

Abstract
The generation and maintenance of immune responses are controlled by both co-stimulatory and co-inhibitory signalling through T cell co-receptors, many of which belong to the immunoglobulin-like superfamily or the tumour necrosis factor receptor superfamily. Agonistic or antagonistic monoclonal antibodies targeting these co-receptors have the potential to enhance immunity. Furthermore, their activity on the immunosuppressive regulatory T cell populations which are prevalent within many tumours provides an additional rationale for their use as anti-cancer therapies. This review summarizes the interactions between cancer and the immune system, highlighting the ways in which these new classes of immunostimulatory antibodies might enhance anti-tumour immunity and summarizing early clinical experience with their use.
Keywords: cancer immunotherapy, co-inhibition, co-stimulation, CTLA-4, regulatory T cells
Cancer and the immune system
The immune surveillance hypothesis posits that the immune system plays a key role in suppressing tumour growth and that the incidence of cancer would be much greater were it not for the ability of the immune system to identify and eliminate nascent tumour cells. Widespread acceptance of this concept was hampered for many years by a lack of firm supportive experimental evidence until the turn of this century, when a series of influential papers demonstrated that lymphocytes and interferon-γ co-operate to inhibit the development of spontaneous and carcinogen-induced tumours in mice engineered genetically to lack a functional immune system [1–4]. While the immune system appears capable of eliminating or containing early tumour growth, some tumour cells escape detection and eventually cause cancer. It is hypothesized that selective pressure exerted by the immune system drives the cellular composition of these tumours to become serially less immunogenic (immunoediting), as demonstrated by the finding that tumour cells from immunodeficient mice are more immunogenic than those from immunocompetent mice [1].
Immunoediting may be considered to consist of three processes occurring either independently or sequentially [5]. First, ‘elimination’, in which immunity functions as an extrinsic tumour suppressor; secondly, ‘equilibrium’, in which cancerous cells survive but are held in check by the immune system [6]; and thirdly, ‘escape’, in which tumour cell variants with either reduced immunogenicity or the capacity to attenuate or subvert immune responses grow into clinically apparent cancers [7]. The changes occurring in the escape phase may be considered broadly as those intrinsic to the tumour cells themselves, including enhanced resistance to apoptosis and down-regulation of co-stimulatory ligands, and those involving the local tumour microenvironment. These mechanisms are neither mutually exclusive nor entirely separable. Anti-tumour responses may be frustrated by regulatory mechanisms which normally act to limit T cell responses following chronic exposure to antigen [e.g. up-regulation of cytotoxic T lymphocyte-associated-antigen 4 (CTLA-4) or programmed cell death-1 (PD-1) receptors], or by tumour-induced subversion of other regulatory pathways [e.g. expression of T cell inhibitory molecules such as PD-ligand 1 (PD-L1), B7-H3 or B7x, or accumulation of immunosuppressive T cell or antigen-presenting cell (APC) populations]. Further proposed mediators of local immune suppression include soluble suppressive factors elaborated by the tumour or parenchyma such as interleukin (IL)-10 or transforming growth factor (TGF)-β, and indoleamine 2,3-dioxygenase (IDO) expressed by tumour cells or IDO-competent APCs, which may cause both direct suppression of T cells and enhancement of local regulatory T cell-mediated suppression [8]. The presence of an array of other cell types capable of actively suppressing immune reactions, such as CD4+CD25+FoxP3+ regulatory T cells (Treg), IL-10-secreting regulatory T cells, CD1d-restricted natural killer (NK) T cells, immature and plasmacytoid dencritic cells (DCs) (iDCs and pDCs) and myeloid-derived suppressor cells within the tumour or tumour-draining lymph nodes is clearly critical to induction and/or maintenance of local immune privilege in a number of systems [9]. Such cells may be recruited preferentially to these sites, or expanded or induced therein.
The apparent confirmation of the validity of the immune surveillance hypothesis led to great enthusiasm for the development of immune-based anti-cancer therapies. On the basis of growing evidence that tumours express antigens that can be presented by professional APCs to induce the generation of tumour-specific cytotoxic T lymphocytes (CTLs), tumour immunotherapists aimed to parallel the successes achieved in developing vaccines for infectious diseases. Strategies included vaccination with peptide, DNA or antigen-pulsed DCs, either alone or coupled with approaches based on directly enhancing effector number or function by adoptive transfer of tumour-reactive T cells. However, attempts to target human cancers have been significantly less successful than was initially envisaged possible. While resulting in some impressive responses it is perhaps, in hindsight, unsurprising that, given the multitude of locally immunosuppressive mechanisms engaged within an actively growing tumour, attainment of clinically significant responses are rare even with therapies that succeed in inducing systemic immunity. The presence of large numbers of T cells capable of recognizing tumours is not singularly sufficient to mediate tumour regression, as evidenced by unrestricted tumour growth in T cell receptor (TCR) transgenic mice in which all the T cells are capable of recognizing the tumour antigen [10]. Clinical studies of active immunization have shown that despite expansion of tumour-reactive T cells to levels of up to 40% of the circulating CD8+ T cell repertoire, tumour growth may continue apparently unimpeded [11]. There is now ample experimental evidence that functional systemic anti-tumour activity may not translate into tumour rejection, either because of lack of infiltration of T cells into the tumour [12] or because of local suppression of function within the tumour microenvironment. Although tumour-specific immunity is compromised in tumour-bearing mice, there is often no generalized immune deficiency [13], indicating that tumours can suppress specifically the induction of effective anti-tumour immunity. This concept is perhaps highlighted most clearly by concomitant immunity, wherein a mouse injected with a tumour will reject a subsequent challenge with the same tumour at a distant site, despite continued growth at the site of initial challenge [14]. Because tumours share many similarities with chronic pathogens such as Mycobacterium tuberculosis, the challenges of delivering effective vaccines or immunotherapies are aligned much more closely with those associated with therapy of these established chronic infections than with acute infectious pathogens, in which the majority of the successes have come with prophylactic vaccination strategies. Addressing the factors conferring local immune privilege will probably prove critical to improving outcomes following anti-cancer immunotherapies.
