

Abstract
Carcinoembryonic antigen (CEA) is over-expressed on various human cancer cells and has been the target of immunotherapies using dendritic cells (DCs) pulsed with CEA-specific RNA or peptides, or transduced by CEA-expressing adenovirus or vaccinia virus. Because activated DCs do not phagocytose soluble protein antigens efficiently and pure immature DCs are not obtained easily ex vivo, an efficacious whole CEA protein-loaded DC vaccine has not been reported. To improve the antigen delivery into DCs, we utilized CEA conjugated to a protein-transduction domain, human immunodeficiency virus transactivating Tat. Furthermore, we purified the truncated non-glycosylated CEA from Escherichia coli to overcome the safety concerns and immunosuppressive functions associated with the native CEA protein. Using confocal microscopy and fluorescence activating cell sorter analysis, we demonstrated that the Tat-CEA protein entered the cytoplasm of DCs efficiently within 10 min of co-culture, compared with the negligible amount of CEA into DCs 30 min later. CEA-specific T cell proliferation and cytotoxic T cell responses were enhanced significantly in mice immunized with Tat-CEA-pulsed DCs [DC (Tat-CEA)] compared with those immunized with CEA-pulsed DCs [DC (CEA)]. T helper type 1 responses were more prominent in the DC (Tat-CEA) immunized mice whose splenocytes secreted more interferon-γ and less interleukin-4 than those from DC (CEA) immunized mice. In vivo, the DC (Tat-CEA) vaccine delayed tumour growth significantly and prolonged survival of tumour-bearing mice. These results suggest that protective epitopes are well preserved on bacteria-derived recombinant Tat-CEA. This strategy may provide a basic platform for DC-based anti-CEA vaccines that could be utilized in combination with advanced immune-enhancing therapeutics.
Keywords: cancer immunotherapy, CEA, DC, HIV Tat, PTD
Introduction
Carcinoembryonic antigen (CEA) is a tumour-associated antigen (TAA) discovered in 1965 [1] and is a well-known oncofetal protein detected in multiple types of cancer [2]. CEA is over-expressed in various human cancers, including 90% of gastrointestinal, colorectal and pancreatic cancers, 70% of non-small cell lung cancer and 50% of breast cancers [3]. Because of its abundant expression on tumour cells and secretion into the serum, CEA has been used widely in clinics as a biomarker to decide tumour stage, presence of residual tumours and/or recurrences [4–6].
The CEA has also been used in active vaccination strategies against diverse CEA-expressing cancers because of its exclusive expression on oncofetal tissues and its role in tumorigenesis [2,7]. Over the past few years, CEA-specific immune responses have been induced in vitro and in vivo that could suppress growth of CEA-positive cancers in mouse studies [8–11] as well as in human trials [12,13]. Among the immunotherapies using CEA, dendritic cell (DC)-based vaccinations showed promising results in mice but were not satisfactory in clinical trials [2]. To date, the best clinical report by Fong et al. showed that only 16·7% (two of 12) of vaccinated patients experienced tumour regression and 25%(three of 12) had a minor response or stable disease [14].
The DCs play a pivotal role in the initiation and regulation of tumour-specific immune responses and have been utilized as potent therapeutic vaccines against human cancers [15,16]. Although early results with ex vivo-generated DCs pulsed with TAA provided a proof-of-principle that therapeutic immunity can be elicited in some cancer patients, efficacy was not sufficiently satisfactory in clinical trials [15,17]. Currently, most efforts are focused upon generating large numbers of effector T cells in vivo and in overcoming the immunosuppressive tumour environment in DC vaccine trials [15,16]. Diverse strategies have been developed to improve the efficacy of DC vaccination such as generation of specific DC subsets, selection of TAA, efficient antigen loading, efficacious delivery of DCs to regional lymph nodes and increasing survival and activation of DCs [15,16,18].
The first goal for improvement of DC vaccinations against cancer is loading sufficient amounts of TAA into DCs. Because activated DCs cannot phagocytose external antigens efficiently, the antigen-loading strategies should be designed carefully [15,18]. Various forms and methods such as recombinant proteins, peptides, viral vectors, RNA, immune complexes and killed tumour cell lysate have been used to load TAA into DCs [15]. Among the antigen-loading strategies, utilization of protein transduction domain (PTD) has been given much attention, as it is safer yet is equally as effective in loading TAA as viruses [19,20]. It was hypothesized that the intracellular delivery of TAA into mature DCs by a PTD, [such as human immunodeficiency virus (HIV) Tat peptide] may allow DCs to process and present the internalized antigens to T cells by major histocompatibility complex (MHC) class I molecules efficiently [21]. Many investigators have now begun to explore the utility of HIV Tat as a PTD to increase antigen loading into DCs [20–23]. It was demonstrated that DCs pulsed with Tat-TAA could induce antigen-specific CD4 T cells as well as cytotoxic lymphocytes (CTL) [22,23].
