

Abstract
Time-resolved excited-state absorption intensities after direct two-photon excitation of the carotenoid S1 state are reported for light-harvesting complexes of purple bacteria. Direct excitation of the carotenoid S1 state enables the measurement of subsequent dynamics on a fs time scale without interference from higher excited states, such as the optically allowed S2 state or the recently discovered dark state situated between S1 and S2. The lifetimes of the carotenoid S1 states in the B800-B850 complex and B800-B820 complex of Rhodopseudomonas acidophila are 7 ± 0.5 ps and 6 ± 0.5 ps, respectively, and in the light-harvesting complex 2 of Rhodobacter sphaeroides ≈1.9 ± 0.5 ps. These results explain the differences in the carotenoid to bacteriochlorophyll energy transfer efficiency after S2 excitation. In Rps. acidophila the carotenoid S1 to bacteriochlorophyll energy transfer is found to be quite inefficient (φET1 <28%) whereas in Rb. sphaeroides this energy transfer is very efficient (φET1 ≈80%). The results are rationalized by calculations of the ensemble averaged time constants. We find that the Car S1 → B800 electronic energy transfer (EET) pathway (≈85%) dominates over Car S1 → B850 EET (≈15%) in Rb. sphaeroides, whereas in Rps. acidophila the Car S1 → B850 EET (≈60%) is more efficient than the Car S1 → B800 EET (≈40%). The individual electronic couplings for the Car S1 → BChl energy transfer are estimated to be approximately 5–26 cm−1. A major contribution to the difference between the energy transfer efficiencies can be explained by different Car S1 energy gaps in the two species.
Plants and some bacteria have achieved remarkably high efficiencies of solar light conversion in photosynthesis. Recently, high-resolution crystal structures of some peripheral light-harvesting complexes of photosynthetic purple bacteria (1–3) have become available, enabling us to study the basic principles of the highly efficient collection of solar energy (4–8). In the light-harvesting complexes of purple bacteria, such as light-harvesting complex 2 (LH2) of Rhodobacter sphaeroides or Rhodopseudomonas acidophila, carotenoids (Cars) play important roles as light-harvesting and photoprotective pigments, as they do in the chloroplasts of higher plants (9, 10). In Rb. sphaeroides >95% of the photons absorbed by the Cars are transferred as excitation energy to the reaction center; in the strain 7050 of Rps. acidophila, which is the strain used in this report, the corresponding number is ≈70% (9, 11, 12). In the subunits of the B800-B850 complex of Rps. acidophila there is one Car (rhodopin glucoside) molecule per two bacteriochlorophyll (BChl) molecules in the B850 ring and one BChl in the B800 ring (1). Nine of these subunits comprise the LH2 ring, B800-B850 complex, of Rps. acidophila. Spectroscopic and biochemical reports suggest that the structures of the B800-B820 complex of Rps. acidophila and LH2 of Rb. sphaeroides (with the Car spheroidene) are similar (13, 14).
Despite intensive investigation of the underlying mechanisms (8, 10, 12, 15–20), many aspects of the electronic energy transfer (EET) from the Cars are still poorly understood. The electronic states of Cars often are described by analogy to polyenes (Fig. 1): Their ground state (S0) and first excited state (S1) both possess Ag− symmetry, their second excited state (S2) possesses Bu+ symmetry in the idealized C2h point group. Only the S0 ↔ S2 transition is optically dipole-allowed. Excitation of S2 is followed by very rapid (τ21 ≈100–200 fs) internal conversion (IC) to S1, which means that a significant fraction of the excitation energy flows through this state (21). However, because the S1 → S0 transition is dipole-forbidden it remains a challenge to elucidate the mechanism that promotes Car S1 to Chl EET. Neither the Förster (22, 23) nor Dexter theories (24) provide satisfactory predictions (15, 20). To explore this issue, several authors have deconvoluted Car S1 → BChl EET time constants, τET1, from pump-probe or up-conversion data obtained after excitation of the Car S2 state in different species. A few examples of time constants derived in this way are: peridinin to chlorophyll a (Chl a) EET in the peridinin-Chl a-protein (PCP) from Amphidinium carterae (PCP; 3.1 ps), spheroidene to BChl EET in Rb. sphaeroides (2.0 ps) and okenone to BChl a ET in Chromaticum purpuratum (3.8 ps and 0.5 ps) (19, 25, 26). However, the best way to find direct evidence for Car S1 → BChl EET and to determine its contribution to the overall Car → BChl EET is to excite Car S1 exclusively and thereby measure τET1 and φET1 directly without any convolution with S2 state dynamics. This approach becomes even more important considering the recent identification of another dark state in spheroidene at ≈17,600 cm−1 between the S1 and the S2 states of Cars (18). This state has Bu− symmetry and is thought to play an important role in facilitating the Car S2 → Car S1 IC (27). However, a transition Ag− ↔ Bu− is neither one-photon allowed nor two-photon allowed and therefore does not contribute to our two-photon results.
