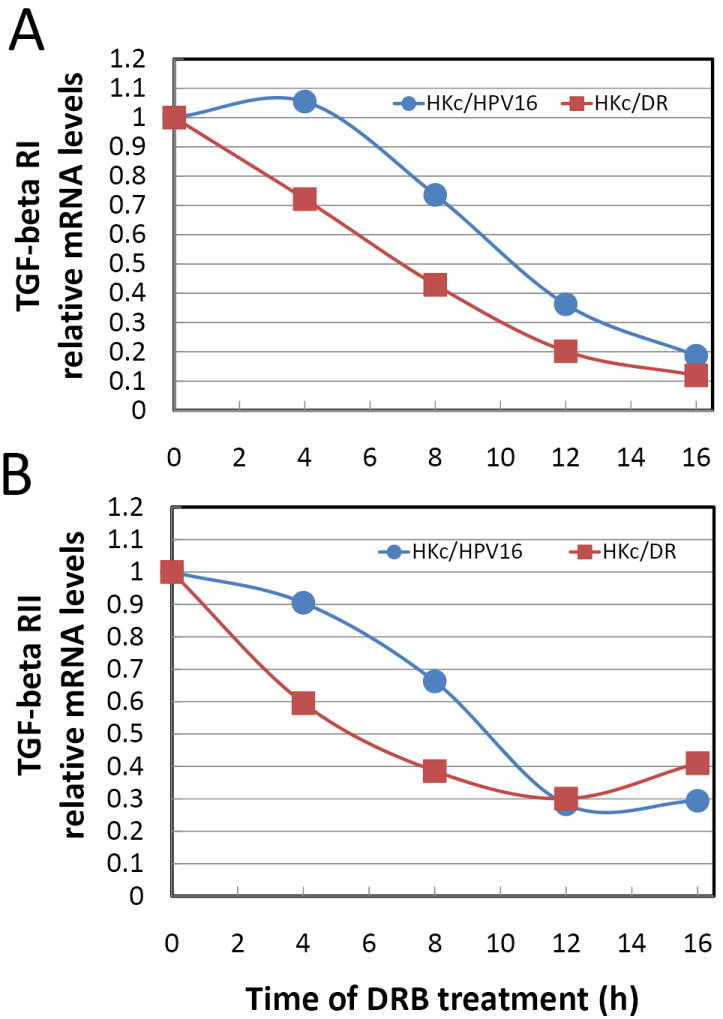
mRNA turnover for TGF- β RI and TGF-β RII in HKc/HPV16d-1 and HKc/DRd-1. The cells were treated with DRB for 0, 4, 8, 12, and 16 h, the RNA was extracted, and the mRNA levels for TGF-β RI (A) and the TGF-β RII (B) were determined using Real Time PCR. The data represent the average of two independent experiments.
4. Discussion
In our in vitro model of human cell carcinogenesis, normal HKc transfected with HPV16 DNA progress towards malignancy through growth factor-independent (HKc/GFI) and differentiation resistant (HKc/DR) stages [10-12]. Our prior studies determined that HKc/HPV16 are initially sensitive to the growth inhibitory effects of TGF-β but become increasingly resistant to the antiproliferative effects of TGF-β during in vitro progression, which we linked to reduced expression of the TGF-β RI but not TGF-β RII [14]. This study sought to explore several possible molecular mechanisms that may result in reduced TGF-β RI expression in HKc/DR.
Several studies have explored alterations in the TGF-β receptors in cervical cancer cell lines, cervical dysplasia, and cervical cancer and some molecular changes have been identified. For example, Kang et al. [23] reported a homologous deletion of the entire TGF-β RII gene in the cervical carcinoma cell line ME-180. However, 7 other cervical cancer cell lines studied that showed resistance or sensitivity to the growth inhibitory effects of TGF-β carry no genetic changes in TGF-β RI or TGF-β RII [23]. This is consistent with our results using Southern blot analysis, where we failed to find any gross structural alterations in TGF-β RI during in vitro progression [14]. Some structural alterations of the TGF-β RI and TGF-β RII genes were identified in paraffin-embedded invasive cervical carcinoma specimens, although these changes were infrequent [24]. Unlike microsatellite instability (MI) positive colorectal cancers, no deletions or insertions were found in the poly (A)10 tract of the TGF-β RII in MI-positive squamous intraepithelial lesions (SIL) and cervical cancer [25]. Using exon-specific PCR and SSCP-DNA sequencing, no aberrations of the TGF-B RII gene were found in 15 cervical cancer tissue samples [25].