The CEA is a glycoprotein known to be involved in carcinogenesis [2]. Because native CEA proteins produced in mammalian cells may be potentially harmful, utilization of recombinant proteins purified from Escherichia coli could be a rational approach to develop a safe and cost-effective DC-based vaccine. It is unknown if the post-translational modification in eukaryotic cells may be necessary to incur protective immunity. However, it has been reported that the glycans of CEA could impair the function and differentiation of DCs through interaction with DC-SIGN expressed on DCs [24,25], suggesting that bacteria-derived non-glycosylated CEA might be a better immunogen if the protective epitopes could be presented effectively on MHC class I and MHC class II molecules.
Woo et al. have reported that recombinant Tat-CEA fusion protein could elicit CEA-specific immunity when the tumour cell lysate expressing Tat-CEA was injected directly into the mice, together with cytosine–guanine dinucleotide (CpG)-oligonucleotides as an adjuvant [26]. In addition, they demonstrated that a marginal anti-tumour immunity could be induced when mice were immunized with DCs electroporated with mRNAs encoding Tat-CEA [8]. However, the survival of tumour-bearing mice was not increased significantly by immunization when compared with control groups. Furthermore, the potential harmful effects of the recombinant CEA proteins expressed in mammalian cells could not be ruled out.
Thus far, only one study using CEA fusion protein derived from E. coli has been reported in which DCs loaded with recombinant protein, when fused with heat shock protein, induced immunity against CEA-positive tumours [9]. However, a strategy is needed to enhance the antigen-loading efficacy of DCs, as mature DCs rarely phagocytose protein antigens. Therefore, we investigated the efficacy and feasibility of a recombinant CEA-PTD fusion protein produced in a prokaryotic system. In the present study, we showed that DCs pulsed with bacterial recombinant Tat-CEA could elicit CEA-specific immune responses and anti-tumour immunity in a mouse model.
Materials and methods
Mice and cell lines
C57BL/10NAGCSnAi-[KO] Rag2 (H-2b) mice were obtained from Taconic Farms, Inc. (Hudson, NY, USA). C57BL/6 (H-2b) mice were purchased from the Center for Animal Resource Development, Seoul National University College of Medicine (SNU, Seoul, Korea). The animal experiments were performed after approval from the SNU animal welfare committee. The MC38 and MC38-expressing human CEA (MC38-CEA2) (H-2b) mouse adenocarcinoma cell lines were kindly provided by Dr J. Schlom (NIH, Bethesda, MD, USA) [27]. Both MC38-CEA2 and MC38 cell lines were cultured and maintained in complete Dulbecco's modified Eagle medium (Welgene Inc., Daegu, Korea) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Grand Island, NY, USA), 50 µg/ml gentamicin (Invitrogen), 100 units/ml penicillin–streptomycin (Invitrogen) and 2 mM L-glutamine (Welgene Inc., Daegu, Korea).
Production of Tat-CEA fusion proteins
A human CEA cDNA was purchased from Origene Technologies, Inc. (Rockville, MD, USA). The N-terminal DNA fragment of CEA corresponding to nucleotide sequences from 103 to 996 (GenBank Accession no. M17303) was amplified using polymerase chain reaction (PCR) with a sense primer 5′-CCGAATTCAAGCTCACTATTGAATCCAC-3′ (CEA103–122, EcoRI site underlined) and an anti-sense primer 5′-CGGCTCGAGGGAGTTGTTGCTGGTG-3′ (CEA981–996, XhoI site underlined). The resulting PCR products were digested with EcoRI and XhoI (Takara, Shiga, Japan) and then gel purified. Purified PCR products were cloned into the EcoRI/XhoI-digested pET-23a vector (Novagen, Darmstadt, Germany) or pET-23a vector containing 11 amino acids of Tat PTD (YGRKKRRQRRR) at the NheI/EcoRI sites to generate pET-CEA and pET-Tat-CEA respectively. E. coli DH5α (Real Biotech Corporation, Taipei, Taiwan) were transformed with the constructs by standard heat-shock procedures and selected on Luria broth (LB) agar plate containing 100 µg/ml of ampicillin (Sigma, St Louis, MO, USA).