Figure 1.

(Upper) Energy diagram for LH2. Black arrows indicate energy pathways occurring in the TPE pump probe experiment. Gray arrows indicate additional energy pathways in conventional one-photon experiments. OPE, One-photon excitation. Fl., Fluorescence. τET2, Time constant for Car S2 → BChl EET [≈50 fs (30)]. τ21, Time constant for Car S2 → S1 IC [≈135 fs (30)]. τET1, Time constant for Car S1 → BChl EET. τ10, Time constant for Car S1 → S0 IC. τS1, (τET1−1 + τ10−1)−1 lifetime of the Car S1 state measured in this work. (Lower) TPE spectrum of LH2 of Rb. sphaeroides (line with symbols), one-photon absorption spectrum (dashed line). Data taken from ref. 28. TPE-wavelengths for the TPE pump probe experiment are indicated with double arrows.
An elegant way to populate S1 directly is by two-photon excitation (TPE) because transitions between states of Ag symmetry are two-photon allowed. Recently we have found evidence for Car S1 → BChl EET in Rb. sphaeroides by measuring the Car TPE spectrum through the steady-state BChl fluorescence (see Fig. 1) (28). We obtained similar results in the light harvesting complex LHCII of higher plants and green algae (29).
In this work we report fs time-resolved excited-state absorption (ESA) of the Car S1 state in three light-harvesting complexes of purple bacteria observed after TPE of this state. We report resuts for the B800-B850 and B800-B820 LH2s of Rps. acidophila and the LH2 of Rb. sphaeroides. To understand the mechanism that promotes this EET, we model the dynamics by using a theory that calculates the ensemble average energy transfer rates using as inputs the site energy disorder and the electron–phonon coupling of the chromophores as well as the excitonic nature of the acceptors.
Experimental Procedures
LH2 of Rb. sphaeroides was prepared from the Rb. sphaeroides strain 2.4.1 by using previously described methods (28). LH2s of Rps. acidophila were isolated from Rps. acidophila strain 7050 by using the methods described in refs. 9 and 30. β-Carotene (Acros, New Jersey) was dissolved in octane by sonicating for ≈10 s and centrifuging for 4 min at ≈8,000 g. The solutions had an optical density of ≈1 per mm at the maximum of the absorption. The samples were flowed through a 1-mm path-length quartz cell and maintained at 4°C under a nitrogen atmosphere. To reduce scattering further, a 0.45 μm filter was added to the flow cycle. The pump-pulses [≈10 nJ, 85 fs full width half maximum (FWHM), 250 kHz] and white-light probe pulses were both provided by a Coherent 9450 optical parametric amplifier. Excitation light intensity fluctuations were monitored with a power meter connected to a computer. To minimize population via one-photon excitation caused by the probe-white light we blocked the dominant 800-nm component with a highly reflective laser mirror and filtered the probe wavelength with a 555 ± 5-nm interference filter before the sample cell. The pump beam was focused with an f = 5-cm achromatic lens and the probe beam with an f = 10 cm. Only about <25% of the intensity of both beams was blocked by a 50-μm pinhole. Our instrument response function had a FWHM of ≈90 fs. The signal was detected by using a photodiode and lock-in amplification. To suppress white-light fluctuations with a frequency similar to the pump-chopper frequency, a part of the white light was measured with a separate photodiode and lock-in amplifier also triggered by the chopper. Subtracting this trace resulted in an improvement of the signal-to-noise ratio of more than a factor of 5. The polarization dependence was measured by using a 1,310-nm λ/4 and λ/2-waveplate in the excitation beam. Results without a waveplate were identical to those with λ/2-waveplate.
Results
β-Carotene in octane solution, a Car with known properties and similarities with spheroidene (31), was used to test the experimental setup. From one-photon pump probe data it is known that ESA from the S1 of Cars has a maximum close to 550 nm, therefore we chose this wavelength as the detection wavelength in all experiments (32). The excitation wavelength was ≈1,310 nm, which is close to the 0–0 transition of the S1 two-photon absorption of the Car (Fig. 1.). The changes in the probe intensity after TPE were typically on the order of 0.1%. A monoexponential fit to the transient absorption data gave a time constant of 9 ± 0.2 ps, in excellent agreement with the reported S1 lifetime of β-carotene (31).