Monoclonal antibodies in cancer immunotherapy
Monoclonal antibodies have revolutionized the treatment of a number of malignancies. Their mechanisms of action vary, but can be categorized broadly as direct or indirect. The former include blockade of function (e.g. hindering ligand binding, increasing internalization of receptors) and stimulation of function (e.g. inducing apoptosis) on the tumour cells. In addition, monoclonal antibodies can be used to target local delivery of conjugated therapeutics such as toxins or radioisotopes. The indirect mode of action is mediated by the immune system and includes the activation of complement-dependent cytoxicity and both complement-dependent and antibody-dependent cellular cytotoxicity involving macrophages and NK cells, effectors of the innate immune system. The development of antibodies that can interfere with the function of co-stimulatory and co-inhibitory pathways on effector T cells has provided a novel mechanism for indirect anti-cancer activity. The primary targets for such interventions were envisaged initially to be the effector T cells, but it is becoming clear that regulatory T cell populations might also be important targets for their overall activity. A number of regulatory CD4+ T cell subtypes are recognized, including those which are produced by the thymus, express CD4, CD25, 4-1BB, OX40, glucocorticoid-induced tumour necrosis factor receptor (TNFR) family-related gene (GITR) and CTLA-4, and appear crucially dependent upon the expression of the forkhead box P3 transcription factor (FoxP3) for their development (so-called ‘naturally occurring’ Treg, nTreg); and those which arise from naive CD4+ T cells as a result of ‘tolerogenic’ encounters in the periphery. The latter ‘inducible’ Treg include IL-10-producing, FoxP3-negative Tr1 cells, TGF-β-producing Th3 cells and extra-thymically generated CD4+CD25+FoxP3+ iTreg cells. The acquisition of regulatory phenotype and suppressive functions by conventional non-regulatory CD4+ T cells following exposure to antigens under certain conditions is now recognized as a major contributor to the maintenance of T cell homeostasis and control of inflammation. Characterization of the conditions that drive such peripheral conversion is ongoing, but factors such as suboptimal antigen stimulation in combination with TGF-β appear to be important, both of which are likely to be relevant within the tumour microenvironment [15]. IDO produced by either tumour cells or parenchyma (eg. pDCs) also favours conversion [16]. The dominant inhibitory potential of Treg cell populations in murine models of malignancy is well established [17], and more recently their potential role in human malignancies has been demonstrated [18]. Their relative abundance predicts for tumour outcomes in murine models [19], and correlates inversely with outcomes in several epithelial carcinomas [20]. Intriguingly, in haematological malignancies this association is reversed and high levels of Treg appear to confer improved prognoses [21,22]. Because the targets of many cancer vaccination strategies are self-antigens, it is perhaps no surprise that ‘therapeutic’ cancer vaccines can induce amplification of tumour-specific Treg[23].
Promoting T cell function via modulation of co-stimulation or co-inhibition
Immune activation is regulated by two major families of co-receptors expressed by T cells: the immunoglobulin-like (Ig) superfamily and the TNFR superfamily [24,25] (Fig. 1). Co-stimulatory members of the former include CD28 and inducible T cell co-stimulator (ICOS), while OX40, CD27, 4-1BB, CD30, GITR and herpes-virus entry mediator (HVEM) are members of the latter. CTLA-4 and PD-1 are the most well-established inhibitory members of the Ig superfamily. The B and T lymphocyte attenuator (BTLA) is the most recently described inhibitory member of this family [26]. The identities of the receptors for the newer B7 ligands (B7-H3 and B7x/B7-H4) remain elusive, but these receptors may also mediate significant inhibitory activity, perhaps more so in the periphery, given the tissue distribution of the ligands. Stimulatory or blocking monoclonal antibodies are being investigated extensively for their abilities to enhance T cell numbers, function and maintenance of immunological memory [27,28].
Fig. 1.
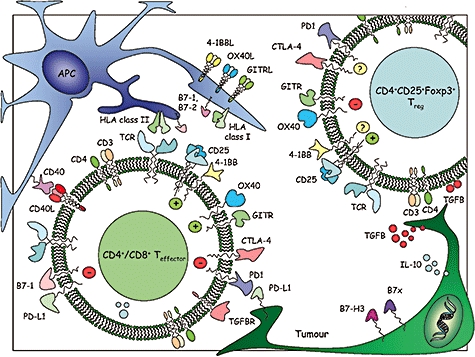
Potential immunomodulatory targets for monoclonal antibody therapy. In the case of effector T cells, agonistic antibodies directed towards co-stimulatory pathways and antagonistic or blocking antibodies directed towards co-inhibitory pathways can directly enhance effector numbers, functions or persistence. The situation with respect to regulatory T cell populations remains less clear, but it appears likely from the current literature that agonists of classic co-stimulatory molecules may abrogate function, whereas blockade of cytotoxic T lymphocyte-associated-antigen 4 may also reduce suppressor function.
Stimulatory antibodies to 4-1BB (CD137), OX40 (CD134) and GITR
The TNFR family members are appealing candidates for the development of targeted therapeutics. The greatest attention has been focused upon 4-1BB and OX40. 4-1BB is expressed on activated T cells (including Treg and NK T cells), activated NK and dendritic cells, eosinophils, mast cells and endothelial cells in some metastatic tumours [25]. Its ligand 4-1BBL is expressed on activated DCs, B cells and macrophages. Ligation on T cells results in up-regulation of anti-apoptotic genes and protection from activation-induced cell death (AICD), enhancing establishment of durable memory CTLs [29]. The up-regulation of 4-1BB on antigen-experienced T cells suggests that anti-4-1BB may target these primed T cells differentially, influencing those T cells preferentially with highest avidity receptors and explaining partially why 4-1BB co-stimulation may be superior to CD28 co-stimulation for the generation of antigen-specific cells for adoptive therapies [30]. While it is assumed that co-stimulation of CD8+ T cells is the principal mechanism of action of anti-4-1BB, various other immunomodulatory activities may contribute. In this respect, a common theme developing in our understanding of the function of immunostimulatory antibodies is their possible multiplicity of function, reflecting the cellular distribution of the receptors. Thus, (i) activation of APCs; (ii) reduction in Treg suppressive capacity or enhancement of effector resistance to suppression; and (iii) co-stimulation of CD4+ and CD8+ T cells are all supported by experimental data [31,32]. Furthermore, activated NK cells and NK T cells may be relevant targets for anti-tumour activity [33,34]. Reverse signalling into cells expressing the ligands for a number of Ig or TNFR superfamily members is another recurring theme of recent investigations into immune modulating functions of these molecules. In the case of 4-1BBL, this may result in enhanced production of inflammatory mediators or enhanced cell adhesion, facilitating egress of immune effectors into sites of inflammation [35,36]. There are conflicting data as to whether 4-1BB ligation on Treg results in enhanced or reduced suppressive capacity [31,37,38], and synergy of anti-tumour activity with approaches that are thought to target Treg number or function has been taken as evidence that any 4-1BB-mediated inhibitory effects on Treg function may be relatively modest [39]. Forced expression of 4-1BBL in murine tumours enhances immunogenicity, reducing engraftment rates in immune-competent recipients, although growth of untransfected cells is affected only modestly in relatively poorly immunogenic tumours [40]. Agonistic anti-4-1BB monoclonal antibodies enhance anti-tumour CTL responses, enabling rejection of established syngeneic tumour cell lines [41–43]. Activity appears critically dependent upon CD8+ T cells and (in most studies) NK cells, with the role of CD4+ T cells varying in different tumour models [34,42–44]. As with other immunostimulatory antibodies, combination with vaccination strategies enhances activity in poorly immunogenic tumour models [45,46]. Co-administration with transgenic tumour-specific CTL enhances anti-tumour activity, apparently via a reduction in AICD rather than enhanced proliferation [47]. Early experience with a humanized clinical grade antibody (BMS-663513) in patients with melanoma and renal cell carcinoma suggests that the antibody is well tolerated with some evidence of activity (6% partial responses in melanoma patients), demonstrating the probable need to evaluate combinatorial approaches [39,48–50]. Two particularly intriguing and seemingly paradoxical features of anti-4-1BB monoclonal antibodies are their ability to ameliorate autoimmunity and to suppress humoral immunity in mice [51–53]. While this suppressive activity has been proposed to be advantageous in terms of limiting possible antibody-mediated toxicities, it is recognized that it might also be deleterious to the development of optimal anti-cancer immunity.