Purification of CEA fusion proteins
For the expression and purification of CEA fusion proteins, E. coli BL21 (DE3) strains (Novagen) were transformed with either pET-CEA or pET-Tat-CEA. Bacteria were grown in LB containing ampicillin (100 µg/ml). Protein expression was induced by adding 0·4 mM isopropyl β-D-thiogalactoside (IPTG; Duchefa, Zwijndrecht, the Netherlands) for 4 h at 37°C. Bacterial cells harvested by centrifugation at 1000 g for 10 min were resuspended in binding buffer (300 mM NaCl, 50 mM sodium phosphate buffer, 10 mM imidazole) containing 1·5% Triton X-100 (Sigma) and then disrupted by sonication on ice for 5 min. Sonicated lysates were centrifuged at 1600 g for 20 min at 4°C, and then the pellets containing CEA or Tat-CEA protein were resuspended in binding buffer containing 6 M urea (Sigma), sonicated and centrifuged at 1600 g for 30 min at 4°C. The supernatant was applied to Ni-NTA His-binding resin (Novagen), which was pre-equilibrated with the binding buffer containing 6 M urea. His-tagged proteins bound to Ni-NTA resin were eluted with elution buffer (300 mM NaCl, 50 mM sodium phosphate buffer, 250 mM imidazole) containing 6 M urea. Purified proteins were dialysed serially against elution buffer to remove imidazole, urea and residual salts. Finally, the identity and purity of purified proteins were assessed by Western blot analysis and Coomassie blue staining respectively.
Western blot assay
Whole bacterial cell lysates and purified proteins were separated in 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and then transferred onto polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA, USA). Membranes were blocked with 5% non-fat milk (BD, San Jose, CA, USA) in Tris-buffered saline Tween-20 (TBST) [20 mM Tris-Cl (pH 7·6), 100 mM NaCl and 0·05% Tween-20] and then incubated with mouse anti-human CD66 (CEA) monoclonal antibody (clone 26/5/1; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or mouse anti-His-Tag monoclonal antibody (clone 27E8; Cell Signaling, Danvers, MA, USA) at 4°C overnight, followed by washing three times with TBST. The membranes were incubated with horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG) antibody (Santa Cruz Biotechnology) at room temperature (RT) for 1 h. After washing, immunoreactive bands were detected using enhanced chemiluminescence solution (Amersham, Piscataway, NJ, USA) and luminescent image analyser-4000 (Fujifilm, Miami Beach, FL, USA).
In vitro generation of bone marrow-derived DCs
The DCs were generated from bone marrow (BM) of 6- to 10-week-old Rag2 knock-out mice. BM cells were flushed out of the femurs and tibias with serum-free CellGro medium (CellGenix, Freiburg, Germany). The single-cell suspension was then filtered through a nylon cell strainer (70-mm Nylon mesh; BD), washed twice with complete CellGro medium [CellGro supplemented with recombinant mouse granulocyte–macrophage colony-stimulating factor (GM-CSF) (0·75 ng/ml) and mouse interleukin (IL)-4 (1·5 ng/ml; PeproTech, Rocky Hill, NJ, USA), penicillin (100 units/ml), streptomycin (100 µg/ml), gentamicin (50 µg/ml), L-glutamine (2 mM) and β-mercaptoethanol (ME) (50 nM; Gibco Invitrogen, Grand Island, NY, USA)] and seeded at a concentration of 1 × 106 cells per well in a 24-well plate in a final volume of 2 ml of complete CellGro medium. Half the medium was replaced every other day with an equal volume of complete CellGro medium for 6 days. For determination of the effect of Tat-CEA or CEA on the ability of DCs to express co-stimulatory molecules and I-Ab, DCs were loaded with 50 µg/ml of recombinant proteins for 16 h. Cell surface molecules were analysed by flow cytometry as described below.