Since our previous studies [14] as well as those discussed above [23-25] suggested that reduced expression of TGF-β RI in HKc/DR was unlikely linked to gross structural alterations of the receptor gene, we explored whether mutations in the promoter of TGF-β RI gene or receptor methylation may lead to altered TGF-β RI expression. Hypermethylation of the TGF-β RI gene in human gastric cancer cell lines [18] and of the TGF-β RII gene in murine renal cell carcinoma cells [26] have been correlated with a loss of receptor expression and TGF-β resistance. However, we found no evidence for promoter methylation or promoter mutations playing any role in reducing TGF-β RI expression in HKc/DR. While some genetic alterations have been identified in the TGF-β RII gene promoter, these alterations are unlikely to play a key role for the decreased expression of TGF-β RII often found in head and neck squamous cell carcinoma [27].
The TGF-β RI gene is approximately 31 kb in length and consists of nine exons [28]. The TGF-β RI promoter is GC-rich, lacks a TATA and CAAT box, contains four consensus binding sites for the Sp1 family of transcription factors, and a consensus AP2 binding site [15]. The Sp1 family of transcription factors, including Sp1 and Sp3, are ubiquitously expressed and share the same DNA binding specificity [29]. Sp1 is generally considered to be a positive regulator of transcription while Sp3 is repressive [20]. However, Sp3 becomes an activator if acetylated [30]. Clearly, transcription factors of the Sp1 family are important regulators of both TGF-β RI and TGF-β RII promoters. Sp1 deficiency has been linked to reduced TGF-β RI expression in the GEO human colon carcinoma cell line [16,31] and TGF-β RII expression in late passage MCF-7L human breast cancer cells [32] and the pancreatic cancer cell line MIA PaCa-2 [33]. Furthermore, Sp3 acts as a transcriptional repressor of TGF-β RI and TGF-β RII in GEO and MCF-7L breast cancer cells and both cell lines have increased levels of Sp3 protein [17,22]. However, treatment of MCF-7L cells with the histone deacetylase inhibitor trichostatin A induces acetylation of Sp3, which then acts as a transcriptional activator of TGF-β RII [30]. Based on these studies we explored in detail whether or not altered Sp1 and Sp3 protein levels in HKc/DR might contribute to reduced expression of TGF-β RI. However, our Western blot studies exploring Sp1 and Sp3 failed to find any differences in the levels of these transcription factors between HKc/HPV16 and HKc/DR that would contribute to reduced TGF-β RI expression in HKc/DR. Furthermore, EMSA/supershift analysis determined that there was no change between HKc/HPV16 and HKc/DR in the binding of Sp1 and Sp3 to the two proximal Sp1 binding sites within the TGF-β RI promoter. Thus the mechanism leading to reduced TGF-β RI expression in HKc/DR must be different than that previously reported in MCF-7L and GEO cells [16,17,22,31,]. We also studied mRNA turnover for TGF-β RI and TGF-β RII in HKc/HPV16 and HKc/DR. We observed an increase in the mRNA turnover for both TGF-β RI and TGF-β RII as HKc/HPV16 progress to the HKc/DR stage. Thus it is unlikely that altered mRNA turnover explains the reduced steady state levels we find for TGF-β RI in HKc/DR even though mRNA levels for TGF-β RII are unchanged. Treatment of MCF-7L with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) results in the accumulation of acetylated histones in the chromatin of the TGF-β RI gene, increases TGF-β RI expression, and restored TGF-β responsiveness in these cells [34]. It will be important to explore whether histone deacetylation may play a role in transcriptional regulation/silencing of TGF-β RI in HKc/DR.
