

Abstract
Immunodeficient non-obese diabetic (NOD)-severe combined immune-deficient (scid) mice bearing a targeted mutation in the gene encoding the interleukin (IL)-2 receptor gamma chain gene (IL2rγnull) engraft readily with human peripheral blood mononuclear cells (PBMC). Here, we report a robust model of xenogeneic graft-versus-host-like disease (GVHD) based on intravenous injection of human PBMC into 2 Gy conditioned NOD-scid IL2rγnull mice. These mice develop xenogeneic GVHD consistently (100%) following injection of as few as 5 × 106 PBMC, regardless of the PBMC donor used. As in human disease, the development of xenogeneic GVHD is highly dependent on expression of host major histocompatibility complex class I and class II molecules and is associated with severely depressed haematopoiesis. Interrupting the tumour necrosis factor-α signalling cascade with etanercept, a therapeutic drug in clinical trials for the treatment of human GVHD, delays the onset and progression of disease. This model now provides the opportunity to investigate in vivo mechanisms of xenogeneic GVHD as well as to assess the efficacy of therapeutic agents rapidly.
Keywords: GVHD, humanized mice, Hu-PBL-scid, IL-2r gamma chain, NOD-scid, xenogeneic
Introduction
Transplantation of haematopoietic stem cells (HSC) is a potential curative therapy for a variety of human diseases, including hereditable diseases of the blood, metabolic disorders, autoimmune diseases and cancer [1–5]. Although HSC transplantation has been used successfully for treatment of these disorders, it is not without risk. The most common complication is the development of graft-versus-host disease (GVHD) [6]. The development of acute GVHD is the most prevalent cause of morbidity in allogeneic HSC transplant recipients and accounts for up to 40% of the mortality in these patients. Additionally, up to 50% of surviving HSC-transplant patients will be affected by chronic GVHD [7]. A small animal model that mimics accurately the clinical presentation of GVHD would aid in the development of improved therapeutics and provide a tool for investigating the underlying mechanisms involved in disease pathogenesis.
Creating a small animal model of the human immune system by injecting human peripheral blood mononuclear cells (PBMC) into immunodeficient mice based on the CB17-severe combined immunedeficient (scid) mouse was first reported in 1988, but engraftment was low (reviewed in [8]). Although the non-obese diabetic (NOD)-scid mouse described in 1995 led to improved human cell engraftment, the level of engrafted human cells following intraperitoneal injection remained suboptimal (typically 0·1–5%), highly variable, and only poor engraftment was observed following intravenous injection of PBMC [9]. Neither CB17- nor NOD-scid mice engrafted with human PBMC provided a reproducible model of xenogeneic GVHD.
To improve human PBMC engraftment several groups have targeted natural killer (NK) cells, which are a major obstacle to human PBMC engraftment [10–13]. An H2d mouse deficient in both the recombination activating 2 gene (Rag2null) and an interleukin-2 receptor γ common chain targeted mutation (IL2rγnull), which completely ablates NK cell activity [8], showed improved engraftment compared with NOD-scid mice. However, even extensive preconditioning of the recipient with total body irradiation and macrophage depletion using chlodronate-containing liposomes did not eliminate the variability in human PBMC engraftment and the development of xenogeneic GVHD [12].
A second approach to inhibit the maturation of NK cells was based on NOD-scid mice genetically deficient in beta-2 microglobulin (β2mnull) [13]. In the absence of β2m, no host major histocompatibility complex (MHC) class I is expressed and NK cell numbers as well as activity are severely depressed [13]. NOD-scid β2mnull mice support higher levels of human PBMC engraftment and develop symptoms of xenogeneic GVHD without the requirement for macrophage depletion. However, retro-orbital injection of human T lymphocytes into lightly irradiated NOD-scid β2mnull mice produced xenogeneic GVHD in only 59% of the animals, whereas PBMC injected intravenously (i.v.) via the tail vein resulted in only transient engraftment and failed to induce xenogeneic GVHD [13]. A third approach has used C57BL/6-Rag2null IL2rγnull mice treated with clondronate-containing liposomes and 4 Gy irradiation, and this model system has been used to examine the in vivo function of human regulatory T cells [14].
We have reported recently the development of NOD-scid IL2rγnull mice [15]. A deficiency in the IL2rγ gene leads to severe defects in innate immunity [15] and a complete lack of NK cells [16]. We demonstrated recently that NOD-scid IL2rγnull mice engraft readily with high numbers of human PBMC following intravenous injection of as few as 5–10 × 106 cells [17]. These PBMC were capable of rejecting human islet allografts within 7–15 days, but between 30 and 45 days after PBMC transfusion we observed the development of GVHD-like symptoms [17].
Here, we report a human into mouse model of xenogeneic GVHD based on intravenous injection of human PBMC into lightly irradiated NOD-scid IL2rγnull mice. These mice develop xenogeneic GVHD consistently (100%) following injection of as few as 5 × 106 PBMC, regardless of the PBMC donor used. The development of xenogeneic GVHD in this model is highly dependent on host expression of MHC and is associated with severely depressed haematopoiesis. Interrupting the tumour necrosis factor (TNF)-α signalling cascade with etanercept, a therapeutic drug in clinical trials for human GVHD, delays the onset and progression of disease.
Materials and methods
Animals
NOD.CB17-Prkdcscid (NOD-scid), NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NOD-scid IL2rγnull), NOD.Cg-PrkdcscidIl2rgtm1WjlB2mtm1Unc (NOD-scid IL2rγnullβ2mnull), NOD.Cg-PrkdcscidIl2rgtm1WjlH2-Ab1tm1Gru/Sz (NOD-scid Abo IL2rγnull), NOD.Cg-Prkdcscid B2mtm1Unc (NOD-scid β2mnull), NOD.Cg-Prkdcscid H2-Ab1tm1Gru/Sz, (NOD-scid Abo) and NOD.Cg-Prkdcscid B2mtm1Unc H2-Ab1tm1Gru/Sz (NOD-scid β2mnull Aβnull) mice were obtained from colonies developed and maintained by L. D. S. at The Jackson Laboratory (Bar Harbor, ME, USA).
The NOD.Cg-PrkdcscidIl2rgtm1WjlB2mtm1Unc/Sz (NOD-scid IL2rγnullβ2m null) genetic stock was developed by first crossing NOD-scid/scid IL2rγnull females with NOD-scid/scid B2mnull males. All the F1 male offspring from this cross were NOD-scid IL2rγnull/Y +/B2mnull. These F1 males were then back-crossed to NOD-scid/scid IL2rγnull females. The NOD-scid/scid IL2rγnull+/B2mnull female offspring were crossed to their NOD-scid/scid IL2rγnull/Y +/B2mnull siblings. The NOD-scid/scid IL2rγnull B2mnull offspring were intercrossed to establish this strain.
The NOD.Cg-PrkdcscidIl2rgtm1WjlH2-Ab1tm1Gru/Sz (NOD-scid Abo IL2rγnull) genetic stock was developed by first crossing NOD-scid/scid IL2rγnull females with NOD-scid/scid Abo males. All the F1 male offspring from this cross were NOD-scid/scid IL2rγnull/Y +/Abo. These F1 males were then back-crossed to NOD-scid/scid IL2rγnull females. The NOD-scid/scid IL2rγnull+/Abo female offspring were crossed to their NOD-scid/scid IL2rγnull/y+/Abo siblings. The NOD-scid/scid IL2rγnull Abo offspring were intercrossed to establish this strain.
All animals were housed in a specific pathogen-free facility in microisolator cages, given autoclaved food and maintained on acidified autoclaved water and sulphamethoxazole–trimethoprim medicated water (Goldline Laboratories, Fort Lauderdale, FL, USA) [18] provided on alternate weeks. All animal use was in accordance with the guidelines of the Animal Care and Use Committee of the University of Massachusetts Medical School and The Jackson Laboratory and conformed to the recommendations in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences, 1996).
Collection of human PBMC
Human PBMC were obtained from healthy volunteers and blood donors under signed informed consent in accordance with the Declaration of Helsinki and approval from the Institutional Review Board of the University of Massachusetts Medical School. PBMC were collected in heparin and purified by Ficoll-Hypaque density centrifugation and suspended in RPMI-1640 for injection into mice at the cell doses indicated.
Xenogeneic GVHD protocol
The NOD-scid IL2rγnull mice were irradiated with 2 Gy unless indicated otherwise and injected i.v. 4 h later with various doses of PBMC. In all experiments, mice were weighed two to three times weekly, and the appearance of xenogeneic GVHD-like symptoms including weight loss (>15%), hunched posture, ruffled fur, reduced mobility and tachypnoea was used to determine time of euthanasia and is indicated as time of survival.