Two experiments were used to confirm that the pump was a two-photon process. First, the power dependence of the signal intensities were found to have a quadratic dependence on the excitation power (Fig. 2b). The fit suggests a power dependence with an exponent of 2.2 ± 0.3. Second, we measured the dependence of the signal on the polarization of the two-photon pump pulse (33). In one-photon spectroscopy the signal depends only on the relative orientation of linear polarized pump and probe fields. In two-photon spectroscopy, the signal is in general different for linear, circular, or any elliptical polarization of the pump beam. The polarization ratio, Ω = Scirc/Slin, defined as the ratio of the signal obtained with circularly and with linearly polarized excitation, depends on the symmetries of the excited- and ground-state wave functions. Therefore this experiment can be used not only to verify the two-photon nature of the signals, but also to identify the symmetry of the two-photon excited molecule via its polarization ratio Ω. This can be used to confirm that we have indeed excited the Car in the light-harvesting complexes. The result, shown in Fig. 2c, gives Ω = 0.84 ± 0.07, which is identical within error to the literature value of Ω ≈ 0.8 for all-trans diphenyloctatetraene and diphenylhexatriene (34).
Figure 2.

(a) Time dependence of ESA (λdet = 550 nm) of β-carotene in octane (squares) observed after TPE (λexc = 1,310 nm). Monoexponential fit: 9 ± 0.2 ps (solid line). (b) Power dependence of ESA. Power fit: Exponent = 2.2 ± 0.3 (solid line). (c) Relative intensity of ESA excited with linear and circular polarized light (Ω = 0.84 ± 0.07). The “coherence spike” is very large in the two-photon experiments (off scale) and has a quadratic dependence on the excitation power and a linear dependence on the Car concentration. No spike was seen with a pure buffer sample. In experiments with chlorophyll a, which has no two-photon allowed states, the coherence spike was found to be substantially smaller in magnitude (data not shown).
In Fig. 3a the time dependence of the ESA is shown for the LH2 of Rb. sphaeroides and the B800-B850 and B800-B820 LH2 of Rps. acidophila observed after TPE at 1,310 nm. All traces show a decay on the ps time scale to a constant offset, which persists over the entire dynamic range accessible with our setup (≈400 ps). The results of fits to OD(t) = y0 + Ae−t/τS1 are summarized in Table 1. A relatively fast decay with a time constant of τS1 ≈ 1.9 ± 0.5 ps is observed for Rb. sphaeroides, whereas both complexes of Rps. acidophila show a significantly slower decay (τS1 ≈ 7 ± 0.5 ps and τS1 ≈ 6 ± 0.5 ps). The observed τS1 for Rb. sphaeroides is in very good agreement with the value recently reported by Koyama and coworkers (19). As Fig. 3b shows, the preexponential factor A has a quadratic dependence on the excitation power, the solid line in Fig. 3b being fit by an exponent of 2.2 ± 0.3. We assign the decay component as the lifetime, τS1, of the S1 state of the Cars in the light-harvesting complexes. Further confirmation of this assignment is provided by the polarization dependence shown in Fig. 3c. In both species the observed polarization ratio is very similar to those observed for β-carotene and polyenes (34) in solution (Ω = 0.84 ± 0.04 for Rb. sphaeroides and Ω = 0.80 ± 0.04 for Rps. acidophila).
Figure 3.

(a) Time dependence of ESA (λdet = 550 nm) along with monoexponential fits with constant offset of the B800-B850 complex (□, τS1 = 7 ± 0.5 ps) and the B800-B820 complex (▪, τS1 = 6 ± 0.5 ps) of Rps. acidophila and LH2 of Rb. sphaeroides (○, τS1 = 1.9 ± 0.5 ps) observed after TPE (λexc = 1,310 nm). The traces have been normalized and offset from each other by 0.2. (b) Power dependence of the prefactor in the monoexponential fit to the data of Rb. sphaeroides (○). Power fit: Exponent = 2.2 ± 0.3 (solid line). (c) Power dependence of the constant offset in the fit to the data of Rb. sphaeroides (○). Solid line: Linear fit. (d) Relative intensity of the fluorescence obtained after TPE (λexc = 1,310 nm) with linear and circular polarized light of B800-B850 of Rps. acidophila (□, Ω = 0.80 ± 0.04) and LH2 of Rb. sphaeroides (○, Ω = 0.84 ± 0.04).