OX40 is expressed transiently on activated CD4+ and CD8+ T cells, functioning as a late co-stimulatory receptor [25,54]. It is also expressed by NK T cells and Treg. Its ligand OX40L is expressed in a similar distribution to 4-1BBL, on activated DCs, B cells and macrophages, as well as activated T cells and endothelial cells [25]. OX40 ligation regulates CD4+ and CD8+ T cell survival and memory generation, preventing T cell tolerance [55–58]. It also impairs the suppressor functions of Treg[59,60], allowing tumour-resident DCs to traffic to draining lymph nodes to prime tumour-specific CD8+ CTL [61]. Furthermore, OX40 triggering appears to be antagonistic for FoxP3 induction in antigen-responding naive CD4+ T cells, effectively suppressing the generation of iTreg[62,63], and blocks the generation of IL-10-producing Tr1 cells [64], suggesting that OX40 may antagonize the generation of a number of different inducible Treg populations. 4-1BB and OX40 act independently to facilitate robust CD8 and CD4 recall responses, overlapping in their intracellular signalling pathways, yet neither 4-1BB nor GITR signalling seem to block the generation of Tr1 [64], and there are currently no reports illustrating whether they influence FoxP3+ iTreg induction. As with 4-1BBL, forced expression of OX40L by tumour cells increases immunogenicity, with tumour rejection dependent upon both CD4+ and CD8+ T cells [65]. Furthermore, intratumoural injection of DCs modified to have enhanced expression of OX40L can effect tumour rejection in murine models that are dependent upon CD8+ CTL responses, themselves dependent upon CD4+ T cells and NK T cells [66]. Agonistic anti-OX40 antibodies increase anti-tumour activity in a number of transplantable tumour models [67]. Concomitant activity on both effector and regulatory compartments may be a prerequisite of effective rejection of established tumours [61]. In preclinical models, OX40 ligation enhances several other immunostimulatory approaches [68–71].
The GITR is also expressed transiently on activated T cells [72,73], and expressed constitutively at high levels on Treg with further induction following activation [74,75]. Its ligand GITRL is expressed at low levels on B cells, macrophages and some DCs, increasing transiently following activation. GITR ligation stimulates both proliferation and function of CD4+ and CD8+ T cells [76]. Its activity on Treg has remained more contentious [77–79]. Anti-GITR antibodies reduce suppression in co-cultures of CD4+CD25– effectors and CD4+CD25+ Treg, but whether this relates to reduced Treg suppressor function, increased resistance of effectors to the preserved suppressor function of Treg, or a combination of both, has yet to be demonstrated definitively. Experiments using mixtures of GITR+/+ and GITR–/– effector and regulatory cells suggest that ligation of GITR on the effector population rather than the regulatory population is critical for abrogating suppression [79], suggesting that enhanced effector resistance to suppression may be key in in vitro assays. Injection of adenovirus expressing recombinant GITRL into B16 melanoma promotes T cell infiltration and reduced tumour volumes [80], while agonistic anti-GITR antibodies have been shown to enhance both rejection of established methylcholanthrene-induced fibrosarcomas, and to enhance systemic anti-tumour responses and concomitant immunity when given following B16 melanoma challenge [81,82]. Furthermore, the same antibody also enhances the impact of DNA-vaccination in terms of generation of systemic immunity and enhancing resistance to challenge with murine melanoma [83].
Stimulation through checkpoint blockade: CTLA-4 (CD152), PD-1 (CD279) and PD-L1 (CD274)
In contrast to the TNFR superfamily, the existence of co-inhibitory receptors mediating direct down-regulation of lymphocyte activation and/or effector function has been a recognized feature of the Ig superfamily for some time. Indeed, the co-inhibitory receptor-ligand members outnumber the co-stimulatory members within this superfamily, engendering the idea of regulatory or inhibitory checkpoint blockade as a therapeutic anti-cancer strategy [84]. Blockade of inhibitory immune checkpoints for therapeutic benefit offers considerable promise, particularly as combination with other treatment modalities that promote cross-priming of anti-tumour immunity may yield additive or synergistic activity. The strategy that is the most advanced in clinical development involves antibodies which block CTLA-4 [28].
The CTLA-4 is expressed by activated CD4+ and CD8+ T cells, although its surface expression is regulated tightly with a short half-life. It influences some of the earliest events in T cell activation, being mobilized rapidly from intracellular vesicles to the immune synapse after TCR engagement, but is endocytosed promptly in the unphosphorylated state. It is expressed constitutively by nTreg and iTreg, although the majority is found intracellularly, even following activation. CTLA-4 shares the B7-1 (CD80) and B7-2 (CD86) ligands with CD28, a critical co-stimulatory molecule. Ligation of CD28 in concert with TCR stimulation enhances T cell proliferation by inducing production of IL-2 and anti-apoptotic factors, decreasing the number of ligated TCR that are required for a given biological response. CTLA-4 engagement blocks augmentation of gene regulations by CD28-mediated co-stimulation, and its function as a negative regulator of CD28-dependent T cell responses is demonstrated strikingly by the phenotype of CTLA-4 knock-out mice, which succumb to a rapidly lethal polyclonal CD4-dependent lymphoproliferation within 3–4 weeks of birth [85,86]. CTLA-4 has significantly higher affinities for both B7 ligands than does CD28. Accumulation of both receptors at the synapse is influenced by ligand binding. CD28 is recruited in the absence of B7-1 and B7-2 binding but is not effectively stabilized there, and its localization can be disrupted by CTLA-4. The latter is dependent more critically upon ligand binding for concentration at the synapse. CTLA-4 may, therefore, both out-compete CD28 for ligand, particularly when ligand densities are limiting, and be able to exclude CD28 from the immunological synapse by virtue of the generation of extended high affinity lattices of alternating CTLA-4 and B7-1 homodimers [87]. For this reason the tight spatial and temporal regulation of CTLA-4 expression is likely to be critical for determining the outcome of CD28-mediated signalling. Furthermore, CTLA-4 ligation induces decreased production of cytokines (particularly IL-2, and its receptor) and cell cycle arrest in G1, suggesting that ligation-dependent mechanisms also contribute to its negative regulatory function. Finally, CTLA-4 has an important role in Treg-mediated suppression, as evidenced by the recent demonstration that Treg-specific CTLA-4 deficiency in conditional knock-out (CKO) mice is associated with a profound reduction in their suppressive capacity [88]. CKO mice developed a lethal autoimmune lymphoproliferative syndrome with a slightly slower tempo than CTLA-4–/– mice. The mechanism(s) by which CTLA-4 mediates these Treg-associated effects remain(s) unclear, but may be dependent upon reverse signalling into B7-expressing cells [89,90]. Furthermore, Treg-mediated suppression during in vitro suppressor assays is associated with reduced activation of APCs (evidenced by reduced surface expression of B7 molecules [88]).