Antibodies and flow cytometry
Fluorescein isothiocyanate-conjugated anti-human CD8 (clone HIT8a), phycoerythrin-conjugated anti-human CD4 (clone RPA-T4), peridinin chlorophyll-conjugated anti-mouse Ly5 (clone 30-F11), allophycocyanin-conjugated anti-human CD45 (clone H130), Alexa Fluor 700-conjugated anti-human CD3 (clone UCHT1) and anti-mouse CD16/32 (clone 2.4G2) monoclonal antibody (mAb) were obtained from BD Pharmingen (San Jose, CA, USA). Alexa Fluor 405-conjugated anti-human CD20 (clone 2H7) was obtained from AbD-Serotec (Oxford, UK).
At times indicated in the text, single-cell suspensions of spleens or bone marrow flushed from the femurs and tibias were prepared in RPMI-1640 and washed in phosphate-buffered saline containing 0·1% sodium azide (Sigma-Aldrich, St Louis, MO, USA) and 1% fetal bovine serum (HyClone, Logan, UT, USA). Blood was collected into ethylenediamine tetraacetic acid.
Blood or single-cell suspensions of splenocytes or bone marrow were incubated with anti-CD16/32 for 5 min at 4°C to block non-specific Fc binding. Antibodies at the appropriate dilutions, as determined by previous titration against human or mouse lymphocytes, were incubated with 106 spleen or bone marrow cells or 100 µl of blood for 30 min at 4°C. Labelled spleen or bone marrow cells were washed and fixed with 1% paraformaldehyde (Polysciences, Warrington, PA, USA) in phosphate-buffered saline. Blood samples were processed with fluorescence activated cell sorter lysing solution (BD Biosciences, San Jose, CA, USA) according to the manufacturer's protocol. At least 20 000 events were acquired with an LSRII instrument (BD Biosciences, San José, CA, USA) and analysed using FlowJo Software (Mac version 8·2; Tree Star, Ashland, OR, USA).
For all analyses, anti-mouse CD45 staining was performed to exclude murine host cells from analysis, and only human CD45+ cells that were mouse CD45-negative were included in the analyses [19]. Matching isotype antibodies were used as negative controls, and the reported values have been corrected using these isotype control values [20]. Levels of human CD45+ cells reaching ≥0·1% in the blood and ≥1·0% in the spleen at 4 weeks were considered as successfully engrafted mice [18,20–22].
Human cytokine production
The NOD-scid IL2rγnull mice received 2 Gy total body irradiation using a Gammacell 40 (Atomic Energy of Canada, Ottawa, Canada) 4 h prior to receiving intravenous injections via the tail vein of 20 × 106 human PBMC. Plasma samples were prepared from blood collected in ethylenediamine tetraacetic acid 1, 4 and 24 h after PBMC injection. Plasma samples were stored at −80°C until use. The Human Inflammatory Cytokine Cytometric Bead Array kit (BD Bioscience) was used to determine the plasma concentration of human cytokines.
Mixed lymphocyte culture
Single-cell suspensions of spleens were recovered from NOD-scid, NOD-scid-β2mnull, NOD-scid Abo or NOD-scid β2mnull Abo mice. Red blood cells were lysed with hypotonic ammonium chloride lysing solution, and the cells were washed, suspended at 10 × 106 cells/ml, exposed to 20 Gy 137Cs radiation using a Gammacell 40 (Atomic Energy of Canada) and used as stimulator cells.
Murine stimulator cells (5 × 106 in 500 µl) were added to each well of a 24-well tissue culture plate (Falcon, Becton Dickinson Labware, Lincoln Park, NJ, USA). Responder human PBMC were labelled with 1 µm carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes, Eugene, OR, USA) [23]. Labelled responder cells (1 × 106 in 500 µl of medium) were added to triplicate wells containing murine stimulator cells, and the plates were incubated for 7 days at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were harvested, stained for cell surface markers and analysed with an LSRII instrument (BD Biosciences). Mod-Fit software (Verity, Topsham, ME, USA) was used to determine precursor frequency.
Haematological analyses
Haematological analyses of peripheral blood collected in heparin were performed on three cohorts of NOD-scid IL2rγnull mice: (i) untreated mice, (ii) mice given only 2 Gy 16–17 days prior to analysis; and (iii) mice given 2 Gy and injected i.v. with 5 × 106 human PBMC 16–17 days prior to analysis. All analyses were performed on a CBC-Diff Veterinary Hematology System (Heska, Fort Collins, CO, USA).
Histopathological analyses
Tissues were recovered from mice at necropsy, fixed in 10% buffered formalin and processed for histology and immunohistology, as described previously [24]. Bones were decalcified and histological sections were prepared. Immunohistochemical staining was performed with mAbs specific for human CD45 using a DakoCytomation EnVision Dual Link system implemented on a Dako Autostainer Universal Staining System (Dako, Glostrup, Denmark). The sections were counterstained with haematoxylin.
Statistics
All measures of variance are presented as standard deviation. Significance of difference of independent means was assumed for P-values of <0·05. Comparisons of two means used the independent samples t-test and comparisons of three or more means used one-way analyses of variance. Survival curves were generated by the Kaplan–Meier method. The equality of allograft survival distributions for animals in different treatment groups was tested using the log rank statistic. Duration of survival is presented as the median. All statistical analysis was performed using GraphPad Prism software (Graphpad Software, San Diego, CA, USA), and values of P < 0·05 are considered to indicate statistical significance.
Results
Low-dose irradiation accelerates xenogeneic GVHD in NOD-scid IL2rγnull mice injected with human PBMC
The NOD-scid IL2rγnull mice engrafted with human PBMC develop a gradual weight loss and exhibit hunched posture, anaemia, decreased mobility and ruffled fur, symptoms that are consistent with xenogeneic GVHD [25]. A recently reported human PBMC into H2d-Rag2null IL2rγnull mouse model required total body irradiation and host macrophage depletion for expression of xenogeneic GVHD [26]. Because unconditioned NOD-scid IL2rγnull mice engraft at higher levels with human PBMC than do BALB/c-Rag1null IL2rγnull mice [27] and eventually develop xenogeneic GVHD [17], we first asked whether low-dose irradiation would accelerate the onset and progression of xenogeneic GVHD in NOD-scid IL2rγnull mice.
The NOD-scid IL2rγnull mice given 2 Gy of total body irradiation and injected i.v. via the tail vein with 20 × 106 human PBMC develop accelerated weight loss and xenogeneic GVHD leading to decreased survival compared with mice that received PBMC in the absence of irradiation preconditioning [Fig. 1a; median survival time (MST) = 11 days versus MST = 34 days respectively; P < 0·0001]. As expected, 2 Gy irradiation had no effect on survival of mice not injected with human PBMC (Fig. 1a). To determine if weight loss was a sensitive predictor of xenogeneic GVHD, irradiated and non-irradiated NOD-scid IL2rγnull mice were injected with 20 × 106 human PBMC and weighed three times weekly for approximately 6 weeks. We found that weight loss was correlated strongly with poor survival in mice engrafted with human PBMC (Fig. 1b).
Fig. 1.
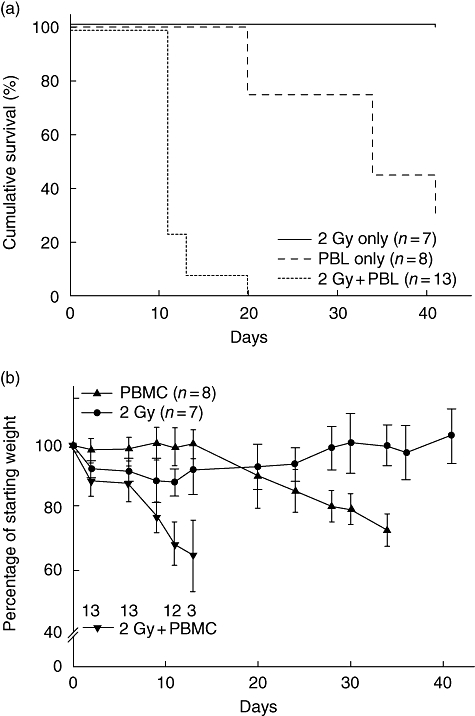
Sublethal irradiation accelerates xenogeneic graft-versus-host disease (GVHD) in Hu-peripheral blood mononuclear cells (PBMC) non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice. NOD-scid IL2rγnull mice received either 2 Gy irradiation (solid line, n = 7), 20 × 106 human PBMC intravenously (i.v.) [dashed line, median survival time (MST) = 34 days, n = 8, 3 PBMC donors] or 2 Gy irradiation followed by PBMC (dotted line, MST = 11, n = 13, three PBMC donors). (a) Survival of irradiated, PBMC-injected mice was significantly shorter than that of non-irradiated mice receiving PBMC (P < 0·0001). (b) The average weight is shown as a percentage of starting weight. Each data point represents the mean ± 1 standard deviation. The numbers on the graph indicate the number of mice remaining alive in the irradiation, PBMC treatment group at each time-point.
We next determined if the increased weight loss and accelerated development of xenogeneic GVHD in irradiated hosts was due to increased human T cell engraftment. We observed that 2 Gy irradiation increased significantly the percentage of human CD45+ cells in the bone marrow, spleen and blood of NOD-scid IL2rγnull mice when analysed at 13 or 14 days after PBMC injection (Fig. 2a–c). Although the percentages of human T cells were increased in irradiated hosts compared with non-irradiated hosts, only slight alterations in the ratio of CD4 : CD8 T cells was observed (Fig. 3a–c).