The HPV16 oncogenes E6 and E7 have been shown to interact with multiple cellular proteins [35], raising the possibility that these interactions could somehow affect TGF-β receptor expression. To explore this possibility we acutely expressed the HPV16 E6 and E7 in normal HKc and measured the promoter activity of TGF-β RI and TGF-β RII using luciferase-based reporter constructs. We observed that acute expression of HPV16 E6 or E7 decreased by 40-60% the promoter activity of both the TGF-β RI and TGF-β RII promoter constructs. At present there is no evidence that the HPV16 E6 or E7 oncoproteins themselves bind directly to sequences within the TGF-β RI or TGF-β RII promoters. Thus, while these experiments show that E6 and E7 expression may influence TGF-β RI and TGF-β RII promoter activity, they do not explain the differential loss of TGF-β RI mRNA expression that we observe in HKc/DR. We have previously reported that basal TGF-β RI promoter activity decreased during in vitro progression of HKc/HPV16 [14] but had not explored TGF-β RII promoter activity. Interestingly in the present study we determined that as HKc/HPV16 progress to the HKc/DR stage the promoter activity of TGF-β RI is consistently decreased to a greater extent than TGF-β RII, indicating that changes in promoter activity coupled with an increased turnover of the RNA, may be at the basis of the loss of TGF-β RI mRNA expression we observe in HKc/DR, and that HPV16 oncoproteins may drive at least the decrease in promoter activity for this receptor. However, the reason for the differential expression of TGF-β RI and TGF-β RII in HKc/DR, and the specific mechanisms by which E6 and E7 influence the activity of their promoters remain to be determined
In summary, we have shown that decreased expression of the TGF-β RI in HKc/DR is not due to mutations in or hypermethylation of the TGF-β RI promoter, nor is it due to changes in the overall protein levels of the transcription factors Sp1 or Sp3, or the binding of these transcription factors to the two proximal Sp1 binding sites within the TGF-β RI promoter. We have uncovered inhibitory effects of the HPV16 oncoproteins E6 and E7 on TGF-β RI and RII promoter activity. Future studies will explore the molecular basis for this inhibition, as well as the role that posttranslational modifications of Sp1 and Sp3 (phosphorylation and acetylation) and chromatin structure surrounding the TGF-β RI gene may play in decreased expression of TGF-β RI as HKc/HPV16 progress in vitro.
Acknowledgments
This research was supported by Grant R01CA89502 from the National Cancer Institute and Grant 5P20MD001770 from the National Center on Minority Health and Health Disparities. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center on Minority Health and Health Disparities of the National Institutes of Health.
Footnotes
Conflict of interest Statement None Declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huang SS, Huang JS. TGF-β control of cell proliferation. J Cell Biochem. 2005;96:447–462. doi: 10.1002/jcb.20558. [DOI] [PubMed] [Google Scholar]
- 2.Massague J, Gomis RR. The logic of TGF-β signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 3.Rahimi RA, Leof EB. TGF-β signaling: A tale of two responses. J Cell Biochem. 2007;102:593–608. doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- 4.Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol Lett. 2002;82:85–91. doi: 10.1016/s0165-2478(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 5.Mehra A, Wrana JL. TGF-beta and the Smad signal transduction pathway. Biochem Cell Biol. 2002;80:605–622. doi: 10.1139/o02-161. [DOI] [PubMed] [Google Scholar]