Flow cytometric analysis
At day 6 of DCs culture, 1 ml of media was removed, and then purified CEA or Tat-CEA proteins were added at a concentration of 50 µg/ml and incubated for 10 min or 30 min. DCs were washed with ice-cold fluorescence activated cell sorter (FACS) buffer [1× phosphate-buffered saline (PBS) (pH 7·2) (Welgene) containing 1% bovine serum albumin (AMResco, Solon, OH, USA) and 1 mM ethylenediamine tetraacetic acid (Sigma)] and then blocked on ice for 30 min with ultra-block solution [a mixture of 10% rat sera, 10% hamster sera, 10% mouse sera and 10 µg/ml 2.4G2 monoclonal antibody (Gibco Invitrogen)]. DCs were stained with fluorescein isothiocyanate (FITC)-conjugated anti-I-antibody (clone AF6-120), phycoerythrin (PE)-conjugated anti-CD40 (clone 3/23; BD Pharmingen, San Diego, CA, USA), allophycocyanin (APC)-conjugated anti-CD80 (clone 16-10A1; eBioscience, San Diego, CA, USA), APC/Cy7-conjugated anti-CD11c (clone N418), PE/Cy7-conjugated anti-CD86 (clone GL-1; BioLegend, San Diego, CA, USA) or anti-CD11c-PE (clone HL3; BioLegend, San Diego, CA, USA) on ice for 30 min. Dead cells were excluded by staining with 7-amino-actinomycin D (7-AAD; BD Pharmingen). To detect intracellular CEA antigens, DCs were fixed and permeabilized with fixation/permeabilization solution (BD Biosciences) on ice for 30 min, washed twice with 1 × Perm/Wash buffer (BD Biosciences) and then incubated with anti-human CEA-FITC (clone B1.1/CD66; BD Pharmingen, San Diego, CA, USA). Cells were analysed by FACS Canto II flow cytometry equipped with FACS Diva software (BD Biosciences, Mountain View, CA, USA).
Confocal laser microscopy
The DCs pulsed with CEA or Tat-CEA protein as above were washed twice in PBS (pH 7·2), transferred onto polylysine-coated microscope slides (Menzel-Gläser, Braunschweig, Germany) and fixed in 4% paraformaldehyde solution at RT for 10 min. Fixed DCs were then permeabilized with permeabilization solution (eBiosciences) at RT for 20 min and non-specific bindings were blocked with 10% goat serum (Invitrogen) at RT for 1 h and stained with anti-human CEA-FITC (clone B1.1/CD66; BD PharMingen) and anti-mouse CD11c-Alexa Fluor 647 (clone N418; eBioscience) at RT for 2 h. The nucleus was stained with 1 mM Lo-Pro3 (Molecular Probe, Eugene, OR, USA). Cells were analysed with a FluoView1000 laser confocal microscope (Olympus, Nagano, Japan).
Lymphocyte proliferation assay
B6 mice at 6–8 weeks old were immunized at the tail base with DCs (1 × 106) transduced with CEA or Tat-CEA. At 10 days post-immunization, lymph node (inguinal and peri-aortic) cells from immunized B6 mice were cultured for 3 days in Iscove's modified Eagle medium (IMDM) (3·5 × 105 per well in 96-well plates) supplemented with 10% heat-inactivated FBS, 50 nM β-ME, 50 µg/ml gentamicin (Invitrogen), 100 units/ml penicillin–streptomycin, 2 mM L-glutamine (Welgene) and various concentrations of purified CEA. Cells were labelled with [3H]-methylthymidine (1 µCi) (Amersham) for an additional 18 h, and then cells were harvested. Incorporation of [3H]-methylthymidine was quantified by using Micro Beta TriLux (PerkinElmer, Waltham, MA, USA).
Cytotoxicity assay
B6 mice at 6–8 weeks old were immunized at the tail base twice at weekly intervals with DCs (1 × 106) transduced with CEA or Tat-CEA. At 7 days after the final immunization, cytotoxic responses of effector cells were assessed by performing the p-JAM (‘just another method’) test as described previously [28]. Briefly, splenocytes harvested from mice were restimulated with 30 µg/ml of CEA proteins for 3 days in complete IMDM and used as effector cells. The effector cells were washed three times with PBS to remove residual proteins completely and then cultured with MC38-CEA2 or MC38 target cells (3 × 105 cells/well) in 96-well plates at various effector/target ratios. After 18 h at 37°C, effector cells were washed three times with PBS and target cells remaining on the bottom of the culture plates were labelled with [3H]-methylthymidine (5 µCi; Amersham) for 3 h at 37°C. Incorporation of [3H]-methylthymidine was quantified by using Micro Beta TriLux (Wallac) after harvesting the cells. Percentage of specific lysis = counts per million (CPM) of target alone − [CPM of (target + killers) − (killer alone)]/CPM of target alone × 100% [28].
Evaluation of cytokine secretion
At 7 days after the final immunization, splenocytes were harvested from B6 mice immunized with DCs (1 × 106) at the tail base twice at weekly intervals and incubated for 3 days in the absence or presence of 30 µg/ml CEA proteins as described above. Secreted cytokines in the culture media were measured by the cytometric bead array kit (BD Pharmingen) according to the manufacturer's instructions using FACS Canto II flow cytometry (BD).