Fig. 2.
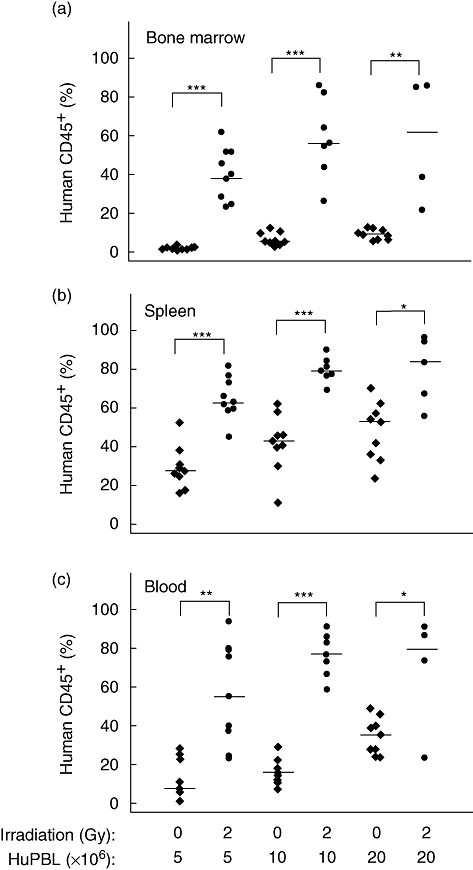
Sublethal irradiation facilitates engraftment of human peripheral blood mononuclear cells (PBMC) in Hu-PBMC non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice. NOD-scid IL2rγnull mice received were either non-irradiated or treated with 2 Gy irradiation 4 h prior to intravenous injection of 5, 10 or 20 million PBMC performed as described in Materials and methods. The percentage of human CD45+ cells in the bond marrow (a), spleen (b) and peripheral blood (c) is shown. Significant differences are noted in the figures. Each symbol represents an individual mouse. Results are compiled from three different experiments using three different PBMC donors. Horizontal lines represent the median of the values for each group. *P < 0·01; **P < 0·001; ***P < 0·0001.
Fig. 3.
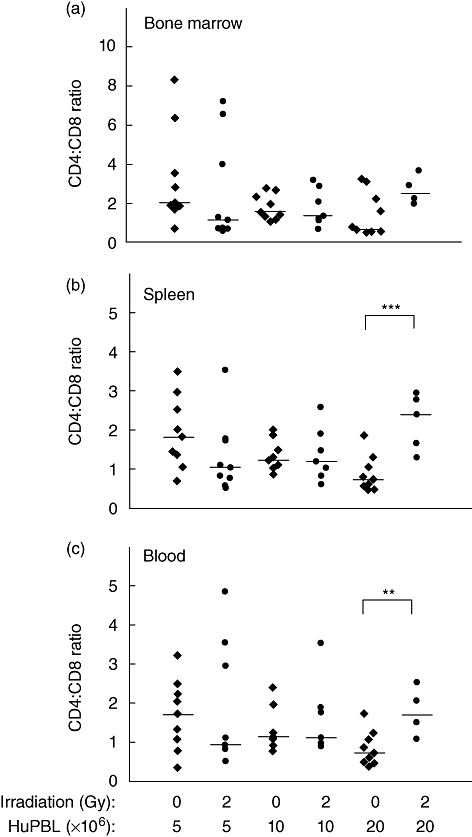
Sublethal irradiation induces only slight alterations in the CD4 : CD8 T cell ratios of the cells that engraft in Hu-peripheral blood mononuclear cells (PBMC) non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice. NOD-scid IL2rγnull mice shown in Fig. 2 were analysed for the percentages of CD4 and CD8 T cells by flow cytometry, as described in Materials and methods. The ratio of human CD4 : CD8 T cells in the bond marrow (a), spleen (b) and peripheral blood (c) is shown. Significant differences are noted in the figures. Each symbol represents an individual mouse. Results are compiled from three different experiments using three different PBMC donors. Horizontal lines represent the median of the values for each group. **P < 0·001; ***P < 0·0001.
Infiltration of human CD45+ cells in peripheral tissues
Previous reports have described infiltration of human cells into various tissues of immunodeficient mice developing xenogeneic GVHD-like symptoms [12]. We next examined histologically the spleen, lung, liver, duodenum, skin and tongue of mice developing clinical symptoms of xenogeneic GVHD. Human CD45+ cells were detected in the spleen, lung, liver and duodenum (Fig. 4), with lower numbers detected in the tongue and skin (data not shown). The spleen exhibited extensive infiltration of human CD45+ mononuclear cells. In the liver there are multifocal aggregates of human CD45+ mononuclear cells, primarily within periportal regions but also scattered throughout the hepatic parenchyma. Occasionally, these aggregates were associated with piecemeal hepatocellular necrosis. The lungs showed minimal multifocal aggregates of CD45+ human mononuclear cells expanding the alveolar septa. The lamina propria of the duodenum was expanded mildly by infiltrates of CD45+ human mononuclear cells (Fig. 4).
Fig. 4.
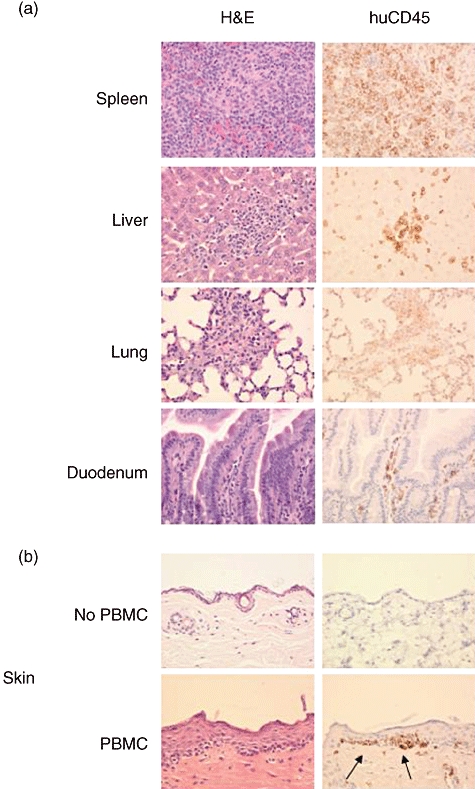
Infiltration of tissues with human cells in peripheral blood mononuclear cells (PBMC)-engrafted non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice. (a) Human CD45+ cells were detected in the spleen, lung, liver and small intestine of NOD-scid IL2rγnull mice that were treated with 2Gy irradiation followed by intravenous injection of 5 × 106 human PBMC, 400×. (b) Upper panels: truncal skin from NOD-scid IL2rγnull mice 50 days after treatment with 2 Gy only. The skin shows normal structure. There were no human CD45+ mononuclear cells present. Lower panels: truncal skin from NOD-scid IL2rγnull mouse 63 days after irradiation with 2 Gy, injection with 5 × 106 human PBMC, and repeated doses of etanercept as described in Materials and methods. Infiltrate of lymphocytes is present at the dermal epidermal junction. Arrows point to human cell infiltration.
No mononuclear infiltrates were seen in mice that were treated with 2 Gy radiation only (Fig. 4b, upper panels). In contrast, in one long-term surviving mouse treated with etanercept (see Materials and methods), a loss of fur was observed and there was a bandlike infiltrate along the dermal–epidermal junction comprised of human CD45+ mononuclear cells (Fig. 4b, lower panels).
Kinetics of xenogeneic GVHD development is dependent on inoculum cell dose
We have observed previously that as few as 5 × 106 PBMC result consistently in engraftment in non-irradiated NOD-scid IL2rγnull mice [17]. To determine the effect of cell dose on disease development, we injected 5 × 106, 10 × 106 or 20 × 106 human PBMC i.v. into 2 Gy irradiated mice. There was a strong correlation between the MST and cell dose (Fig. 5; MST = 22 days, 17 days and 12 days respectively). All cohorts of mice given even low dose of 5 × 106 PBMC from different donors developed xenogeneic GVHD.
Fig. 5.
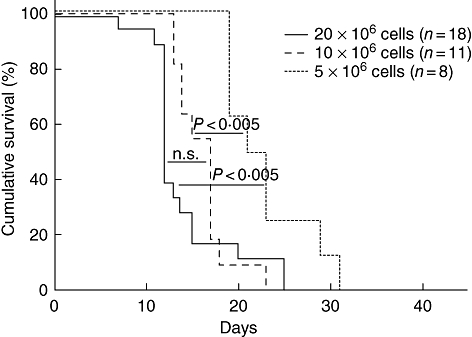
Xenogeneic graft-versus-host disease (GVHD) progression is dependent on peripheral blood mononuclear cells (PBMC) cell dose. Survival of non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice that received 2 Gy irradiation prior to injection of 20 × 106[solid line, median survival time (MST) = 12 days, n = 18, four PBMC donors], 10 × 106 (dashed line, MST = 17 days, n = 11, four PBMC donors) or 5 × 106 (dotted line, MST = 22 days, n = 8, three PBMC donors), human PBMC intravenously (i.v.).