- 6.Feng X-H, Derynck R. Specificity and versatility in TGF-β signaling through smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 7.Nakao A, Imamura T, Souchelnytskyi S, Kawabata M, Ishisaki A, Oeda E, Tamaki K, Hanai J, Heldin CH, Miyazono K, ten Dijke P. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. Embo J. 1997;16:5353–5362. doi: 10.1093/emboj/16.17.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardali K, Moustakas A. Actions of TGF-β as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Jakowlew SB. Transforming growth factor-β in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–457. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- 10.Creek KE, Geslani G, Batova A, Pirisi L. Progressive loss of sensitivity to growth control by retinoic acid and transforming growth factor-beta at late stages of human papillomavirus type 16-initiated transformation of human keratinocytes. Adv Exp Med Biol. 1995;375:117–135. doi: 10.1007/978-1-4899-0949-7_11. [DOI] [PubMed] [Google Scholar]
- 11.Pirisi L, Creek KE, Doniger J, DiPaolo JA. Continuous cell lines with altered growth and differentiation properties originate after transfection of human keratinocytes with human papillomavirus type 16 DNA. Carcinogenesis. 1988;9:1573–1579. doi: 10.1093/carcin/9.9.1573. [DOI] [PubMed] [Google Scholar]
- 12.Creek KE, Jenkins GR, Khan MA, Batova A, Hodam JR, Tolleson WH, Pirisi L. Retinoic acid suppresses human papillomavirus type 16 (HPV16)-mediated transformation of human keratinocytes and inhibits the expression of the HPV16 oncogenes. Adv Exp Med Biol. 1994;354:19–35. doi: 10.1007/978-1-4899-0939-8_2. [DOI] [PubMed] [Google Scholar]
- 13.Borger DR, Mi Y, Geslani G, Zyzak LL, Batova A, Engin TS, Pirisi L, Creek KE. Retinoic acid resistance at late stages of human papillomavirus type 16-mediated transformation of human keratinocytes arises despite intact retinoid signaling and is due to a loss of sensitivity to transforming growth factor-beta. Virology. 2000;270:397–407. doi: 10.1006/viro.2000.0282. [DOI] [PubMed] [Google Scholar]
- 14.Mi Y, Borger DR, Fernandes PR, Pirisi L, Creek KE. Loss of transforming growth factor-beta (TGF-beta) receptor type I mediates TGF-beta resistance in human papillomavirus type 16-transformed human keratinocytes at late stages of in vitro progression. Virology. 2000;270:408–416. doi: 10.1006/viro.2000.0283. [DOI] [PubMed] [Google Scholar]
- 15.Bloom BB, Humphries DE, Kuang PP, Fine A, Goldstein RH. Structure and expression of the promoter for the R4/ALK5 human type I transforming growth factor-β receptor: regulation by TGF-β. Biochim Biophys Acta. 1996;1312:243–248. doi: 10.1016/0167-4889(96)00043-2. [DOI] [PubMed] [Google Scholar]
- 16.Periyasamy S, Ammanamanchi S, Tillekeratne MP, Brattain MG. Repression of transforming growth factor-beta receptor type I promoter expression by Sp1 deficiency. Oncogene. 2000;19:4660–4667. doi: 10.1038/sj.onc.1203822. [DOI] [PubMed] [Google Scholar]
- 17.Ammanamanchi S, Brattain MG. Sp3 is a transcriptional repressor of transforming growth factor-beta receptors. J Biol Chem. 2001;276:3348–3352. doi: 10.1074/jbc.M002462200. [DOI] [PubMed] [Google Scholar]
- 18.Kang SH, Bang YJ, Im YH, Yang HK, Lee DA, Lee HY, Lee HS, Kim NK, Kim SJ. Transcriptional repression of the transforming growth factor-β type I receptor gene by DNA methylation results in the development of TGF-β resistance in human gastric cancer. Oncogene. 1999;18:7280–7286. doi: 10.1038/sj.onc.1203146. [DOI] [PubMed] [Google Scholar]
- 19.Baldwin A, Pirisi L, Creek KE. NFI-Ski interactions mediate transforming growth factor beta modulation of human papillomavirus type 16 early gene expression. J Virol. 2004;78:3953–3964. doi: 10.1128/JVI.78.8.3953-3964.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouwman P, Philipsen S. Regulation of the activity of Sp1-related transcription factors. Mol Cell Endocrinol. 2002;195:27–38. doi: 10.1016/s0303-7207(02)00221-6. [DOI] [PubMed] [Google Scholar]
- 21.Jackson SP, MacDonald JJ, Lees-Miller S, Tjian R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell. 1990;63:155–165. doi: 10.1016/0092-8674(90)90296-q. [DOI] [PubMed] [Google Scholar]