Antibody-mediated blockade of CTLA-4 is particularly effective at enhancing secondary immune responses, more markedly in CD4+ T cells. While often having only modest effects as a monotherapy in preclinical tumour models of poorly immunogenic tumours, anti-CTLA-4 synergizes with a number of other anti-tumour immunotherapies [24,28]. Furthermore, early clinical studies have shown that CTLA-4 blockade has activity as a monotherapy (5–15% objective response rates in melanoma and renal carcinoma) and, in keeping with murine models, enhanced activity in combination with a number of other therapies in the treatment of human malignancies including melanoma, renal, ovarian and prostatic carcinomas [24,91–95]. More than 4000 patients have been treated to date with anti-CTLA-4 (ipilimumab or tremelimumab). Adverse immunological events have been a feature of some of the early studies, often associating with clinical responses, although they have generally proved manageable and the majority reversible, allaying some of the concerns that the use of therapeutics designed to enhance immune reactivity non-specifically and to interfere with tumour-induced tolerance might uncouple mechanisms of self-tolerance systemically, resulting in uncontrolled autoimmunity. This is a theoretical concern for agents inducing immunostimulation either by agonism of co-stimulatory pathways, antagonism of co-inhibitory pathways or subversion of Treg-mediated suppression. The severe toxicity experienced by normal volunteers receiving a ‘super-agonistic’ co-stimulatory antibody directed towards CD28 (TGN1412) highlights the need for careful evaluation of these new therapeutics [96], although other targets which do not obviate the requirement for TCR signalling in inducing T cell activation (a feature of super-agonists) will probably have more favourable toxicity profiles, as is the case with CTLA-4 blockade. The association between adverse immune events and responses with anti-CTLA-4 is apparent across tumour types. For example, in patients with enterocolitis, response rates of 45% and 46% have been reported for metastatic melanoma and renal cell cancer respectively [97]. What remains less clear is whether this association is an inevitable outcome of the mechanism of action of this new class of immunotherapeutics, or whether a narrow therapeutic window exists in which beneficial anti-tumour activity can be dissociated from adverse immune events, as has been hinted in some studies.
The PD-1 is expressed by activated CD4+ and CD8+ T cells, as well as B cells, monocytes and at lower levels on NK T cells. It binds to two separate ligands, PD-L1 and PD-L2, which exhibit distinct expression profiles [98]. PD-L1 is expressed broadly, and can be detected on resting and activated T cells (including nTreg), B cells, macrophages, DCs and mast cells. In addition, its expression on non-haematopoietic cells may have physiological relevance, suggesting that inhibition through PD-L1/PD-1 may not be restricted solely to the interaction of T cells and professional APCs, but that it may also occur during the effector phase of the immune response in peripheral tissues, perhaps helping to prevent immune-mediated tissue damage directly at the tissue interface. By comparison, PD-L2 has a much more limited expression profile. It is not expressed on naive or activated T cells, but is instead restricted to activated macrophages, myeloid dendritic cells and mast cells, suggesting that it fulfils a role that differs from that of PD-L1. PD-1–/– mice develop an array of autoimmune pathologies characterized by high titres of autoantibodies, in keeping with a negative regulatory effect on T and/or B cells [99,100]. PD-L1 and PD-L2 may also regulate T cell responses through reverse signalling. Cross-linking antibodies against PD-L2 directly induce dendritic cells to produce immunomodulatory cytokines such as IL-6 and tumour necrosis factor-α, at the same time as protecting them from cell death [101,102]. Furthermore, PD-1-Ig inhibits DC activation and increases IL-10 production independently of any influence on IDO. PD-L1 binds B7-1 with an affinity intermediate between those of CTLA-4 and CD28 for B7-1, allowing suppression of T cell proliferation and cytokine production through either B7-1 or PD-L1 signalling [103]. Thus, blockade of both CTLA-4 and PD-L1 might eliminate simultaneously cell intrinsic negative signalling through CTLA-4, B7-1, PD-L1 and PD-1, while favouring positive signalling through CD28. Recent data highlight the relevance of this pathway to chronic T cell responses to pathogens, wherein ‘exhausted’ T cells demonstrate a selective up-regulation of PD-1 and administration of anti-PD-L1 antibodies in vivo restores their activity [104,105]. Together, these data suggest that blockade of PD-1 and/or PD-L1 can restore functionality of the T cell compartment and might be applied to enhance T cell activity towards other chronic pathologies such as cancer.
The PD-L1 is expressed by a variety of human and murine tumours, and PD-1 is expressed by tumour-infiltrating lymphocytes, leading to the hypothesis that they may be important in restricting intratumour effector T cell responses [106]. In humans, myeloid DCs isolated from tumour or lymph nodes from ovarian carcinoma patients express high levels of PD-L1, and are capable of enhancing T cell activity only following PD-L1 blockade [107]. Similarly, plasmacytoid DCs in tumour-draining lymph nodes produce high levels of IDO which results in Treg activation, up-regulation of PD-L1 on the DCs and negative regulation of T cell responses [108]. PD-L1 expression has been correlated directly with poor prognosis in a number of human cancers [109–111]. Forced expression of PD-L1 on murine tumour lines diminished T cell activation and tumour killing in vitro, and enhanced tumour growth markedly in vivo, while anti-PD-L1 antibodies blocked these effects [112,113]. In the 4T1 mammary carcinoma model PD-L1 is up-regulated in vivo by the tumour, making it refractory to immunotherapy with the anti-4-1BB antibody. Co-administration with anti-PD-L1 resulted in dramatic tumour rejection [114]. Similarly, anti-PD-L1 antibody delayed in vivo growth of PD-L1-expressing murine myeloma cell lines. PD-L1 blockade has also been shown to synergize with adoptive cellular therapy to induce rejection of squamous cell carcinoma [113]. Furthermore, adoptive transfer of PD1–/– tumour-reactive CD8+ T cells caused rejection of B16 melanoma, while neither wild-type nor CTLA-4–/– tumour-reactive CD8+ T cells were capable of inducing rejection [115].
Very few studies have examined the ability of PD-1 blockade to directly promote anti-tumour T cell responses in vivo. Anti-PD-1 antibodies reduce dissemination of B16 melanoma and CT26 colon carcinoma [116], and induce a small but significant decrease in growth of murine pancreatic carcinoma [117]. A fully human anti-PD-1 monoclonal antibody has been developed recently and its ability to enhance the function of human tumour-specific T cells has been tested in vitro[118]. Vaccine-induced melanoma-reactive CD8+ T cells showed an increase in number and function upon restimulation in vitro with the blocking anti-PD-1 antibody. There were no apparent effects on cell death, suggesting that the augmented in vitro responses were due mainly to increased proliferation of tumour-reactive T cells rather than decreased cell death. While these results are promising, a better understanding of the mechanisms underpinning bidirectional regulation through PD-L1 and B7-1 may help to inform future trials and perhaps to help decide which molecule (PD-L1 or PD-1) makes the optimal target for cancer immunotherapy.