Haematopoietic hypoplasia in PBMC-engrafted mice developing xenogeneic GVHD
Haematopoietic hypoplasia has been observed in mice [28–30] and humans [31] that develop GVHD. To determine if haematopoietic hypoplasia also occurred in NOD-scid IL2rγnull mice developing xenogeneic GVHD following engraftment of human PBMC, haematological analyses were performed on peripheral blood. In untreated mice, the haematocrit, haemoglobin, red blood cell count and platelet counts were within normal range and were slightly but significantly higher than values observed in mice treated with only 2 Gy irradiation 16–17 days earlier (Fig. 6). In contrast, by day 16 after irradiation and injection of 5 × 106 human PBMC, severely depressed haematopoiesis was readily apparent (Fig. 6).
Fig. 6.
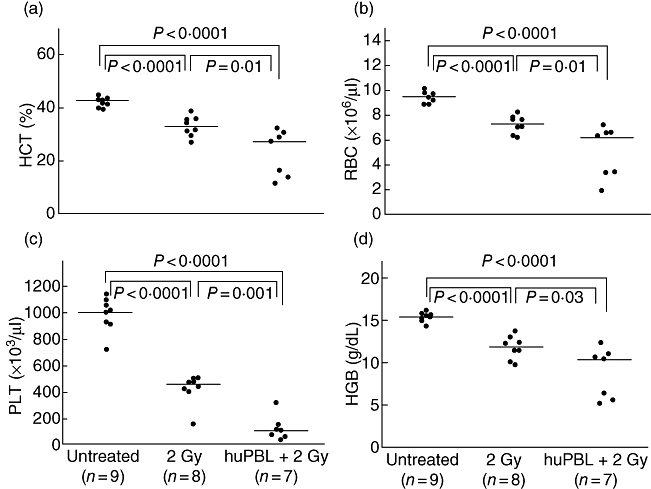
Haematological analyses of blood from non-irradiated non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice, or blood collected 16–17 days after treatment with either 2 Gy irradiation alone, or 2 Gy irradiation followed by an intravenous injection of 5 × 106 peripheral blood mononuclear cells (PBMC) were performed as described in Materials and methods. Significant differences are noted in the figures. Each symbol represents an individual mouse. Horizontal lines represent the median of the values for each group. HCT, haematocrit; PLT, platelets, HGB, haemoglobin; RBC, red blood cells.
Histologically, mice irradiated with 2 Gy and engrafted with human PBMC showed widespread diffuse necrosis of haematopoietic tissue in the medullary cavity of femurs (Fig. 7a), ribs and vertebral bodies (not shown). Haematopoietic tissue in the bone marrow cavities was replaced by microhaemorrhages, abundant fibrin strands and necrotic debris. Remaining erythroid and myeloid cells often had pyknotic nuclei and were undergoing necrosis with occasional intact mononuclear cells admixed within the necrotic debris. Human CD45+ staining of the femur showed numerous intact positive staining mononuclear cells scattered throughout the medullary cavity (Fig. 7b). Mice treated with 2 Gy of irradiation alone (Fig. 7c and d) and untreated mice (Fig. 7e and f) showed normal bone marrow morphology. No cells staining with anti-human CD45 antibody were present within the medullary cavity of mice not injected with PBMC.
Fig. 7.
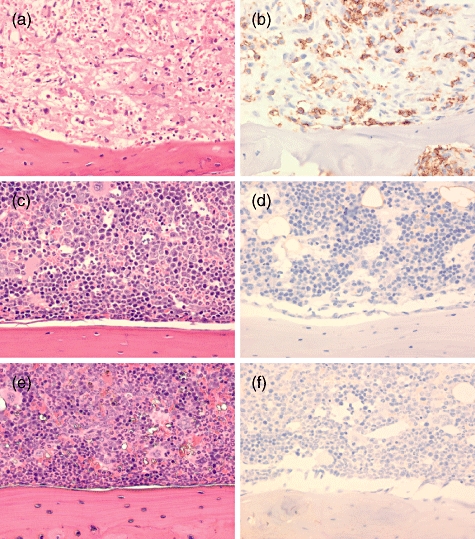
Femur medullary cavities of human peripheral blood mononuclear cells (PBMC) recipients and control mice. Histological analyses of bone marrow from non-irradiated non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice, or from mice necropsied at 16–17 days after treatment with either 2 Gy irradiation alone or 2 Gy irradiation followed by an intravenous injection of 5 × 106 PBMC as described in Materials and methods. (a,c,e) Haematoxylin and eosin; (b,d,f) human CD45 immunoperoxidase staining, ×400. (a,b) Mouse treated with 2 Gy and injected with human PBMC. Bone marrow shows diffuse severe necrosis of haematopoietic components. Human CD45+ cells are present throughout the medullary cavity. (c,d) Mouse treated with 2 Gy without PBMC engraftment. Bone marrow medullary compartment appears largely intact. No human CD45+ cells are present. (e,f) Untreated mouse exhibiting normal bone marrow. No human CD45+ cells are present.
Murine MHC stimulates human PBMC proliferation in vitro
It has been hypothesized that human CD4+ T cell expansion in immunodeficient mice is driven by human anti-mouse MHC class II reactivity [32]. However, in previous models using CB17-scid or NOD-scid mice, CD8+ rather than CD4+ T cells predominate in the engrafted recipient [20–22], suggesting a strong reactivity to host MHC class I. Both CD4+ and CD8+ T cells engraft at high levels in NOD-scid IL2rγnull mice [17], suggesting that reactivity to mouse MHC class I as well as class II may occur.
To investigate the role of host MHC in the development of xenogeneic GVHD in mice given human PBMC, we first determined the percentage of human T cells that proliferate in vitro in response to murine MHC class I and class II molecules using a CFSE-based assay that analysed human T cell proliferation in response to cells from NOD-scid mice that were genetically deficient in MHC class I or class II molecules.
As expected [32], a lower proportion of human CD4+ cells proliferated in response to NOD-scid Abo and NOD-scid β2mnull Abo (both MHC class II-deficient) splenocytes compared with NOD-scid splenocytes (P = 0·025 and 0·05 respectively, Fig. 8a, left panel). In contrast, a lower proportion of human CD8+ cells proliferated in response to NOD-scid β2mnull and NOD-scid β2mnull Abo (both MHC class I-deficient) splenocytes compared with NOD-scid splenocytes (both P = 0·025, Fig. 8a, right panel). These data suggest that much but not all proliferation of human T cells engrafted into immunodeficient NOD-scid mice is in response to stimulation by host MHC class I and class II molecules.
Fig. 8.
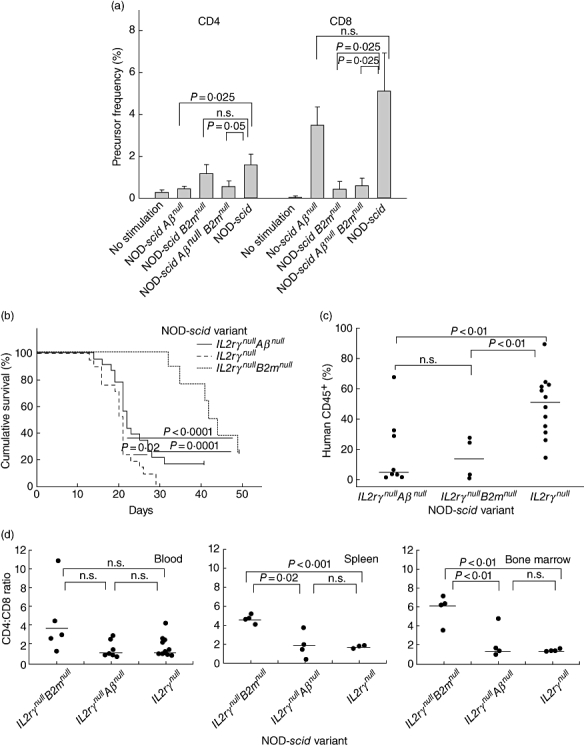
Human peripheral blood mononuclear cells (PBMC) xenoreactivity to mice deficient in major histocompatibility complex (MHC) class I and class II. (a) Carboxyfluorescein succinimidyl ester (CFSE)-labelled human PBMC were cultured alone or in the presence of antigen-presenting cell splenocytes obtained from non-obese diabetic (NOD)-severe combined immunedeficient (scid), NOD-scid β2mnull, NOD-scid Aβnull or NOD-scid β2mnull Aβnull mice for 7 days. The precursor frequency index is shown of one experiment representative of three independent experiments (± 1 standard deviation). (b) Survival of NOD-scid IL2rγnull[dashed line, median survival time MST = 21 days, n = 21, six PBMC donors], NOD-scid IL2rγnullAβnull (solid line, MST = 22 days, n = 23, six PBMC donors) and NOD-scid IL2rγnullB2mnull (dotted line, MST = 46·5 days, n = 10, four PBMC donors) following 2 Gy irradiation and intravenous injection of 5 × 106 cells. (c) Engraftment of human CD45+ cells in the peripheral blood of mice at time of killing. (d) Ratio of the percentage of human CD4+ : human CD8+ cells recovered from the peripheral blood, spleen and bone marrow of mice at time of killing. Each symbol represents an individual mouse. Horizontal lines represent the mean of the values of each group.