Table 1.
| λexc/nm
|
1310
|
1200
|
|||||
|---|---|---|---|---|---|---|---|
| Species | β-carotene | Rb. sphaeroides | Rps. acidophila B800–B850 | Rps. acidophila B800–B820 | Rb. sphaeroides | Rps. acidophila B800–B850 | Rps. acidophila B800–B820 |
| τS1/ps | 9 ± 0.5 | 1.9 ± 0.5 | 7 ± 0.5 | 6 ± 0.5 | 3 ± 0.5 | 7 ± 0.5 | 7.5 ± 0.5 |
| A/% | 100 | 41 | 38 | 34 | 46 | 45 | 46 |
| y0/% | 0 | 59 | 62 | 66 | 54 | 55 | 54 |
| Ω | 0.84 ± 0.07 | 0.84 ± 0.04 | 0.80 ± 0.04 | — | — | — | — |
OD(t) = y0 +
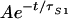 ;
Ω = Scirc/Slin.
;
Ω = Scirc/Slin.
The constant offset y0 has a linear dependence on the excitation power (Fig. 3c). No constant offset can be seen with pure buffer as sample. This result can be explained as a direct excitation of triplet states. It is not surprising that, with the given field intensities, the probability of directly exciting a spin forbidden transition via one-photon excitation is similar to the probability of exciting the S1 state via TPE. However, because no offset has been observed with Cars in solution, we must assume that it is the triplet state of the BChl that can be seen in the transient absorption of the light-harvesting complexes. This interpretation is supported by the fact that the ESA of the triplet states of BChl has a maximum close to the detection wavelength of 550 nm and that the triplet states are the only states with an energy corresponding to one-photon excitation at 1,310 nm (35). For the interpretation of the S1 state dynamics the triplet state excitation can be neglected, because the overall population of excited states is very small.
In Fig. 4 the time-resolved data of the Car S1 ESA for all species are shown analogously to Fig. 3a but with TPE at 1,200 nm. As can been seen from Fig. 1, an excitation wavelength of 1,200 nm corresponds to the blue edge of the TPE spectrum of the light-harvesting complexes. The kinetics are very similar to the kinetics observed after TPE at 1,310 nm. We observe a relatively fast decay with the time constant τS1 ≈ 3 ± 0.5 ps for Rb. sphaeroides, whereas both complexes of Rps. acidophila show a significantly slower decay: τS1 ≈ 7 ± 0.5 ps and τS1 ≈ 7.5 ± 0.5 ps. However, the data, especially from Rb. sphaeroides, are not fit as well by a monoexponential decay as the data for TPE at 1,310 nm. This is possibly related to the 2,000 cm−1 of excess vibrational energy resulting from 1,200-nm excitation compared with 1,310 nm. Another difference between the data observed with TPE at 1,200 nm and that at 1,310 nm is the significantly smaller relative magnitude of the constant offset (see Table 1). We will use the time constants observed with excitation at 1,310 nm for our subsequent analysis.
Figure 4.

Time dependence of ESA (λdet = 550 nm) along with monoexponential fits with constant offset of the B800-B850 complex (□, τS1 = 7 ± 0.5 ps) and the B800-B820 complex (■, τS1 = 7.5 ± 0.5 ps) of Rps. acidophila and LH2 of Rb. sphaeroides (○, τS1 = 3 ± 0.5 ps) observed after TPE (λexc = 1,200 nm). The traces have been normalized and offset from each other by 0.2.
Discussion
Efficiencies of the Energy Transfer Via the Car S2 and the Car S1 States.
The significantly quenched Car S1 lifetime observed after direct TPE of Rb. sphaeroides, τS1 ≈ 1.9 ± 0.5 ps, compared with the lifetime of spheroidene in solution, τ10 ≈ 9 ps (31), provides clear evidence for efficient Car S1 → BChl EET. We calculate τET1 for Rb. sphaeroides, assuming τ10 is unchanged, to be τET1 = (τS1−1 − τ10−1)−1 ≈ 2.4 ± 0.5 ps. The unquenched S1 lifetime of the Car in Rps. acidophila, rhodopin glucoside, has been determined by Frank and coworkers (36) to be τ10 ≈ 4.1 ps. We measured τS1 ≈ 6–7 ps in the antenna complex, indicating that Car S1 → BChl EET is negligible in Rps. acidophila. Even if the time constant for IC of rhodopin glucoside in the protein is as long as τ10 ≈ 9 ps, like the corresponding value for β-carotene or spheroidene in solution, the Car S1 → BChl EET time τET1 could not be faster than 25 ps, and the Car S1 → BChl EET efficiency φET1 then would be φET1 = (τS1−1 − τ10−1) τS1 < 28%.† The same calculation for Rb. sphaeroides, using the value τ10 = 9 ps for spheroidene, yields a Car S1 → BChl EET efficiency of φET1 ≈ 80%.