Other inhibitory members of the Ig superfamily offer possible targets for co-inhibitory blockade, although the impact such interventions would have on anti-tumour activity remains more speculative at present. Thus, the as-yet unidentified receptor for the B7x (B7-H4) ligand offers one such possibility. The current literature on B7-H3 and anti-tumour responses remains somewhat contradictory [24]. The BTLA (CD272) is also a potential target, offering the unique example of an Ig superfamily member whose ligand is a member of the TNFR family (HVEM).
Redefining response criteria
The mechanisms underlying the anti-tumour activity of immunostimulatory therapies are indirect, relying upon the activation of tumour-reactive immune effector cells, and contrasting with the direct activity of most conventional chemotherapeutics. The kinetics of clinical responses may therefore differ significantly, potentially taking longer to become manifest. This issue is well illustrated by early experience with CTLA-4 blockade. While the development and application of response evaluation criteria in solid tumours (RECIST) has greatly enhanced objectivity in clinical trials reporting, it is now clear that the tempo of responses with anti-CTLA-4 is such that disease stabilization over several months, or possibly even progression, may occur before the development of a clinical response [119–121]. RECIST criteria were not designed to allow for this pattern of response. In a report summarizing the pooled experience from six of the larger ipilimumab studies in stage III–IV melanoma (including monotherapy and combination with peptide vaccine, dacarbazine or IL-2), 46 of 356 (12·9%) patients receiving 0·1–20 mg/kg in single or multiple doses achieved a response [11 complete (CR) and 35 partial (PR) responses][119]. Many responses occurred later than is typical with cytotoxic agents. In 28 of 46 patients CR/PR was documented at time-points beyond 12 weeks from initiation of treatment. In four of 46 patients (or 1·1% of the total number treated) responses occurred following initial progression. Delayed response onset occurred irrespective of dose, regimen and therapeutic partner. The results suggest that continued treatment/observation may be beneficial despite initial progressive or stable disease. In addition, this pattern does not appear to be confined to patients with melanoma, as similarly delayed responses have been reported with renal cancer [120]. It is therefore important to accept this new concept of delayed response assessment so that clinical activity is not rejected prematurely and falsely.
Combinatorial immunotherapeutics
It is clear from preclinical models and early clinical experience that multi-modal approaches may be required to eradicate poorly immunogenic tumours. The recent literature demonstrates the potential for many combinations to give synergistic or additive effects. Attempting to choose rationally which are likely to be the best approaches remains challenging, but considering approaches under a number of basic headings allows the identification of potentially attractive combinations. Thus, modern immunotherapeutic strategies can be divided according to those which: (i) improve antigen presentation or immunogenicity (e.g. vaccines, CpG oligodeoxynucleotides); (ii) improve T effector function, numbers or persistence directly (e.g. agonistic anti-TNFR antibodies, cytokines); (iii) remove or disable immunological checkpoints, either cell intrinsic or cell extrinsic (e.g. CTLA-4 or PD-1 blockade, Treg depletion and possibly agonistic anti-TNFR antibodies); (iv) ‘reset’ the system taking advantage of proliferative advantages in a lymphopenic environment (e.g. adoptive cellular therapy); and (v) improve antigen specificity or TCR avidity for tumour antigens (e.g. TCR gene therapies). It has also become apparent that some agents bridge these categories, so the duality of enhancing effector function and reducing suppression afforded by, e.g. CTLA-4 blockade, or OX40 stimulation, may be achieved with one agent. Because recent data highlight the ability of regulatory checkpoints to limit the efficacy of any directly stimulatory strategy, the inclusion of at least one therapy aimed at disabling immune checkpoints is theoretically attractive. So, for example, the combination of anti-CTLA-4 with vaccines, CpG oligodeoxynucleotides, regulatory T cell depletion or anti-4-1BB enhances activity markedly [122–125]. Similarly diverse synergy is seen when combining anti-4-1BB antibodies with other modalities [48,49,68,126]. One potential advantage of approaches relying on the synergy of multiple components is that they might reduce the toxicity induced by higher doses of each agent administered as monotherapy (e.g. immune responses may be constrained towards tumour-related antigens if combined with antigen-specific vaccination or adoptive cellular therpaies rather than ubiquitous self-antigens). Appropriate timing of sequential therapies is likely to become an important factor in such combinatorial approaches.
Conclusions
Major challenges remain, including identification of the best combinatorial strategies. These will continue to be informed by careful mechanistic studies in mouse models, while recognizing potential differences compared with humans. The identification of robust predictors of response may parallel attempts to tailor chemotherapeutics according to the genetic profile of the tumour or of tumour infiltrates. Ultimately, attempts to manipulate the host in order to achieve such favourable immunological profiles will need to demonstrate improved clinical outcomes in comparative studies, and in this regard the application of novel classes of immunostimulatory antibodies offers great promise.
Acknowledgments
Karl S. Peggs is currently an investigator at the Department of Haematology, UCL Cancer Institute, University College London, UK, and receives funding from the Leukaemia Research Fund, UK. Sergio A. Quezada is a Research Fellow funded by the Irvington Institute Fellowship Program of the Cancer Research Institute USA, and a junior member of the Millennium Nucleus on Immunology and Immunotherapy, Pontifícia Universidad Católica de Chile. James P. Allison is an investigator of the Howard Hughes Medical Institute and holds the David H. Koch Chair in Immunological Studies at the Memorial Sloan-Kettering Cancer Center.
Disclosure
James P. Allison is co-inventor of intellectual property concerning CTLA-4 that is held by University of California, Berkeley and is a consultant for Medarex and Bristol Meyers Squibb, who are involved in the clinical development of anti-CTLA-4.