Recognition of host MHC drives the development of xenogeneic GVHD
Based on this observation, we hypothesized that NOD-scid IL2rγnull mice lacking MHC class I or class II expression would exhibit delayed development of xenogeneic GVHD. To test this, MHC class I-deficient NOD-scid IL2rγnullβ2mnull or MHC class II-deficient NOD-scid IL2rγnull Abo mice were irradiated with 2 Gy and injected with 5 × 106 human PBMC. NOD-scid-IL2rγnull Abo mice appeared to be highly susceptible to GVHD, although disease progression was slightly but significantly delayed compared with that observed in NOD-scid IL2rγnull mice expressing both MHC molecules (Fig. 8b, MST = 22 versus 21 days respectively, P = 0·02). In contrast, NOD-scid IL2rγnullβ2mnull mice were relatively resistant to disease development (MST = 44 days), a survival time which was significantly longer than that observed in NOD-scid IL2rγnull Abo (P = 0·001) and NOD-scid IL2rγnull (P < 0·0001) mice.
We next analysed the levels of human CD45+ cell engraftment in the blood at the time of euthanasia in animals exhibiting xenogeneic GVHD. Decreased human CD45+ cell engraftment was observed in both NOD-scid IL2rγnullβ2mnull mice (13·9 ± 6·9%, P < 0·01) and NOD-scid IL2rγnull Abo mice (18·1 ± 8·3%, P < 0·01) compared with mice expressing both MHC molecules (53·6 ± 4·4%, Fig. 8c). Surprisingly, levels of human CD45+ cell engraftment between the mice deficient in MHC class I and MHC class II were not significantly different (P = not significant). As expected, we observed that NOD-scid IL2rγnullβ2mnull mice have significantly higher CD4 : CD8 ratios in spleen (P = 0·02) and bone marrow (P < 0·01) than the other two strains (Fig. 8d).
Soluble TNF-α receptor delays progression of xenogeneic GVHD
The TNF-α has been reported to have an important role in the pathogenesis of GVHD [33–36]. In Phases I and II clinical trials, anti-TNF-α antibody treatment decreased the severity of disease [37]. Similarly, blocking TNF-α with etanercept (Enbrel®, Immunex Corp., Thousand Oaks, CA, USA; a soluble TNF-α decoy receptor) as part of a GVHD treatment regimen was shown to be efficacious [38].
To determine if etanercept would delay or prevent xenogeneic GVHD in our model system, we first treated NOD-scid IL2rγnull mice with two doses of etanercept injected intraperitoneally prior to irradiation and intravenous injection of 20 × 106 human PBMC. Pretreatment with etanercept increased significantly the survival of mice from a MST of 12–16 days, with one mouse surviving to day 41 (Fig. 9a, P < 0·001). To determine if a more aggressive drug regimen would prolong survival further, mice were injected with etanercept every 3 days following injection of 20 × 106 human PBMC in addition to the pretreatment dosing. This dosing regimen increased survival (MST = 23 days) significantly over the two-dose regimen (Fig. 9a, P = 0·01). For all groups, weight loss correlated consistently with disease progression (Fig. 9b).
Fig. 9.
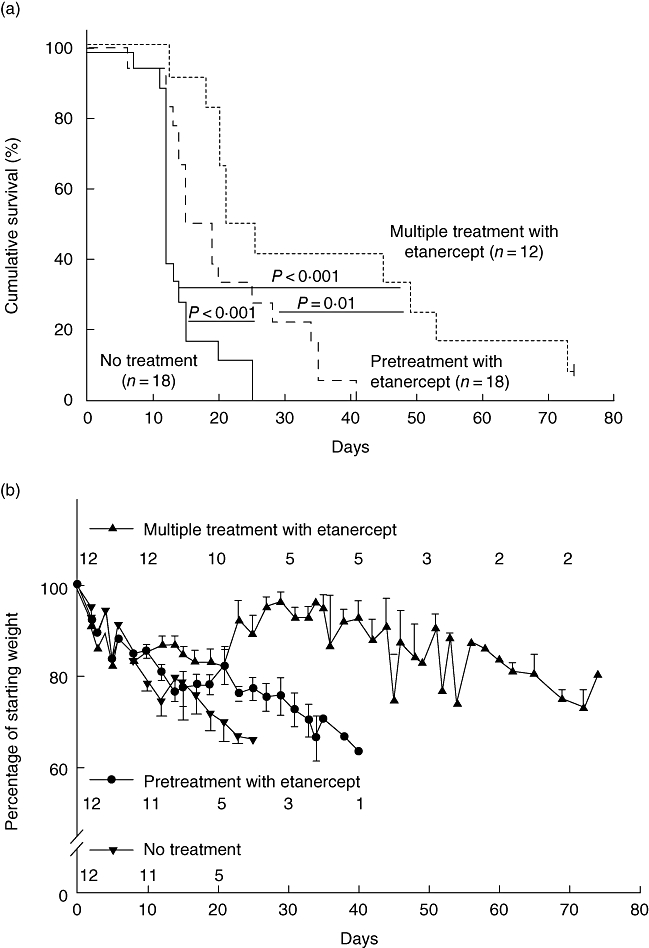
Soluble tumour necrosis factor (TNF)-α receptor delays progression of xenogeneic graft-versus-host disease (GVHD). (a) Survival of non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice that received 20 × 106 peripheral blood mononuclear cells (PBMC) intravenously (i.v.) 4 h after 2 Gy irradiation [solid line, median survival time (MST) = 12 days, n = 18, seven PBMC donors], with etanercept pretreatment of 100 µg/injection on days −3 and −1 prior to injection of PBMC (dashed line, MST = 16 days, n = 18, seven PBMC donors) or etanercept pretreatment plus 100 µg every 3 days (dotted line, MST = 23 days, n = 12, four PBMC donors). (b) The average weight in irradiated mice that received PBMC alone, PBMC plus etanercept pretreatment, or etanercept pretreatment plus 100 µg every 3 days is shown as the percentage of starting weight. The numbers shown correspond with the number of mice that remain alive at each time-point.
Etanercept-dependent disease prevention at lower PBMC doses
Injection of 20 × 106 human PBMC leads consistently to a robust xenogeneic GVHD, with no mouse surviving past day 25 (Fig. 9a). Although there was no significant improvement in survival with the two-dose etanercept pretreatment, the repeated-dose etanercept treatment increased survival significantly compared with mice receiving only 5 × 106 PBMC (P < 0·001) or the two-dose treatment (Fig. 10a, P = 0·034). Interestingly, three of 25 mice that were pretreated with the two doses of etanercept as well as six of 24 mice that received repeat doses of etanercept survived to the end of the observation period.
Fig. 10.
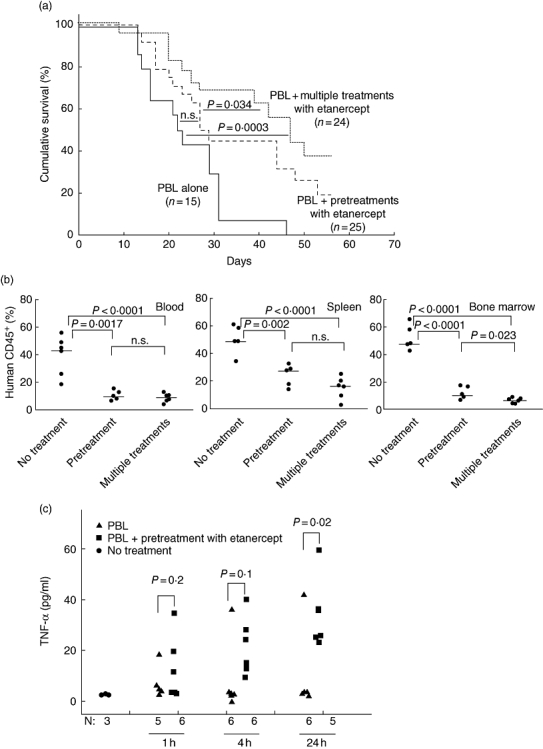
Etanercept-dependent prevention of xenogeneic graft-versus-host disease (GVHD) at lower peripheral blood mononuclear cells (PBMC) cell doses. (a) Survival of non-obese diabetic (NOD)-severe combined immunedeficient (scid) IL2rγnull mice given 5 × 106 PBMC intravenously (i.v.). Four h after 2 Gy irradiation [solid line, median survival time (MST) = 22 days, n = 15, seven PBMC donors], with etanercept pretreatment of 100 µg/injection on days −3 and −1 prior to injection of PBMC (dashed line, MST = 28 days, n = 25, seven PBMC donors) or etanercept pretreatment plus additional 100 µg etanercept injections every 3 days (dotted line, MST = 47 days, n = 24, seven PBMC donors). (b) Mice received 2 Gy irradiation 4 h prior to 5 × 106 PBMC; 14 days later, human CD45+ cell engraftment in the blood (left panel), spleen (middle panel) and bone marrow (right panel) was evaluated by flow cytometry. Etanercept treatment decreased human CD45+ engraftment in all three tissues tested. Horizontal lines represent the median of the values of each group. (c) Levels of human tumour necrosis factor (TNF)-α in plasma samples collected from untreated or etanercept-pretreated mice 1, 4 and 24 h after PBMC injection. All mice received 2 Gy irradiation 4 h prior to intravenous (i.v.) injection of 5 × 106 PBMC from one of two PBMC donors. Each symbol represents an individual mouse.