Tumour growth
To establish a CEA-positive tumour-bearing mouse model, 6-week-old B6 mice were injected in the right flank with MC38-CEA2 cells (1 × 106) each. At 7 days after tumour cell injection, mice were immunized at the tail base with DCs pulsed with CEA or Tat-CEA proteins (1 × 106 cells/mouse in 100 µl PBS) at weekly intervals for 4 weeks. Control mice were injected with PBS only. Five mice were used for each group. Tumour volume (mm3) = (A × B2)/2, where A is the long diameter and B is the short diameter [29–31]. Percentage survival of mice in each group was also measured during 30 days after tumour inoculation.
Statistical analysis
Statistical analyses were performed by a Student's t-test or Kaplan–Meier test (log-rank test) for survival data using SigmaPlot software (Jandel, San Rafael, CA, USA). The data are presented as mean ± standard error and considered statistically significant at P < 0·05.
Results
Expression and purification of CEA fusion proteins
The 894 base pair N-terminal region (103–996) of the human cea gene was PCR amplified using full-length human CEA cDNA as templates. PCR products were cloned in-frame to generate pET-CEA and pET-Tat-CEA (Fig. 1a). The expression of recombinant proteins in E. coli BL21 (DE3) transformed with pET-CEA or pET-Tat-CEA was induced by IPTG and verified by Western blot analysis. As shown in Fig. 1b (upper panel), both CEA and Tat-CEA were detected as 33 kDa and 36 kDa proteins, respectively, by anti-human CEA antibody. The levels of CEA expression were highly increased upon IPTG induction, indicating that human CEA can be expressed readily in E. coli. As CEA was cloned in-frame into the upstream of 6× His-tag sequences of pET-23a vector, expression of His-tagged CEA or Tat-CEA fusion proteins was then confirmed by Western blot analysis using anti-His antibody. As shown in the lower panel of Fig. 1b, both CEA and Tat-CEA were expressed in E. coli as His-fusion proteins. We purified recombinant fusion proteins from bacterial pellets under denaturing conditions, followed by a Ni-NTA affinity chromatography. Coomassie blue staining of the purified proteins revealed that the eluted proteins were highly pure (Fig. 1c, left panel). From a 1-l culture, we obtained 1·7 ± 0·3 mg or 0·5 ± 0·06 mg of purified recombinant CEA or Tat-CEA respectively. The purified proteins were identified as His-tagged CEA proteins by Western blot analysis using anti-His antibody (Fig. 1c, right panel).
Fig. 1.

Expression and purification of Tat-carcinoembryonic antigen (CEA) fusion proteins. (a) Schematic diagram of pET-Tat-CEA vector, which carries Tat-CEA fused to the 6× His tag. (b) Expression of Tat-CEA fusion proteins. Expression of CEA proteins were assessed by Western blot analysis with anti-human CEA antibody (upper panel) or anti-His antibody (lower panel). Transformed Escherichia coli BL21 (DE3) was cultured in the presence (+) or absence (−) of isopropyl β-D-thiogalactoside (IPTG). (c) Purification of Tat-CEA fusion proteins. Recombinant CEA proteins were purified from E. coli lysate using Ni-NTA resin column chromatography and analysed by Coomassie blue stain (left panel) and Western blot analysis with anti-His antibody (right panel).
Protein transduction of DCs with CEA fusion proteins
To investigate the transduction efficiency of purified CEA fusion proteins into DCs, either CEA or Tat-CEA fusion proteins were incubated with DCs for 10 min or 30 min, followed by intracellular staining of CEA for FACS analysis. As shown in Fig. 2a, percentage (%) CEApos DCs (b + c in Fig. 2a) was enhanced markedly when incubated with Tat-CEA (94·6%) compared with those incubated with CEA (15·0%) for the first 10 min after transduction. Longer incubation (30 min) resulted in only a marginal increase, representing 17·9% and 97·0% CEApos DCs transduced with CEA or Tat-CEA respectively. The mean fluorescence intensity (MFI) of DC (Tat-CEA, 31·6) was significantly higher than that (20·7) of DC (CEA). Interestingly, a portion (region c in Fig. 2a) of DC (CEA) showed higher MFI (419·8) than that (178·0) of DC (Tat-CEA). Again, confocal microscopic analysis showed efficient delivery of antigen when CEA was fused with Tat PTD (Fig. 2b). As shown in Fig. 2b, the level of intracellular staining of CEA was greater in DCs transduced with Tat-CEA than that of CEA. These results suggest that Tat PTD-mediated delivery of CEA to DCs is fast and efficient.
Fig. 2.