References
- 1.Shankaran V, Ikeda H, Bruce AT, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 2.Smyth MJ, Crowe NY, Godfrey DI. NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol. 2001;13:459–63. doi: 10.1093/intimm/13.4.459. [DOI] [PubMed] [Google Scholar]
- 3.Girardi M, Oppenheim DE, Steele CR, et al. Regulation of cutaneous malignancy by gammadelta T cells. Science. 2001;294:605–9. doi: 10.1126/science.1063916. [DOI] [PubMed] [Google Scholar]
- 4.Gao Y, Yang W, Pan M, et al. Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J Exp Med. 2003;198:433–42. doi: 10.1084/jem.20030584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 6.Koebel CM, Vermi W, Swann JB, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007 doi: 10.1038/nature06309. [DOI] [PubMed] [Google Scholar]
- 7.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–6. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 8.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–54. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6:715–27. doi: 10.1038/nri1936. [DOI] [PubMed] [Google Scholar]
- 10.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosenberg SA, Sherry RM, Morton KE, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 12.Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma. J Exp Med. 2008;205:2125–38. doi: 10.1084/jem.20080099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Radoja S, Rao TD, Hillman D, Frey AB. Mice bearing late-stage tumors have normal functional systemic T cell responses in vitro and in vivo. J Immunol. 2000;164:2619–28. doi: 10.4049/jimmunol.164.5.2619. [DOI] [PubMed] [Google Scholar]
- 14.Kurt RA, Park JA, Panelli MC, et al. T lymphocytes infiltrating sites of tumor rejection and progression display identical V beta usage but different cytotoxic activities. J Immunol. 1995;154:3969–74. [PubMed] [Google Scholar]
- 15.Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol. 2005;6:1219–27. doi: 10.1038/ni1265. [DOI] [PubMed] [Google Scholar]
- 16.Fallarino F, Grohmann U, You S, et al. The combined effects of tryptophan starvation and tryptophan catabolites down-regulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol. 2006;176:6752–61. doi: 10.4049/jimmunol.176.11.6752. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu J, Yamazaki S, Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999;163:5211–18. [PubMed] [Google Scholar]
- 18.Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–9. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 19.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935–45. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf D, Wolf AM, Rumpold H, et al. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res. 2005;11:8326–31. doi: 10.1158/1078-0432.CCR-05-1244. [DOI] [PubMed] [Google Scholar]
- 21.Carreras J, Lopez-Guillermo A, Fox BC, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood. 2006;108:2957–64. doi: 10.1182/blood-2006-04-018218. [DOI] [PubMed] [Google Scholar]
- 22.Alvaro T, Lejeune M, Salvado MT, et al. Outcome in Hodgkin's lymphoma can be predicted from the presence of accompanying cytotoxic and regulatory T cells. Clin Cancer Res. 2005;11:1467–73. doi: 10.1158/1078-0432.CCR-04-1869. [DOI] [PubMed] [Google Scholar]
- 23.Zhou G, Drake CG, Levitsky HI. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood. 2006;107:628–36. doi: 10.1182/blood-2005-07-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peggs KS, Quezada SA, Allison JP. Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol Rev. 2008;224:141–65. doi: 10.1111/j.1600-065X.2008.00649.x. [DOI] [PubMed] [Google Scholar]
- 25.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe N, Gavrieli M, Sedy JR, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–9. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 27.Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95–106. doi: 10.1038/nrc2051. [DOI] [PubMed] [Google Scholar]
- 28.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. 2006;90C:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myers L, Lee SW, Rossi RJ, et al. Combined CD137 (4-1BB) and adjuvant therapy generates a developing pool of peptide-specific CD8 memory T cells. Int Immunol. 2006;18:325–33. doi: 10.1093/intimm/dxh371. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Snyder KM, Suhoski MM, et al. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol. 2007;179:4910–18. doi: 10.4049/jimmunol.179.7.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris GP, Chen L, Kong YC. CD137 signaling interferes with activation and function of CD4+CD25+ regulatory T cells in induced tolerance to experimental autoimmune thyroiditis. Cell Immunol. 2003;226:20–9. doi: 10.1016/j.cellimm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Robertson SJ, Messer RJ, Carmody AB, Mittler RS, Burlak C, Hasenkrug KJ. CD137 costimulation of CD8+ T cells confers resistance to suppression by virus-induced regulatory T cells. J Immunol. 2008;180:5267–74. doi: 10.4049/jimmunol.180.8.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim DH, Chang WS, Lee YS, et al. 4-1BB engagement costimulates NKT cell activation and exacerbates NKT cell ligand-induced airway hyperresponsiveness and inflammation. J Immunol. 2008;180:2062–8. doi: 10.4049/jimmunol.180.4.2062. [DOI] [PubMed] [Google Scholar]
- 34.Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol. 1998;190:167–72. doi: 10.1006/cimm.1998.1396. [DOI] [PubMed] [Google Scholar]
- 35.Kang YJ, Kim SO, Shimada S, et al. Cell surface 4-1BBL mediates sequential signaling pathways ‘downstream’ of TLR and is required for sustained TNF production in macrophages. Nat Immunol. 2007;8:601–9. doi: 10.1038/ni1471. [DOI] [PubMed] [Google Scholar]
- 36.Drenkard D, Becke FM, Langstein J, et al. CD137 is expressed on blood vessel walls at sites of inflammation and enhances monocyte migratory activity. FASEB J. 2007;21:456–63. doi: 10.1096/fj.05-4739com. [DOI] [PubMed] [Google Scholar]
- 37.Elpek KG, Yolcu ES, Franke DD, Lacelle C, Schabowsky RH, Shirwan H. Ex vivo expansion of CD4+CD25+FoxP3+ T regulatory cells based on synergy between IL-2 and 4-1BB signaling. J Immunol. 2007;179:7295–304. doi: 10.4049/jimmunol.179.11.7295. [DOI] [PubMed] [Google Scholar]
- 38.Choi BK, Bae JS, Choi EM, et al. 4-1BB-dependent inhibition of immunosuppression by activated CD4+CD25+ T cells. J Leukoc Biol. 2004;75:785–91. doi: 10.1189/jlb.1003491. [DOI] [PubMed] [Google Scholar]
- 39.Choi BK, Kim YH, Kang WJ, et al. Mechanisms involved in synergistic anticancer immunity of anti-4-1BB and anti-CD4 therapy. Cancer Res. 2007;67:8891–9. doi: 10.1158/0008-5472.CAN-07-1056. [DOI] [PubMed] [Google Scholar]
- 40.Melero I, Bach N, Hellstrom KE, Aruffo A, Mittler RS, Chen L. Amplification of tumor immunity by gene transfer of the co-stimulatory 4-1BB ligand: synergy with the CD28 co-stimulatory pathway. Eur J Immunol. 1998;28:1116–21. doi: 10.1002/(SICI)1521-4141(199803)28:03<1116::AID-IMMU1116>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 41.Shuford WW, Klussman K, Tritchler DD, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. 1997;186:47–55. doi: 10.1084/jem.186.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melero I, Shuford WW, Newby SA, et al. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat Med. 1997;3:682–5. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- 43.Lynch DH. The promise of 4-1BB (CD137)-mediated immunomodulation and the immunotherapy of cancer. Immunol Rev. 2008;222:277–86. doi: 10.1111/j.1600-065X.2008.00621.x. [DOI] [PubMed] [Google Scholar]