One possible mechanism by which etanercept could delay disease development would be to reduce human PBMC engraftment. We observed that the percentage of human CD45+ cells 14 days after irradiation with 2 Gy and injection of 5 × 106 human PBMC was decreased significantly in the peripheral blood (P < 0·0001), spleen (P < 0·0001) and bone marrow (P < 0·0001) in mice given either etanercept dosing regimen compared with non-drug-treated mice (Fig. 10b).
A second possible mechanism by which etanercept modulates xenogeneic GVHD in this model could be the reduction of bioavailable human TNF-α. Plasma levels of human TNF-α in mice that received PBMC alone were low (Fig. 10c). Human TNF-α levels in mice treated with etanercept were not statistically different from mice given PBMC without drug treatment at 1 (12·7 ± 12·4 versus 7·2 ± 6·4 pg/ml; n = 6, P = 0·39) or 4 h post-PBMC injection (22·4 ± 14·8 versus 9·5 ± 14·6 pg/ml; P = 0·2). Surprisingly, 24 h after PBMC injection, plasma levels of human TNF-α were increased significantly in etanercept-treated mice compared with mice that received PBMC alone (33·9 ± 15·1 versus 9·4 ± 16·0 pg/ml; P < 0·05, Fig. 10c).
Discussion
In this study, we have developed a robust model of xenogeneic GVHD based on the NOD-scid IL2rγnull strain of mice. NOD-scid IL2rγnull mice given a low dose of irradiation develop xenogeneic GVHD consistently, following intravenous injection of as few as 5 × 106 PBMC. Disease was associated with weight loss, anaemia and decreased platelet counts. Using this model, we documented the relative contribution of host MHC class I and class II to human cell engraftment and disease expression using unique newly generated stocks of MHC-deficient NOD-scid IL2rγnull mice. This new model of xenogeneic GVHD was then used to demonstrate that etanercept mediates its protective effect in part by decreasing the engraftment of human PBMC and in part by neutralizing TNF-α.
The clinical relevance of this model system is documented by our observation, in vitro and in vivo, that the majority of the ‘xenoreactivity’ in this GVHD model system is directed against host MHC class I and class II of the host, i.e. similar to the MHC reactivity observed in alloresponses in human GVHD [6]. The therapeutic relevance of this model system is shown by the ability of TNF blockade to block xenogeneic GVHD, similar to the clinical observation of the utility of TNF blockade as a therapeutic for allogeneic GVHD in mice [39–41] and humans [38,42].
We have shown NOD-scid IL2rγnull mice previously to be superior hosts in their ability to support human PBMC engraftment relative to NOD-scid mice or BALB/c-Rag1null IL2rγnull mice [17,27]. In unconditioned NOD-scid IL2rγnull mice injected i.v. with 20 × 106 human PBMC, we observed the gradual development of phenotypic changes consistent with xenogeneic GVHD over the course of 4–8 weeks, similar to recently reported results following injection of 10 × 106 human PBMC into unconditioned NOD-scid IL2rγnull mice [25]. To investigate whether we could optimize this model for the study of xenogeneic GVHD, we first determined the effects of preconditioning with low-dose irradiation. It has been reported that the development of xenogeneic GVHD in other model systems is enhanced by administration of sublethal whole body irradiation [12,13,26]. However, in those models [12,26], GVHD was observed following only very specific injection routes, high cell doses [13] or macrophage depletion [12]. For example, a xenogeneic GVHD model based on NOD-scid β2mnull recipient mouse exhibited disease only after retro-orbital but not tail vein injection of human PBMC [13]. Furthermore, a minimum inoculum of 10 × 106 purified T cells was required for mice given 2·5 Gy of irradiation, and then only about 60% of the mice developed the disease. In contrast, we observed that 100% of NOD-scid IL2rγnull mice treated with only 2 Gy irradiation and injected with 20 × 106 human PBMC developed xenogeneic GVHD rapidly and consistently. Furthermore, we observed that 100% of mice developed xenogeneic GVHD following intravenous injection with as few as 5 × 106 PBMC obtained from multiple different donors.
Using this optimized model, we investigated the mechanism driving the development of xenogeneic GVHD. We observed that a severe anaemia and thrombocytopenia developed rapidly following injection of PBMC and that these deficiencies were observed prior to the appearance of clinical symptoms of GVHD. Haematopoietic hypoplasia has been observed in mice [28–30] and humans [31] that develop GVHD, although these abnormalities cannot be used in isolation to establish the diagnosis of GVHD [39]. However, thrombocytopenia at the time of GVHD diagnosis in the clinic has been associated with a poor prognosis [39]. The rapid development of anaemia and thrombocytopenia was not due entirely to the irradiation conditioning, as mice given 2 Gy irradiation but no PBMC exhibited slight anaemia and somewhat depressed platelet cell counts. This rapid onset of anaemia in mice injected with PBMC could be due to internal bleeding or severely depressed haematopoiesis in the marrow. To begin to investigate this, histological analyses of bone from 2 Gy-treated mice with or without injection of PBMC revealed a severe reduction in marrow haematopoiesis in mice given PBMC but not in mice treated with irradiation alone. Furthermore, although no gross evidence of internal bleeding was observed on necropsy, additional analyses are being performed to address this possibility.
It has been suggested previously that the development of xenogeneic GVHD in immunodeficient mice is dependent on the relative engraftment of human T lymphocytes which is probably driven by recognition of host MHC [40]. In murine allo-GVHD models, mice deficient in MHC class I or class II develop delayed CD8+ or CD4+ mediated disease respectively [41], suggesting that alloreactivity to both host MHC alleles is important in disease pathogenesis. In human–mouse xenoreactive models, early experiments evaluating human T cells isolated from CB17-scid mice demonstrated that human CD4+ T cells recovered from engrafted mice were specific for murine MHC class II [32]. However, the predominance of CD8+ cells in all models of human PBMC engraftment suggests that a strong reactivity against host MHC class I is also present [20–22]. Furthermore, the high levels of engraftment of both CD4+ and CD8+ human cells observed in PBMC-engrafted NOD-scid IL2rγnull mice [17] suggest that reactivity to both host MHC class I and class II may occur.
To investigate the relative role of murine host MHC class I and class II in human cell proliferation, we first stimulated human PBMC in vitro with murine splenocytes isolated from mice genetically deficient in each of these molecules. As expected [32], a lower precursor frequency of human CD8+ cells was observed following stimulation by splenocytes deficient in murine MHC class I, and lower precursor frequency of human CD4+ cells was observed following stimulation by splenocytes deficient in murine MHC class II. In vivo, the delay in xenogeneic GVHD observed in human PBMC-engrafted mice lacking MHC class I or class II was also consistent with host MHC playing a major role in triggering disease development. Comparison of human cell engraftment in these strains revealed a decrease in human CD45+ cells in both MHC class I- or class II-deficient mice compared with NOD-scid IL2rγnull mice that express both sets of molecules. We observed significant differences in the level of PBMC engraftment in irradiated versus unirradiated recipients, but the differences in engraftment induced only slight alterations in the CD4 : CD8 T cell ratios of the cells that engrafted. Surprisingly, however, there was no difference in the levels of CD45+ cell engraftment between MHC class I- and class II-deficient recipients even though there was a significant difference in the kinetics of xenogeneic GVHD development. This observation suggests that human CD45+ cell engraftment is not the only determinant of xenogeneic GVHD expression and that the engraftment and activation of CD8+ cells appear to play a primary role in the pathogenesis. This is also reflected in the CD4+ : CD8+ ratio, which was highly skewed towards CD4 cells in the MHC class I-deficient mice that exhibited delayed development of xenogeneic GVHD.