Using the known values for the overall Car → BChl EET quantum yield, φOA, together with the values of φET1 we are able to calculate the energy transfer efficiencies for Car S2 → BChl EET, φET2. The overall EET efficiency is the sum of both efficiencies, weighted by their population probabilities: φOA = φET2 + φET1φ21, where φ21 is the quantum efficiency for Car S2 → Car S1 IC, φ21= τ21−1/(τET2−1 + τ21−1) = 1 − φET2 (see Fig. 1). We now assume that φOA differs from 100% entirely owing to Car S1 → Car S0 IC. The quantum yield for Car S1 → Car S0 IC is φ10 = (1 − φET1)φ21. Therefore φOA also can be written as φOA = 1 −[(1 − φET1) × φ21] = 1 + (φET1 − 1) × φ21. Substituting φ21 = 1 − φET2 into this equation gives φET2 = 1 − [(φOA − 1)/(φET1 −1)]. Thus we estimate φET2 > 75% for Rb. sphaeroides and φET2 > 60% for Rps. acidophila. The contributions of the Car S1 → BChl EET to φOA are φET1 × φ21 < 20% for Rb. sphaeroides and φET1 × φ21 < 10% for Rps. acidophila. The results of these quantum efficiency calculations are summarized in Table 2.
Table 2.
Calculated Car S1 → BChl energy transfer time constants and efficiencies
| Species | τS1/ps* | τET1/ps | φET1 | φOA(9,11) | φET2 | φET1φ21 | φ21 |
|---|---|---|---|---|---|---|---|
| Rb. sphaeroides | 1.9 ± 0.5 | 2.4 ± 0.5 | 80% | >95% | >75% | <20% | <25% |
| Rps. acidophila | 6.5 ± 0.5 | >25 | <28% | ∼70% | >60% | <10% | <40% |
φOA = φET2 + φET1φ21.
Measured in this work.
The Mechanism of Car S1 → BChl Energy Transfer.
The mechanism of electronic coupling that promotes Car S1 → BChl EET usually has been discussed in light of the relative merits of Förster versus Dexter energy transfer theories (22, 24, 37). In general, the coupling may be partitioned into a Coulombic contribution VCoul, operative at all separations, and a short-range contribution Vshort that explicitly depends on interpenetration of the donor and acceptor molecular orbitals. Thus V = VCoul + Vshort. However, Vshort has been shown previously to be insignificant for interactions between the pigments in LH2 (20, 38).
The transition density cube (TDC) (17) method is very useful for calculating VCoul when the donor and acceptor molecules are closely proximate relative to molecular dimensions, because local interactions between the donor and acceptor transition densities determine the overall coupling. Such details are lost when averaging over wavefunctions to give transition moments. In algebraic form the difference between the dipole approximation and the TDC method is given by
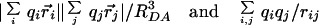 |
1 |
for discrete charges qi at position ri on donor molecule D and charges qj at position rj on acceptor A. rij = ri − rj, and RDA is the center-to-center separation of D and A.
Zhang et al. (19) suggested that the Car S1 → BChl couplings are dominated by Coulombic interactions, VCoul, determined by mixing between the Car S1 and Car S2 states. This would result in a Car S1 → BChl transition density with spatial properties resembling the transition density of the Car S2 → BChl transition. If this is the case, scaling down of the Car S2 → BChl electronic couplings, determined previously using the transition density cube method (17), should provide a reasonable estimate of the Car S1 → BChl couplings. The scaling factor is determined by the state mixing between the Car S1 and Car S2 states. If Coulombic interactions dominate the Car S1 → BChl couplings, small differences in the positions of the pigments will not affect the couplings as much as for short-range interactions. We therefore assume for the species investigated in this work that the estimated couplings are similar.
In recent work Scholes and Fleming (8) showed that the BChl B800 → B850 EET can only be elucidated by introduction of site energy disorder before solving the eigenvalue problem for the B850 acceptor aggregate in each individual LH2, because the assumption of identical B850 eigenstates for each LH2 gives incorrect values for the ensemble average rate, τET−1. We anticipate that a quantitative description of the Car S1 → BChl EET will require a similarly detailed theory.
The model we have used for our calculation is described in detail in ref. 8. Briefly, the ensemble average rate of energy transfer is given by
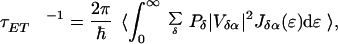 |
2 |
where Vδα is the electronic coupling between the donor state δ and eigenstate α of the acceptor in an individual LH2, calculated by using the electronic couplings in the monomeric site representation, Vest.Coul(m). Pδ is a normalized population factor, and Jδα(ɛ) is the spectral overlap factor for states δ and α, Jδα(ɛ) = aαhom(ɛ)fδhom(ɛ). The superscript “hom” specifies that these line shapes are determined only by fluctuations of the bath that have characteristic frequencies faster than that of the EET. Slower fluctuations (e.g., static disorder) are accounted for by the Monte Carlo averaging procedure denoted by the angular brackets in Eq. 2. The bandwidth of aαhom(ɛ) and the disorder parameters were derived from three pulse-echo peak shift measurements.