Transduction of carcinoembryonic antigen (CEA) into dendritic cells (DCs). (a) Flow cytometric analysis of CEA in DCs transduced with CEA or Tat-CEA. DCs incubated with either purified CEA or Tat-CEA protein for 10 min (left panel) or 30 min (right panel) were stained with anti-mouse CD11c-phycoerythrin (PE), followed by intracellular staining of CEA with anti-human CEA-fluorescein isothiocyanate (FITC) for fluorescence activated cell sorter (FACS) analysis. CD11cpos cells were gated for the histograms. Percentage (%) of CD11cposCEApos cells were analysed using gates (b + c) indicated. Filled histograms indicate isotype controls. (b) Confocal microscopic analysis of DCs transduced with recombinant CEA or Tat-CEA. DCs incubated with recombinant antigens were stained with anti-human CEA-FITC (green), anti-CD11c-Alexa Fluor 647 (blue) and Lo-Pro3 (red, to stain nucleus). Autofluorescence of cells for FITC channel (anti-CEA) was confirmed by staining DC (medium) with anti-CEA-FITC (boxed figure in CEA-30 min-merge panel).
To determine whether Tat-mediated protein transduction and CEA itself could affect DC maturation, we examined the expression level of surface activation markers of DC including I-Ab, CD40, CD80 and CD86 by FACS analysis (Fig. 3). Without LPS, maturation of DCs was observed consistently in the presence of the recombinant proteins, suggesting that transduction of DCs with CEA or Tat-CEA did not affect DC maturation. DC (Tat-CEA) showed a slightly higher level of co-stimulatory molecule expression compared with DC (CEA) or DC (LPS). We attempted to remove the LPS from recombinant proteins by using a Polymixin B column. However, the protein preparations still contained more than 0·5 EU of residual LPS, which may be sufficient to activate DCs. For this reason, without additional LPS, the DCs showed increased expression of co-stimulatory molecules and I-Ab upon incubation with recombinant protein for 16 h. However, after washing DCs with PBS for injection, residual LPS was less than 0·1 EU.
Fig. 3.
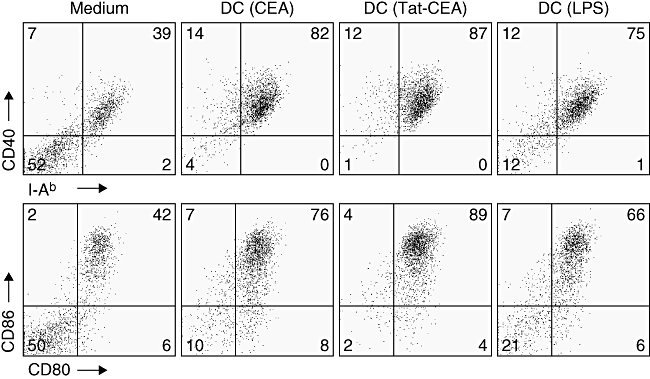
Maturation of dendritic cells (DCs) transduced with recombinant carcinoembryonic antigen (CEA) or Tat-CEA proteins. Expression of co-stimulatory molecules and I-Ab on DCs were analysed after 16 h of incubation with recombinant proteins or lipopolysaccharide (LPS). DCs were stained with antibodies against with I-Ab, CD40, CD86, CD80 and CD11c. Live DCs (7AADneg/CD11cpos) were analysed for the surface molecule expression using a FACS Canto II flow cytometer.
Priming lymphocytes in vivo
To assess whether CEA or Tat-CEA pulsed DCs can prime lymphocytes specific to CEA, we immunized B6 mice with DC (CEA) or DC (Tat-CEA) (1 × 106 cells) at the tail base. Ten days later, lymphocytes were harvested from inguinal and peri-aortic lymph nodes and restimulated with various amounts of CEA in vitro. As shown in Fig. 4, lymphocytes from the mice immunized with DC (Tat-CEA) proliferated in response to CEA in a dose-dependent manner. The proliferation was significantly higher than those obtained from mice immunized with DC (CEA) (P < 0·05). Significant CEA-specific lymphocytes proliferation was not detected from mice immunized with DC alone or PBS. These results suggest that DC (Tat-CEA) prime CEA-specific lymphocytes more efficiently than DC (CEA) in vivo.
Fig. 4.