The role of the inflammatory cytokines, particularly TNF-α in GVHD pathogenesis, has been investigated both in experimental systems and in the clinic. Murine studies of GVHD have shown that TNF-α has a key role in intestinal damage [42–44] and skin pathology [45,46]. Clinical data have shown that high levels of TNF-α production during conditioning regimens prior to allogeneic HSC transplantation were highly predictive of the subsequent severity of acute GVHD [47–49]. Neutralizing TNF-α activity decreased pathology in target organs and improved survival in murine models of allogeneic GVHD [41–43]. Similarly, clinical trials in which etanercept was used to neutralize TNF-α in patients with acute GVHD have proved this approach efficacious when used in conjunction with methylprednisone [38,50].
In agreement with published studies in murine models of GVHD [41–43], treatment with etanercept, a soluble TNF-α decoy receptor, was able to delay progression of xenogeneic GVHD in our humanized mouse model system. This may be due in part to the induction of TNF-α by irradiation [51], leading to the enhancement of recognition of host MHC and minor antigens by the infused PBMC, effectively inducing a more robust xenogeneic GVHD. Furthermore, we observed that etanercept mediates its effects in part by reducing the engraftment of human CD45+ cells, a sensitive indicator of GVHD. Surprisingly, despite the efficacy of etanercept in preventing xenogeneic GVHD, we were unable to detect TNF-α in the serum of mice not treated with etanercept, whereas readily detectable levels of serum TNF-α were detected in mice treated with etanercept. Although counter-intuitive, elevated levels of TNF-α have been reported previously in patients treated with etanercept [52–54] and in mouse model systems [55] where drug therapy was beneficial. These data suggest that although etanercept binds to and neutralizes the biological activity of TNF-α, it also prolongs its half-life in the serum. The detection of TNF-α in the enzyme-linked immunosorbent assay (ELISA) assay in etanercept-treated mice may be due to the ability of TNF-α to dissociate rapidly from etanercept [55–57], and it has been suggested that a high affinity and irreversible binding of etanercept with the anti-TNF-α antibody used in the ELISA assay competes effectively in vitro for the TNF-α[52].
In summary, we have developed a robust model of xenogeneic GVHD disease based on the injection of human PBMC into NOD-scid IL2rγnull mice. Our data highlight the ‘proof of principle’ concept of using this model system to determine rapidly whether proposed therapeutics for GVHD are efficacious in vivo and will permit the underlying mechanism of drug action to be investigated. This model system represents a powerful tool for understanding mechanisms of disease pathogenesis in human GVHD and for evaluating potential therapeutic interventions on a human immune system in a preclinical setting.
Acknowledgments
We thank Linda Paquin, Amy Cuthbert, Keith Daniels, Celia Hartigan, Amy Sands and Candy Knoll for their technical assistance. We also thank the participation of the Clinical Trials Unit at the University of Massachusetts Medical School. This work was supported by National Institutes of Health Grants AI46629, DK53006, HL077642, an institutional Diabetes Endocrinology Research Center (DERC) grant DK32520, a Cancer Center Core Grant CA34196, a Grant from Viacell, Inc., the Beta Cell Biology Consortium and grants from the Juvenile Diabetes Foundation, International. M. K. is supported by a pre-doctoral fellowship from the American Diabetes Association. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Disclosure
None.
References
- 1.Bhatia M, Walters MC. Hematopoietic cell transplantation for thalassemia and sickle cell disease: past, present and future. Bone Marrow Transplant. 2008;41:109–17. doi: 10.1038/sj.bmt.1705943. [DOI] [PubMed] [Google Scholar]
- 2.Burt RK, Verda L, Oyama Y, Statkute L, Slavin S. Non-myeloablative stem cell transplantation for autoimmune diseases. Springer Semin Immunopathol. 2004;26:57–69. doi: 10.1007/s00281-004-0162-6. [DOI] [PubMed] [Google Scholar]
- 3.Prasad VK, Kurtzberg J. Emerging trends in transplantation of inherited metabolic diseases. Bone Marrow Transplant. 2008;41:99–108. doi: 10.1038/sj.bmt.1705970. [DOI] [PubMed] [Google Scholar]
- 4.Welniak LA, Blazar BR, Murphy WJ. Immunobiology of allogeneic hematopoietic stem cell transplantation. Annu Rev Immunol. 2007;25:139–70. doi: 10.1146/annurev.immunol.25.022106.141606. [DOI] [PubMed] [Google Scholar]
- 5.Petersen SL. Alloreactivity as therapeutic principle in the treatment of hematologic malignancies. Studies of clinical and immunologic aspects of allogeneic hematopoietic cell transplantation with nonmyeloablative conditioning. Dan Med Bull. 2007;54:112–39. [PubMed] [Google Scholar]
- 6.Ferrara JL, Levine JE. Graft-versus-host disease in the 21st century: new perspectives on an old problem. Semin Hematol. 2006;43:1–2. doi: 10.1053/j.seminhematol.2005.11.028. [DOI] [PubMed] [Google Scholar]
- 7.Lee SJ. New approaches for preventing and treating chronic graft-versus-host disease. Blood. 2005;105:4200–6. doi: 10.1182/blood-2004-10-4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–30. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 9.Greiner DL, Shultz LD. Use of NOD/LtSz-scid/scid mice in biomedical research. In: Leiter EH, Atkinson MA, editors. NOD mice and related strains: research applications in diabetes, AIDS, cancer and other diseases. Austin, TX: R.G. Landes Co; 1998. pp. 173–203. [Google Scholar]
- 10.Shpitz B, Chambers CA, Singhal AB, et al. High level functional engraftment of severe combined immunodeficient mice with human peripheral blood lymphocytes following pretreatment with radiation and anti-asialo GM1. J Immunol Methods. 1994;169:1–15. doi: 10.1016/0022-1759(94)90119-8. [DOI] [PubMed] [Google Scholar]
- 11.Cao T, Leroux-Roels G. Antigen-specific T cell responses in human peripheral blood leucocyte (hu-PBL)-mouse chimera conditioned with radiation and an antibody directed against the mouse IL-2 receptor beta-chain. Clin Exp Immunol. 2000;122:117–23. doi: 10.1046/j.1365-2249.2000.01340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Rijn RS, Simonetti ER, Hagenbeek A, et al. A new xenograft model for graft-versus-host disease by intravenous transfer of human peripheral blood mononuclear cells in RAG2−/− gammac−/− double-mutant mice. Blood. 2003;102:2522–31. doi: 10.1182/blood-2002-10-3241. [DOI] [PubMed] [Google Scholar]
- 13.Nervi B, Rettig MP, Ritchey JK, et al. Factors affecting human T cell engraftment, trafficking, and associated xenogeneic graft-vs-host disease in NOD/SCID beta2mnull mice. Exp Hematol. 2007;35:1823–38. doi: 10.1016/j.exphem.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hippen KL, Harker-Murray P, Porter SB, et al. Umbilical cord blood regulatory T-cell expansion and functional effects of tumor necrosis factor receptor family members OX40 and 4-1BB expressed on artificial antigen-presenting cells. Blood. 2008;112:2847–57. doi: 10.1182/blood-2008-01-132951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao X, Shores EW, Hu-Li J, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity. 1995;2:223–38. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 16.Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2rγnull mice engrafted with mobilized human hematopoietic stem cell. J Immunol. 2005;174:6477–89. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 17.King M, Pearson T, Shultz LD, et al. A new Hu-PBL model for the study of human islet alloreactivity based on NOD-scid mice bearing a targeted mutation in the IL-2 receptor gamma chain gene. Clin Immunol. 2008;126:303–14. doi: 10.1016/j.clim.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Christianson SW, Greiner DL, Hesselton RM, et al. Enhanced human CD4+ T cell engraftment in β2-microglobulin-deficient NOD-scid mice. J Immunol. 1997;158:3578–86. [PubMed] [Google Scholar]
- 19.Wagar EJ, Cromwell MA, Shultz LD, et al. Regulation of human cell engraftment and development of EBV-related lymphoproliferative disorders in Hu-PBL-scid mice. J Immunol. 2000;165:518–27. doi: 10.4049/jimmunol.165.1.518. [DOI] [PubMed] [Google Scholar]
- 20.Shultz LD, Banuelos S, Lyons B, et al. NOD/LtSz-Rag1null Prf1null mice: a new model system with increased levels of human peripheral leukocyte and hematmopoietic stem cell engraftment. Transplantation. 2003;76:1036–42. doi: 10.1097/01.TP.0000083041.44829.2C. [DOI] [PubMed] [Google Scholar]