Estimation of the Electronic Couplings Mediating the Car S1 → BChl EET.
We modified the B800 → B850 EET model by extending the acceptor Hamiltonian by two diagonal elements representing the two B800 BChl in the closest neighborhood to the Car. We used the mirror image of the Car S1 TPE spectrum of Rb. sphaeroides (28) for the donor-fluorescence, fδhom(ɛ). To estimate the electronic couplings in the monomeric site representation, Vest.Coul(m), we varied the scaling factor for the electronic coupling and the Stokes shift of the mirror image in such a way that the calculation always produced the observed time constant of τET1 ≈ 2.4 ps, Inset in Fig. 5a. The Car S1 state 0–0 transition in Rb. sphaeroides has been determined to be around 14,000 cm−1 (28, 31, 39–41). As can be seen from the inset in Fig. 5a, the electronic couplings scaling factor remains very similar (≈0.15) when the 0–0-transition is varied over a wide range. The resulting individual couplings range from 5 cm−1 to 26 cm−1 (monomeric site representation, see Table 3). The range of these values is in very good agreement with estimates for the necessary couplings from Zhang et al. (19) and Damjanovic et al. (20). Recently, one of us (C.-P.H.) calculated the Coulombic Car S1-BChl couplings by using the transition density cube method and time–dependent density functional wavefunctions. The individual couplings obtained in the ab initio calculation are in very good agreement with the couplings estimated by the scaling-down procedure (Table 3). Analysis of these results provides strong evidence that scaling down the Car S2-BChl couplings is appropriate because the Car S1-BChl Coulomb couplings are dominated by a small mixing of a strongly dipole allowed Car S2-like configuration as proposed by Zhang et al. (19).
Figure 5.

(a) Acceptor DOS in LH2 of Rb. sphaeroides calculated with (solid line) and without introducing disorder (short dashed line) before solving the eigenvalue problem for the acceptor aggregate along with Car S1 emission (long dashed line). Note that the shown DOS is not weighed by electronic coupling. For details see text. (Inset) Electronic coupling scaling factor (see text) needed to reproduce the experimentally observed time constant τET1 as a function of the 0–0 transition of the Car S1 emission. (b) Same results for the B800–B850 complex of Rps. acidophila. (Inset) Calculated time constants τET1 as a function of the Car S1 state 0–0 transition.
Table 3.
Car S1-BChl Qy couplings in the monomeric site representation estimated by scaling down Car S2-BChl Qy couplings, Vest.Coul, and calculated via ab initio methods, VTDDFTCoul
In Fig. 5a the calculated density of states (DOS) is shown together with the S1-emission spectrum. Note that the DOS does not necessarily represent the acceptor states in Förster overlap calculations, because each state is coupled differently to the donor states. However, as can be seen in Fig. 5, for Rb. sphaeroides there is excellent overlap of donor and acceptor states, which explains the efficient Car S1 → BChl EET observed in this species. Disorder causes the couplings and state orderings in the B850 excitonic states to differ significantly between individual LH2s. Nonetheless, the calculated time constant τET1 ≈ 2.4 ps can be separated into a time constant for Car S1 → B850 EET, τB850 ≈ 6.7 ps (15%), and a time constant for Car S1 → B800 EET, τB800 ≈ 2.8 ps (85%). The corresponding time constants for Rps. acidophila are τB850 ≈ 11.7 ps (60%) and τB800 ≈ 17.5 ps (40%).
The Car S1 State in Rps. acidophila.
We estimate the 0–0 transition energy of the Cars in Rps. acidophila by assuming that the S1 emission spectrum only differs from that in Rb. sphaeroides by a shift to lower energies and that the couplings in both species are similar. For Rps. acidophila we used the B850 Hamiltonian reported previously, which was obtained by modeling the 77K absorption and CD spectra (8). To determine the 0–0 transition of Car S1 in Rps. acidophila, we shifted the Car S1 mirror image and calculated the ensemble average time constant by using the same electronic couplings obtained from our Rb. sphaeroides calculations. As can be seen from the Inset in Fig. 5b, time constants larger than the experimentally observed lifetime, τ10 ≈ 7 ps, are found for 0–0 transitions ≈<12,800 cm−1. This value is in very good agreement with S1 energies of other Cars with 11 conjugated double bonds. However, because the Car S1 lifetime, τ10, is not known for rhodopin glucoside, this estimate only gives an upper limit, which is valid in the case τ10 >> τET1. Of course, differences in the electronic coupling also could contribute to the differences in the EET efficiencies, and in this case the 0–0 transition energy will be underestimated. As in the calculations for Rb. sphaeroides, the introduction of disorder did not result in a large difference in the calculated time constant provided we assume that electron–phonon coupling dominates the broadening of the Car S1 spectrum. However, via some model calculations we found that if the heterogeneous broadening were to contribute significantly to the S1 spectra, the time constants may change by more than a factor of 2. We also expect that the temperature dependence of the Car S1 → BChl EET can be modeled only by considering Eq. 2.