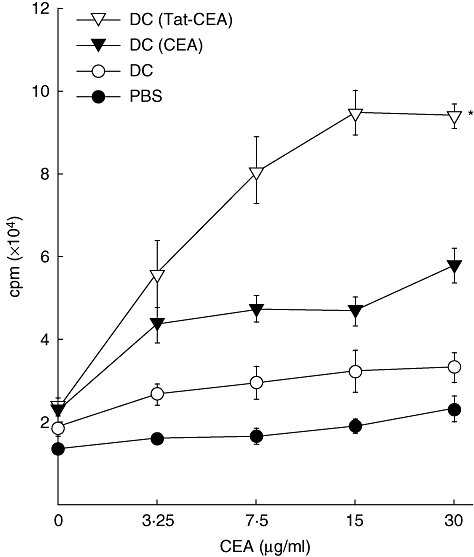
Proliferation of lymphocytes from mice immunized with dendritic cells (DCs). DCs (1 × 106 cells) pulsed with carcinoembryonic antigen (CEA) or Tat-CEA proteins were injected into the tail bases of B6 mice (three mice per group) at day 0. Lymphocytes were harvested from inguinal and periaortic lymph nodes 10 days later and then co-cultured with indicated amounts of CEA proteins for 3 days. The cells were then labelled with [3H]-methylthymidine (1 µCi) for an additional 18 h. Proliferation of lymphocytes was determined by measurement of [3H]-methylthymidine uptake. Representative data from three independent experiments are presented as mean ± standard error. *Statistically significant at P < 0·05 using a Student's t-test compared with other groups.
Induction of CEA-specific cytotoxic splenocytes in vivo
To assess CEA-specific cytotoxic effector cells by immunization of CEA-pulsed DCs, the p-JAM test was performed [28]. As shown in Fig. 5a, splenocytes from the mice immunized with DC (Tat-CEA) lysed MC38-CEA2 target cells more efficiently than those obtained from the mice immunized with DC (CEA). Immunization of mice with DCs alone, however, induced a CEA-specific CTL response as great as the PBS control group against MC38-CEA target cells (Fig. 5a). Cytotoxicity of splenocytes against CEA-negative MC38 target cells was not significant in all four groups (Fig. 5b). These results suggest that immunization of mice with DC (Tat-CEA) could induce a potent CTL response specific to CEApos tumours in vivo.
Fig. 5.
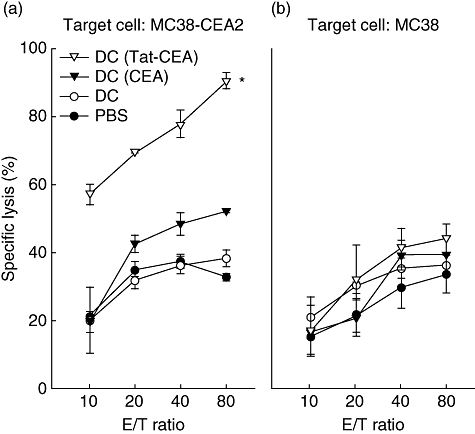
Carcinoembryonic antigen (CEA)-specific cytotoxic lymphocytes from mice immunized with dendritic cells (DCs). DCs (1 × 106 cells) pulsed with either CEA or Tat-CEA proteins were injected twice at weekly intervals to B6 mice (three mice per group). At 7 days post-second immunization, splenocytes were harvested and restimulated with CEA proteins for 3 days to generate effector cells. Effector cells were incubated with target cells (either MC38 or MC38-CEA2 cells) for 18 h and washed away. The remaining target cells on the culture plate were labelled with [3H]-methylthymidine (5 µCi) for 3 h. Specific lysis (%) was determined by the measurement of [3H]-methylthymidine uptake. Representative data from three independent experiments are presented as mean ± standard error of triplicate cultures. *Statistically significant at P < 0·05 using at a Student's t-test compared with other groups.
Cytokine production of splenocytes
We also measured cytokine production of the splenocytes from immunized mice after in vitro restimulation with CEA, as CTL is known to secrete T helper type 1 (Th1) cytokine interferon (IFN)-γ in an antigen-specific manner. As shown in Fig. 6a, IFN-γ production of splenocytes from the mice immunized by DC (Tat-CEA) was slightly higher than that from the mice immunized with DC (CEA), with marginal significance. Interestingly, the splenocytes from mice immunized with DC (CEA) secreted significantly higher levels of IL-4, a representative Th2 cytokine, when compared with that of splenocytes from mice immunized with DC (Tat-CEA) (Fig. 6b, P < 0·05), indicating that the immunity against CEA in mice immunized with DC (Tat-CEA) was more biased to Th1 responses.
Fig. 6.
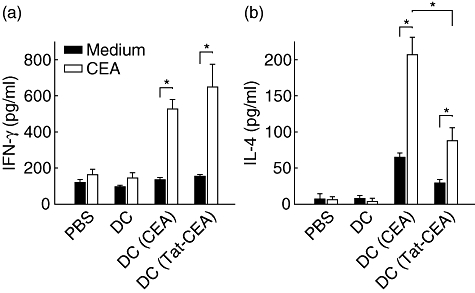
Interferon (IFN)-γ and interleukin (IL)-4 secretion of splenocytes in response to carcinoembryonic antigen (CEA) antigen. The mice were immunized and splenocytes were harvested as in Fig. 5. Splenocytes were cultured for 3 days in the presence of CEA (empty bar) or medium alone (filled bar). Cytokines in the culture supernatant were measured by cytometric bead array. Representative data from three independent experiments are presented as mean ± standard error of triplicate cultures from three mice in each group. *Statistically significant at P < 0·05 using a Student's t-test.