- 44.Miller RE, Jones J, Le T, et al. 4-1BB-specific monoclonal antibody promotes the generation of tumor-specific immune responses by direct activation of CD8 T cells in a CD40-dependent manner. J Immunol. 2002;169:1792–800. doi: 10.4049/jimmunol.169.4.1792. [DOI] [PubMed] [Google Scholar]
- 45.Wilcox RA, Flies DB, Zhu G, et al. Provision of antigen and CD137 signaling breaks immunological ignorance, promoting regression of poorly immunogenic tumors. J Clin Invest. 2002;109:651–9. doi: 10.1172/JCI14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito F, Li Q, Shreiner AB, et al. Anti-CD137 monoclonal antibody administration augments the antitumor efficacy of dendritic cell-based vaccines. Cancer Res. 2004;64:8411–19. doi: 10.1158/0008-5472.CAN-04-0590. [DOI] [PubMed] [Google Scholar]
- 47.May KF, Jr, Chen L, Zheng P, Liu Y. Anti-4-1BB monoclonal antibody enhances rejection of large tumor burden by promoting survival but not clonal expansion of tumor-specific CD8+ T cells. Cancer Res. 2002;62:3459–65. [PubMed] [Google Scholar]
- 48.Shi W, Siemann DW. Augmented antitumor effects of radiation therapy by 4-1BB antibody (BMS-469492) treatment. Anticancer Res. 2006;26:3445–53. [PubMed] [Google Scholar]
- 49.Kim YH, Choi BK, Kim KH, Kang SW, Kwon BS. Combination therapy with cisplatin and anti-4-1BB: synergistic anticancer effects and amelioration of cisplatin-induced nephrotoxicity. Cancer Res. 2008;68:7264–9. doi: 10.1158/0008-5472.CAN-08-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Molckovsky A, Siu LL. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American Society of Clinical Oncology meeting. J Hematol Oncol. 2008;1:20. doi: 10.1186/1756-8722-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun Y, Chen HM, Subudhi SK, et al. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat Med. 2002;8:1405–13. doi: 10.1038/nm1202-796. [DOI] [PubMed] [Google Scholar]
- 52.Seo SK, Choi JH, Kim YH, et al. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004;10:1088–94. doi: 10.1038/nm1107. [DOI] [PubMed] [Google Scholar]
- 53.Mittler RS, Bailey TS, Klussman K, Trailsmith MD, Hoffmann MK. Anti-4-1BB monoclonal antibodies abrogate T cell-dependent humoral immune responses in vivo through the induction of helper T cell anergy. J Exp Med. 1999;190:1535–40. doi: 10.1084/jem.190.10.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–20. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 55.Bansal-Pakala P, Jember AG, Croft M. Signaling through OX40 (CD134) breaks peripheral T-cell tolerance. Nat Med. 2001;7:907–12. doi: 10.1038/90942. [DOI] [PubMed] [Google Scholar]
- 56.Bansal-Pakala P, Halteman BS, Cheng MH, Croft M. Costimulation of CD8 T cell responses by OX40. J Immunol. 2004;172:4821–5. doi: 10.4049/jimmunol.172.8.4821. [DOI] [PubMed] [Google Scholar]
- 57.Dawicki W, Bertram EM, Sharpe AH, Watts TH. 4-1BB and OX40 act independently to facilitate robust CD8 and CD4 recall responses. J Immunol. 2004;173:5944–51. doi: 10.4049/jimmunol.173.10.5944. [DOI] [PubMed] [Google Scholar]
- 58.Mousavi SF, Soroosh P, Takahashi T, et al. OX40 costimulatory signals potentiate the memory commitment of effector CD8+ T cells. J Immunol. 2008;181:5990–6001. doi: 10.4049/jimmunol.181.9.5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takeda I, Ine S, Killeen N, et al. Distinct roles for the OX40–OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol. 2004;172:3580–9. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 60.Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood. 2005;105:2845–51. doi: 10.1182/blood-2004-07-2959. [DOI] [PubMed] [Google Scholar]
- 61.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205:825–39. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.So T, Croft M. Cutting edge: OX40 inhibits TGF-beta- and antigen-driven conversion of naive CD4 T cells into CD25+Foxp3+ T cells. J Immunol. 2007;179:1427–30. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 63.Vu MD, Xiao X, Gao W, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood. 2007;110:2501–10. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ito T, Wang YH, Duramad O, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci USA. 2006;103:13138–43. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andarini S, Kikuchi T, Nukiwa M, et al. Adenovirus vector-mediated in vivo gene transfer of OX40 ligand to tumor cells enhances antitumor immunity of tumor-bearing hosts. Cancer Res. 2004;64:3281–7. doi: 10.1158/0008-5472.can-03-3911. [DOI] [PubMed] [Google Scholar]
- 66.Zaini J, Andarini S, Tahara M, et al. OX40 ligand expressed by DCs costimulates NKT and CD4+ Th cell antitumor immunity in mice. J Clin Invest. 2007;117:3330–8. doi: 10.1172/JCI32693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weinberg AD, Rivera MM, Prell R, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol. 2000;164:2160–9. doi: 10.4049/jimmunol.164.4.2160. [DOI] [PubMed] [Google Scholar]
- 68.Lee SJ, Myers L, Muralimohan G, et al. 4-1BB and OX40 dual costimulation synergistically stimulate primary specific CD8 T cells for robust effector function. J Immunol. 2004;173:3002–12. doi: 10.4049/jimmunol.173.5.3002. [DOI] [PubMed] [Google Scholar]
- 69.Murata S, Ladle BH, Kim PS, et al. OX40 costimulation synergizes with GM-CSF whole-cell vaccination to overcome established CD8+ T cell tolerance to an endogenous tumor antigen. J Immunol. 2006;176:974–83. doi: 10.4049/jimmunol.176.2.974. [DOI] [PubMed] [Google Scholar]
- 70.Lee SJ, Rossi RJ, Lee SK, et al. CD134 costimulation couples the CD137 pathway to induce production of supereffector CD8 T cells that become IL-7 dependent. J Immunol. 2007;179:2203–14. doi: 10.4049/jimmunol.179.4.2203. [DOI] [PubMed] [Google Scholar]
- 71.Gri G, Gallo E, Di Carlo E, Musiani P, Colombo MP. OX40 ligand-transduced tumor cell vaccine synergizes with GM-CSF and requires CD40-Apc signaling to boost the host T cell antitumor response. J Immunol. 2003;170:99–106. doi: 10.4049/jimmunol.170.1.99. [DOI] [PubMed] [Google Scholar]
- 72.Nocentini G, Giunchi L, Ronchetti S, et al. A new member of the tumor necrosis factor/nerve growth factor receptor family inhibits T cell receptor-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:6216–21. doi: 10.1073/pnas.94.12.6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kwon B, Yu KY, Ni J, et al. Identification of a novel activation-inducible protein of the tumor necrosis factor receptor superfamily and its ligand. J Biol Chem. 1999;274:6056–61. doi: 10.1074/jbc.274.10.6056. [DOI] [PubMed] [Google Scholar]
- 74.Kanamaru F, Youngnak P, Hashiguchi M, et al. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol. 2004;172:7306–14. doi: 10.4049/jimmunol.172.12.7306. [DOI] [PubMed] [Google Scholar]
- 75.McHugh RS, Whitters MJ, Piccirillo CA, et al. CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity. 2002;16:311–23. doi: 10.1016/s1074-7613(02)00280-7. [DOI] [PubMed] [Google Scholar]
- 76.Tone M, Tone Y, Adams E, et al. Mouse glucocorticoid-induced tumor necrosis factor receptor ligand is costimulatory for T cells. Proc Natl Acad Sci USA. 2003;100:15059–64. doi: 10.1073/pnas.2334901100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol. 2002;3:135–42. doi: 10.1038/ni759. [DOI] [PubMed] [Google Scholar]
- 78.Ji HB, Liao G, Faubion WA, et al. Cutting edge: the natural ligand for glucocorticoid-induced TNF receptor-related protein abrogates regulatory T cell suppression. J Immunol. 2004;172:5823–7. doi: 10.4049/jimmunol.172.10.5823. [DOI] [PubMed] [Google Scholar]