Conclusions
By measuring the dynamics of the Car S1 ESA after TPE, we determined Car S1 → BChl EET time constants in LH2 of the purple bacteria Rb. sphaeroides and Rps. acidophila without any convolution with dynamics associated with the Car S2 state or the recently discovered dark state between S1 and S2. The energy transfer efficiency was found in Rps. acidophila to be φET1 < 28% and in Rb. sphaeroides φET1 ≈ 80%. The Car S1 → BChl EET was modeled by using a detailed theory that suggests that for Rb. sphaeroides the time constant for Car S1 → B850 EET is τB850 ≈ 6.7 ps (15%) and for Car S1 → B800 EET τB800 ≈ 2.8 ps (85%). The corresponding time constants for Rps. acidophila are τB850 ≈ 11.7 ps (60%) and τB800≈ 17.5 ps (40%). The individual electronic couplings for the Car S1 → BChl EET are in the order of 5–26 cm−1. A major contribution to the drastic difference in the efficiency φET1 can be explained by a shift of the Car S1 energy from ≈14,000 cm−1 in Rb. sphaeroides to a value of ≈<12,800 cm−1 in Rps. acidophila.
Acknowledgments
This work was supported by the Director, Office of Science, of the U.S. Department of Energy under Contract No. DE-AC03–76SF00098. P.J.W. is grateful for financial support from the Deutsche Forschungsgemeinschaft.
Abbreviations
- EET
electronic energy transfer
- Car
carotenoid
- BChl
bacteriochlorophyll
- Chl
chlorophyll
- ESA
excited-state absorption
- TPE
two-photon excitation
- LH2
light-harvesting complex 2 of purple bacteria
- IC
internal conversion
- DOS
density of states
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.190230097.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.190230097
Note that this definition of φET1 is different from the definition of φET1 in our former work on LHCII of higher plants and green algae (29). In this work φET1 has to be multiplied with the quantum efficiency for Car S2 → Car S1 internal conversion after excitation into S2, φ21, to calculate the contribution of Car S1 → BChl EET to φOA, which is the definition of φET1 in ref. 29.
References
- 1.McDermott G, Prince S M, Freer A A, Hawthornthwaitelawless A M, Papiz M Z, Cogdell R J, Isaacs N W. Nature (London) 1995;374:517–521. [Google Scholar]
- 2.McDermott G, Prince S M, Freer A A, Isaacs N W, Papiz M Z, Hawthornthwaitelawless A M, Cogdell R J. Protein Eng. 1995;8:43–43. [Google Scholar]
- 3.Koepke J, Hu X C, Muenke C, Schulten K, Michel H. Structure (London) 1996;4:581–597. doi: 10.1016/s0969-2126(96)00063-9. [DOI] [PubMed] [Google Scholar]
- 4.Sundstrom V, Pullerits T, van Grondelle R. J Phys Chem B. 1999;103:2327–2346. [Google Scholar]
- 5.Nagarajan V, Johnson E T, Williams J C, Parson W W. J Phys Chem B. 1999;103:2297–2309. [Google Scholar]
- 6.Zhao Y, Meier T, Zhang W M, Chernyak V, Mukamel S. J Phys Chem B. 1999;103:3954–3962. [Google Scholar]
- 7.van Oijen A M, Ketelaars M, Kohler J, Aartsma T J, Schmidt J. Science. 1999;285:400–402. doi: 10.1126/science.285.5426.400. [DOI] [PubMed] [Google Scholar]
- 8.Scholes G D, Fleming G R. J Phys Chem B. 2000;104:1854–1868. [Google Scholar]
- 9.Angerhofer A, Cogdell R J, Hipkins M F. Biochim Biophys Acta. 1986;848:333–341. [Google Scholar]
- 10.Koyama Y, Kuki M, Andersson P O, Gillbro T. Photochem Photobiol. 1996;63:243–256. [Google Scholar]
- 11.Frank H A, Cogdell R J. In: Carotenoids in Photosynthesis. Young A, Britton G, editors. London: Chapman & Hall; 1993. pp. 252–326. [Google Scholar]