- 21.Greiner DL, Shultz LD, Yates J, et al. Improved engraftment of human spleen cells in NOD/LtSz-scid/scid mice as compared with C.B-17-scid/scid mice. Am J Pathol. 1995;146:888–902. [PMC free article] [PubMed] [Google Scholar]
- 22.Hesselton RM, Greiner DL, Mordes JP, Rajan TV, Sullivan JL, Shultz LD. High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-scid/scid mice. J Infect Dis. 1995;172:974–82. doi: 10.1093/infdis/172.4.974. [DOI] [PubMed] [Google Scholar]
- 23.Whalen BJ, Love LC, Mordes JP, Rossini AA, Greiner DL. Intravital dye-labeled diabetogenic rat T cells retain dye, home to the pancreas, and induce diabetes. Transplant Proc. 1999;31:1611–4. doi: 10.1016/s0041-1345(99)00057-3. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt MR, Appel MC, Giassi LJ, Greiner DL, Shultz LD, Woodland RT. Human BLyS facilitates engraftment of human PBL derived B cells in immunodeficient mice. PLoS ONE. 2008;3:e3192. doi: 10.1371/journal.pone.0003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Golovina TN, Mikheeva T, Suhoski MM, et al. CD28 costimulation is essential for human T regulatory expansion and function. J Immunol. 2008;181:2855–68. doi: 10.4049/jimmunol.181.4.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Rijn RS, Simonetti ER, Hagenbeek A, et al. Quantitative assessment of human T lymphocytes in RAG2(−/−)gammac(−/−) mice: the impact of ex vivo manipulation on in vivo functionality. Exp Hematol. 2007;35:117–27. doi: 10.1016/j.exphem.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 27.King M, Pearson T, Rossini AA, Shultz LD, Greiner DL. Humanized mice for the study of type 1 diabetes and beta cell function. Ann NY Acad Sci. 2008;1150:46–53. doi: 10.1196/annals.1447.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirabayashi N. Studies on graft versus host (GvH) reactions. I. Impairment of hemopoietic stroma in mice suffering from GvH disease. Exp Hematol. 1981;9:101–10. [PubMed] [Google Scholar]
- 29.Kanamaru A, Okamoto T, Matsuda K, Hara H, Nagai K. Elevation of erythroid colony-stimulating activity in the serum of mice with graft-versus-host disease. Exp Hematol. 1984;12:763–7. [PubMed] [Google Scholar]
- 30.Gleichmann E, Gleichmann H. Pathogenesis of graft-versus-host reactions (GVHR) and GVH-like diseases. J Invest Dermatol. 1985;85:115s–20s. doi: 10.1111/1523-1747.ep12275619. [DOI] [PubMed] [Google Scholar]
- 31.Godder K, Pati AR, Abhyankar SH, Lamb LS, Armstrong W, Henslee-Downey PJ. De novo chronic graft-versus-host disease presenting as hemolytic anemia following partially mismatched related donor bone marrow transplant. Bone Marrow Transplant. 1997;19:813–7. doi: 10.1038/sj.bmt.1700746. [DOI] [PubMed] [Google Scholar]
- 32.Tary-Lehmann M, Lehmann PV, Schols D, Roncarolo MG, Saxon A. Anti-SCID mouse reactivity shapes the human CD4+ T cell repertoire in hu-PBL-SCID chimeras. J Exp Med. 1994;180:1817–27. doi: 10.1084/jem.180.5.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill GR, Teshima T, Gerbitz A, et al. Differential roles of IL-1 and TNF-alpha on graft-versus-host disease and graft versus leukemia. J Clin Invest. 1999;104:459–67. doi: 10.1172/JCI6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90:3204–13. [PubMed] [Google Scholar]
- 35.Xun CQ, Thompson JS, Jennings CD, Brown SA, Widmer MB. Effect of total body irradiation, busulfan-cyclophosphamide, or cyclophosphamide conditioning on inflammatory cytokine release and development of acute and chronic graft-versus-host disease in H-2-incompatible transplanted SCID mice. Blood. 1994;83:2360–7. [PubMed] [Google Scholar]
- 36.Sun Y, Tawara I, Toubai T, Reddy P. Pathophysiology of acute graft-versus-host disease: recent advances. Transplant Res. 2007;150:197–214. doi: 10.1016/j.trsl.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herve P, Flesch M, Tiberghien P, et al. Phase I–II trial of a monoclonal anti-tumor necrosis factor alpha antibody for the treatment of refractory severe acute graft-versus-host disease. Blood. 1992;79:3362–8. [PubMed] [Google Scholar]
- 38.Uberti JP, Ayash L, Ratanatharathorn V, et al. Pilot trial on the use of etanercept and methylprednisolone as primary treatment for acute graft-versus-host disease. Biol Blood Marrow Transplant. 2005;11:680–7. doi: 10.1016/j.bbmt.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Filipovich AH, Weisdorf D, Pavletic S, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005;11:945–56. doi: 10.1016/j.bbmt.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 40.Hoffmann-Fezer G, Gall C, Zengerle U, Kranz B, Thierfelder S. Immunohistology and immunocytology of human T-cell chimerism and graft-versus-host disease in SCID mice. Blood. 1993;81:3440–8. [PubMed] [Google Scholar]
- 41.Teshima T, Ordemann R, Reddy P, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med. 2002;8:575–81. doi: 10.1038/nm0602-575. [DOI] [PubMed] [Google Scholar]
- 42.Hattori K, Hirano T, Miyajima H, et al. Differential effects of anti-Fas ligand and anti-tumor necrosis factor alpha antibodies on acute graft-versus-host disease pathologies. Blood. 1998;91:4051–5. [PubMed] [Google Scholar]
- 43.Cooke KR, Hill GR, Crawford JM, et al. Tumor necrosis factor-alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102:1882–91. doi: 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brown GR, Lee EL, Thiele DL. TNF enhances CD4+ T cell alloproliferation, IFN-gamma responses, and intestinal graft-versus-host disease by IL-12-independent mechanisms. J Immunol. 2003;170:5082–8. doi: 10.4049/jimmunol.170.10.5082. [DOI] [PubMed] [Google Scholar]
- 45.Gilliam AC, Whitaker-Menezes D, Korngold R, Murphy GF. Apoptosis is the predominant form of epithelial target cell injury in acute experimental graft-versus-host disease. J Invest Dermatol. 1996;107:377–83. doi: 10.1111/1523-1747.ep12363361. [DOI] [PubMed] [Google Scholar]
- 46.Piguet PF, Grau GE, Allet B, Vassalli P. Tumor necrosis factor/cachectin is an effector of skin and gut lesions of the acute phase of graft-vs.-host disease. J Exp Med. 1987;166:1280–9. doi: 10.1084/jem.166.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Remberger M, Ringden O, Markling L. TNF alpha levels are increased during bone marrow transplantation conditioning in patients who develop acute GVHD. Bone Marrow Transplant. 1995;15:99–104. [PubMed] [Google Scholar]
- 48.Holler E, Kolb HJ, Moller A, et al. Increased serum levels of tumor necrosis factor alpha precede major complications of bone marrow transplantation. Blood. 1990;75:1011–6. [PubMed] [Google Scholar]
- 49.Holler E, Kolb HJ, Hintermeier-Knabe R, et al. Role of tumor necrosis factor alpha in acute graft-versus-host disease and complications following allogeneic bone marrow transplantation. Transplant Proc. 1993;25:1234–6. [PubMed] [Google Scholar]
- 50.Levine JE, Paczesny S, Mineishi S, et al. Etanercept plus methylprednisolone as initial therapy for acute graft-versus-host disease. Blood. 2008;111:2470–5. doi: 10.1182/blood-2007-09-112987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferrara JL. Pathogenesis of acute graft-versus-host disease: cytokines and cellular effectors. J Hematother Stem Cell Res. 2000;9:299–306. doi: 10.1089/15258160050079407. [DOI] [PubMed] [Google Scholar]
- 52.Nowlan ML, Drewe E, Bulsara H, et al. Systemic cytokine levels and the effects of etanercept in TNF receptor-associated periodic syndrome (TRAPS) involving a C33Y mutation in TNFRSF1A. Rheumatology (Oxf) 2006;45:31–7. doi: 10.1093/rheumatology/kei090. [DOI] [PubMed] [Google Scholar]
- 53.Tsimberidou AM, Waddelow T, Kantarjian HM, Albitar M, Giles FJ. Pilot study of recombinant human soluble tumor necrosis factor (TNF) receptor (p75) fusion protein (TNFR : Fc; Enbrel) in patients with refractory multiple myeloma: increase in plasma TNF alpha levels during treatment. Leuk Res. 2003;27:375–80. doi: 10.1016/s0145-2126(02)00082-6. [DOI] [PubMed] [Google Scholar]
- 54.Eason JD, Pascual M, Wee S, et al. Evaluation of recombinant human soluble dimeric tumor necrosis factor receptor for prevention of OKT3-associated acute clinical syndrome. Transplantation. 1996;61:224–8. doi: 10.1097/00007890-199601270-00011. [DOI] [PubMed] [Google Scholar]
- 55.Mohler KM, Torrance DS, Smith CA, et al. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol. 1993;151:1548–61. [PubMed] [Google Scholar]
- 56.Scallon B, Cai A, Solowski N, et al. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–26. doi: 10.1124/jpet.301.2.418. [DOI] [PubMed] [Google Scholar]
- 57.Evans TJ, Moyes D, Carpenter A, et al. Protective effect of 55- but not 75-kD soluble tumor necrosis factor receptor-immunoglobulin G fusion proteins in an animal model of Gram-negative sepsis. J Exp Med. 1994;180:2173–9. doi: 10.1084/jem.180.6.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]