- 79.Stephens GL, McHugh RS, Whitters MJ, et al. Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol. 2004;173:5008–20. doi: 10.4049/jimmunol.173.8.5008. [DOI] [PubMed] [Google Scholar]
- 80.Calmels B, Paul S, Futin N, Ledoux C, Stoeckel F, Acres B. Bypassing tumor-associated immune suppression with recombinant adenovirus constructs expressing membrane bound or secreted GITR-L. Cancer Gene Ther. 2005;12:198–205. doi: 10.1038/sj.cgt.7700781. [DOI] [PubMed] [Google Scholar]
- 81.Ko K, Yamazaki S, Nakamura K, et al. Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med. 2005;202:885–91. doi: 10.1084/jem.20050940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Turk MJ, Guevara-Patino JA, Rizzuto GA, Engelhorn ME, Sakaguchi S, Houghton AN. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–82. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cohen AD, Diab A, Perales MA, et al. Agonist anti-GITR antibody enhances vaccine-induced CD8(+) T-cell responses and tumor immunity. Cancer Res. 2006;66:4904–12. doi: 10.1158/0008-5472.CAN-05-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 85.Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 86.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 87.Greene JL, Leytze GM, Emswiler J, et al. Covalent dimerization of CD28/CTLA-4 and oligomerization of CD80/CD86 regulate T cell costimulatory interactions. J Biol Chem. 1996;271:26762–71. doi: 10.1074/jbc.271.43.26762. [DOI] [PubMed] [Google Scholar]
- 88.Wing K, Onishi Y, Prieto-Martin P, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–5. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 89.Fallarino F, Grohmann U, Hwang KW, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–12. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 90.Bodor J, Fehervari Z, Diamond B, Sakaguchi S. ICER/CREM-mediated transcriptional attenuation of IL-2 and its role in suppression by regulatory T cells. Eur J Immunol. 2007;37:884–95. doi: 10.1002/eji.200636510. [DOI] [PubMed] [Google Scholar]
- 91.Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006;18:206–13. doi: 10.1016/j.coi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 92.Hodi FS, Mihm MC, Soiffer RJ, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci USA. 2003;100:4712–17. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Attia P, Phan GQ, Maker AV, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol. 2005;23:6043–53. doi: 10.1200/JCO.2005.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ribas A, Bozon VA, Lopez-Berestein G, et al. Phase 1 trial of monthly doses of the human anti-CTLA4 monoclonal antibody CP-675,206 in patients with advanced melanoma. ASCO Annual Meeting, Abstract 7524.
- 95.Fong L, Kavanagh B, Rini BI, Shaw V, Weinberg V, Small EJ. A phase I trial of combination immunotherapy with CTLA-4 blockade and GM-CSF in hormone-refractory prostate cancer. J Clin Oncol. 2006;24:2508. 2006 ASCO Annual Meeting Proceedings. [Google Scholar]
- 96.Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–28. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 97.Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24:2283–9. doi: 10.1200/JCO.2005.04.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008 doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 100.Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–22. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 101.Radhakrishnan S, Nguyen LT, Ciric B, et al. Immunotherapeutic potential of B7-DC (PD-L2) cross-linking antibody in conferring antitumor immunity. Cancer Res. 2004;64:4965–72. doi: 10.1158/0008-5472.CAN-03-3025. [DOI] [PubMed] [Google Scholar]
- 102.Radhakrishnan S, Nguyen LT, Ciric B, Van Keulen VP, Pease LR. B7-DC/PD-L2 cross-linking induces NF-kappaB-dependent protection of dendritic cells from cell death. J Immunol. 2007;178:1426–32. doi: 10.4049/jimmunol.178.3.1426. [DOI] [PubMed] [Google Scholar]
- 103.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barber DL, Wherry EJ, Masopust D, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–7. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 105.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 106.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 107.Curiel TJ, Wei S, Dong H, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–7. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 108.Sharma MD, Baban B, Chandler P, et al. Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117:2570–82. doi: 10.1172/JCI31911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thompson RH, Gillett MD, Cheville JC, et al. Costimulatory B7-H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci USA. 2004;101:17174–9. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hamanishi J, Mandai M, Iwasaki M, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA. 2007;104:3360–5. doi: 10.1073/pnas.0611533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Inman BA, Sebo TJ, Frigola X, et al. PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata: associations with localized stage progression. Cancer. 2007;109:1499–505. doi: 10.1002/cncr.22588. [DOI] [PubMed] [Google Scholar]
- 112.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Strome SE, Dong H, Tamura H, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–5. [PubMed] [Google Scholar]
- 114.Hirano F, Kaneko K, Tamura H, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–96. [PubMed] [Google Scholar]
- 115.Blank C, Brown I, Peterson AC, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–5. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- 116.Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–44. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- 117.Nomi T, Sho M, Akahori T, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–7. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 118.Wong RM, Scotland RR, Lau RL, et al. Programmed death-1 blockade enhances expansion and functional capacity of human melanoma antigen-specific CTLs. Int Immunol. 2007;19:1223–34. doi: 10.1093/intimm/dxm091. [DOI] [PubMed] [Google Scholar]
- 119.Hamid O, Urba WJ, Yellin M, et al. Kinetics of response to ipilimumab (MDX-010) in patients with stage III/IV melanoma. J Clin Oncol. 2007;25:Abstract 8525. ASCO Annual Meeting Proceedings 2007. [Google Scholar]
- 120.Yang JC, Hughes M, Kammula U, et al. Ipilimumab (anti-CTLA4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. 2007;30:825–30. doi: 10.1097/CJI.0b013e318156e47e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saenger YM, Wolchok JD. The heterogeneity of the kinetics of response to ipilimumab in metastatic melanoma: patient cases. Cancer Immunol. 2008;8:1. [PMC free article] [PubMed] [Google Scholar]
- 122.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–66. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Davila E, Kennedy R, Celis E. Generation of antitumor immunity by cytotoxic T lymphocyte epitope peptide vaccination, CpG-oligodeoxynucleotide adjuvant, and CTLA-4 blockade. Cancer Res. 2003;63:3281–8. [PubMed] [Google Scholar]
- 124.Sutmuller RP, van Duivenvoorde LM, van Elsas A, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–32. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kocak E, Lute K, Chang X, et al. Combination therapy with anti-CTL antigen-4 and anti-4-1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res. 2006;66:7276–84. doi: 10.1158/0008-5472.CAN-05-2128. [DOI] [PubMed] [Google Scholar]
- 126.Uno T, Takeda K, Kojima Y, et al. Eradication of established tumors in mice by a combination antibody-based therapy. Nat Med. 2006;12:693–8. doi: 10.1038/nm1405. [DOI] [PubMed] [Google Scholar]