- 12.Frank H A, Cogdell R J. Photochem Photobiol. 1996;63:257–264. doi: 10.1111/j.1751-1097.1996.tb03022.x. [DOI] [PubMed] [Google Scholar]
- 13.Walz T, Jamieson S J, Bowers C M, Bullough P A, Hunter C N. J Mol Biol. 1998;282:833–845. doi: 10.1006/jmbi.1998.2050. [DOI] [PubMed] [Google Scholar]
- 14.Oling F, Boekema E J, Dezarate I O, Visschers R, Vangrondelle R, Keegstra W, Brisson A, Picorel R. Biochim Biophys Acta. 1996;1273:44–50. [Google Scholar]
- 15.Scholes G D, Harcourt R D, Fleming G R. J Phys Chem B. 1997;101:7302–7312. [Google Scholar]
- 16.Scholes G D, Gould I R, Cogdell R J, Fleming G R. J Phys Chem B. 1999;103:2543–2553. [Google Scholar]
- 17.Krueger B P, Scholes G D, Fleming G R. J Phys Chem B. 1998;102:5378–5386. [Google Scholar]
- 18.Sashima T, Nagae H, Kuki M, Koyama Y. Chem Phys Lett. 1999;299:187–194. [Google Scholar]
- 19.Zhang J P, Fujii R, Qian P, Inaba T, Mizoguchi T, Koyama Y, Onaka K, Watanabe Y, Nagae H. J Phys Chem B. 2000;104:3683–3691. [Google Scholar]
- 20.Damjanovic A, Ritz T, Schulten K. Phys Rev E. 1999;59:3293–3311. [Google Scholar]
- 21.Macpherson A N, Gillbro T. J Phys Chem A. 1998;102:5049–5058. [Google Scholar]
- 22.Forster T. Ann Phys. 1948;2:55–75. [Google Scholar]
- 23.Forster T. In: Modern Quantum Chemistry. Sinanoglu O, editor. III. New York: Academic; 1965. p. 93. [Google Scholar]
- 24.Dexter D L. J Chem Phys. 1953;21:836–850. [Google Scholar]
- 25.Andersson P O, Cogdell R J, Gillbro T. Chem Phys. 1996;210:195–217. [Google Scholar]
- 26.Bautista J A, Hiller R G, Sharples F P, Gosztola D, Wasielewski M, Frank H A. J Phys Chem A. 1999;103:2267–2273. [Google Scholar]
- 27.Tavan P, Sculten K. Phys Rev B. 1987;36:4337–4358. doi: 10.1103/physrevb.36.4337. [DOI] [PubMed] [Google Scholar]
- 28.Krueger B P, Yom J, Walla P J, Fleming G R. Chem Phys Lett. 1999;310:57–64. [Google Scholar]
- 29.Walla P J, Yom J, Krueger B P, Fleming G R. J Phys Chem B. 2000;104:4799–4806. [Google Scholar]
- 30.Krueger B P, Scholes G D, Jimenez R, Fleming G R. J Phys Chem B. 1998;102:2284–2292. [Google Scholar]
- 31.Chynwat V, Frank H A. Chem Phys. 1995;194:237–244. [Google Scholar]
- 32.Frank H A, Bautista J A, Josue J, Pendon Z, Hiller R G, Sharples F P, Gosztola D, Wasielewski M R. J Phys Chem B. 2000;104:4569–4577. [Google Scholar]
- 33.McClain W M. J Chem Phys. 1971;55:2789–2796. [Google Scholar]
- 34.Holtom G R, McClain W M. Chem Phys Lett. 1976;44:436–439. [Google Scholar]
- 35.Limantara L, Fujii R, Zhang J P, Kakuno T, Hara H, Kawamori A, Yagura T, Cogdell R J, Koyama Y. Biochemistry. 1998;37:17469–17486. doi: 10.1021/bi981803q. [DOI] [PubMed] [Google Scholar]
- 36.Frank H A, Chynwat V, Desamero R Z B, Farhoosh R, Erickson J, Bautista J. Pure Appl Chem. 1997;69:2117–2124. [Google Scholar]
- 37.Kallmann H, London F. Z Physik Chem. 1929;2:207–243. [Google Scholar]
- 38.Nagae H, Kakitani T, Katoh T, Mimuro M. J Chem Phys. 1993;98:8012–8023. [Google Scholar]
- 39.Frank H A, Desamero R Z B, Chynwat V, Gebhard R, van der Hoef I, Jansen F J, Lugtenburg J, Gosztola D, Wasielewski M R. J Phys Chem A. 1997;101:149–157. [Google Scholar]
- 40.Shashima T, Shiba M, Hashimoto H, Nagae H, Koyama Y. Chem Phys Lett. 1998;290:36–42. [Google Scholar]
- 41.Fujii R, Onaka K, Kuki M, Koyama Y, Watanabe Y. Chem Phys Lett. 1998;288:847–853. [Google Scholar]