- 22.Ammanamanchi S, Brattain MG. 5-azaC treatment enhances expression of transforming growth factor-beta receptors through down-regulation of Sp3. J Biol Chem. 2001;276:32854–32859. doi: 10.1074/jbc.M103951200. [DOI] [PubMed] [Google Scholar]
- 23.Kang SH, Won K, Chung HW, Jong HS, Song YS, Kim SJ, Bang YJ, Kim NK. Genetic integrity of transforming growth factor β (TGF-β) receptors in cervical carcinoma cell lines: loss of growth sensitivity but conserved transcriptional response to TGF-β. Int J Cancer. 1998;77:620–625. doi: 10.1002/(sici)1097-0215(19980812)77:4<620::aid-ijc23>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 24.Chen T, de Vries EG, Hollema H, Yegen HA, Vellucci VF, Strickler HD, Hildesheim A, Reiss M. Structural alterations of transforming growth factor-β receptor genes in human cervical carcinoma. Int J Cancer. 1999;82:43–51. doi: 10.1002/(sici)1097-0215(19990702)82:1<43::aid-ijc9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 25.Chu TY, Lai JS, Shen CY, Liu HS, Chao CF. Frequent aberration of the transforming growth factor-β receptor II gene in cell lines but no apparent mutation in pre-invasive and invasive carcinomas of the uterine cervix. Int J Cancer. 1999;80:506–510. doi: 10.1002/(sici)1097-0215(19990209)80:4<506::aid-ijc4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Q, Rubenstein JN, Liu VC, Park I, Jang T, Lee C. Restoration of expression of transforming growth factor-β type II receptor in murine renal cell carcinoma (renca) cells by 5-aza-2’-deoxycytidine. Life Sciences. 2005;76:1159–1166. doi: 10.1016/j.lfs.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 27.Seijo ER, Song H, Lynch MA, Jennings R, Qong X, Lazaridis E, Muro-Cacho C, Weghorst CM, Munoz-Antonia T. Identification of genetic alterations in the TGFβ type II receptor gene promoter. Mut Research. 2001;483:19–26. doi: 10.1016/s0027-5107(01)00217-2. [DOI] [PubMed] [Google Scholar]
- 28.Vellucci VF, Reiss M. Cloning and genomic organization of the human transforming growth factor-β type I receptor gene. Genomics. 1997;46:278–283. doi: 10.1006/geno.1997.5023. [DOI] [PubMed] [Google Scholar]
- 29.Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- 30.Ammanamanchi S, Freeman JW, Brattain MG. Acetylated Sp3 is a transcriptional activator. J Biol Chem. 2003;278:35775–35780. doi: 10.1074/jbc.M305961200. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Han W, Zborowska E, Liang J, Wang X, Willson JKV, Sun L, Brattain MG. Reduced expression of transforming growth factor β type I receptor contributes to the malignancy of human colon carcinoma cells. J Biol Chem. 1996;271:17366–17371. doi: 10.1074/jbc.271.29.17366. [DOI] [PubMed] [Google Scholar]
- 32.Ammanamanchi S, Kim S-J, Sun L-Z, Brattain MG. Induction of transforming growth factor-β receptor type II expression in estrogen receptor-positive breast cancer cells through Sp1 activation by 5-aza-2’-deoxycytidine. J Biol Chem. 1998;273:16527–16534. doi: 10.1074/jbc.273.26.16527. [DOI] [PubMed] [Google Scholar]
- 33.Venkatasubbarao K, Ammanamanchi S, Brattain MG, Mimari D, Freeman JW. Reversion of transcriptional repression of Sp1 by 5 aza-2’ deoxycytidine restores TGF-beta type II receptor expression in the pancreatic cancer cell line MIA PaCa-2. Cancer Res. 2001;61:6239–6247. [PubMed] [Google Scholar]
- 34.Ammanamanchi S, Brattain MG. Restoration of transforming growth factor-β signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J Biol Chem. 2004;279:32620–32625. doi: 10.1074/jbc.M402691200. [DOI] [PubMed] [Google Scholar]
- 35.Thomas M, Narayan N, Pim D, Tomaic V, Massimi P, Nagasaka K, Kranjec C, Gammoh N, Banks L. Human papillomavirus, cervical cancer and cell polarity. Oncogene. 2008;27:7018–7030. doi: 10.1038/onc.2008.351. [DOI] [PubMed] [Google Scholar]