Tumour growth and survival
Tumour growth was suppressed significantly more in the mice immunized with DC (Tat-CEA) than those in the mice immunized with DC (CEA), DCs alone or PBS (Fig. 7a, P < 0·05). Furthermore, 80% of mice immunized with DC (Tat-CEA) survived for longer than 30 days after tumour inoculation. In contrast, all mice in control groups died of tumours before 30 days (Fig. 7b, P < 0·05). These data suggest strongly that DCs (Tat-CEA) induced more potent protective immune responses against CEA-positive tumours in vivo than DC (CEA).
Fig. 7.

Tumour growth and survival of mice immunized with dendritic cells (DCs). B6 mice (five mice per group) were inoculated on the right flank with MC38-carcinoembryonic antigen (CEA)-2 tumour cells (1 × 106) at 7 days prior to immunization. The mice were immunized with DC (Tat-CEA), DC (CEA), DCs or phosphate-buffered saline (PBS) at weekly intervals for 4 weeks at the tail base. Tumour volume (a) and survival (b) was monitored every other day. Data are presented as mean ± standard error of five mice. *Statistically significant at P < 0·05 using a Student's t-test (a) and Kaplan–Meier test (b) for survival compared with other groups.
Discussion
Therapeutic DC vaccines have been studied extensively during the last decade and their potential for cancer immunotherapy has been well demonstrated in murine models and in human clinical trials with more than 1000 patients [2,18]. For the treatment of CEA-positive tumours, various strategies have been developed to load CEA into the ex vivo-generated DCs such as MHC class I peptide [32], tumour cell fusion [33], viral transduction [34], RNA [8] and anti-idiotype antibody [35].
Among these techniques, DC vaccines loaded with MHC class I CEA peptide have shown more progress than others because of its simplicity, safety and applicability in clinical trials. However, the clinical responses thus far from these trials are not satisfactory [2]. Only marginal immunity against CEA has been reported in multiple studies without significant tumour regression [7,32,36,37]. In a recent clinical trial using DCs pulsed with multiple CEA peptides, only two of 11 colorectal cancer patients showed IFN-γ-secreting T cell responses after in vitro restimulation, while all the vaccinated patients showed progressive disease [32]. The poor outcome may be due to an unsuccessful induction of CTLs as a result of DCs loaded with a restricted repertoire of exogenous peptides [15].
A clinical trial utilizing DCs pulsed with CEApos tumour cell lysates also showed limited immunity, including IFN-γ- or IL-4-producing T cells in the vaccinated patients [38]. All patients showed progressive disease in the clinical settings. The failure may be due to the inefficient antigen presentation as ex vivo-generated DCs usually show activated phenotypes, and activated DCs rarely phagocytose exogenous antigens [39]. Therefore, the antigen-loading strategy is one of the most important items to consider when designing efficient DC vaccines because it determines the efficacy of antigen presentation and subsequent induction of effector T cell responses [18].
In addition, purified recombinant CEA proteins also have been administered into mice [26] and humans [40]. Recombinant CEA proteins produced in insect cells, which lack complex glycosylation, induced both humoral and T cell responses in colorectal carcinoma patients when the purified CEA was vaccinated directly [40]. Anti-CEA IgG titres and T cell response were augmented in all patients when vaccinated together with GM-CSF and the IgG titres were associated with increased survival. However, the vaccine did not elicit sufficient protective immunity against the CEApos cancer, and the anti-CEA immune responses were biased largely to Th2 responses in that study. Nevertheless, the cumulative experiences with the DC-based or non-DC-based CEA vaccines support the idea that CEA can serve as an effective target for immune therapy and such failures underscore the need to develop potent anti-CEA therapeutic vaccines [2,18].
In this study, we examined the efficacy of DC vaccine pulsed with E. coli-derived recombinant Tat-CEA in a murine tumour model. Our strategy may have several advantages over the former trials using ex vivo-manipulated DCs. First, by using the recombinant proteins spanning a long peptide sequence (34–332), we could expect that a broader range of CEA epitopes could be presented by the diverse sets of MHC haplotypes [7,18]. The N-terminal region of CEA used in this study covers several known epitopes that could bind efficiently to MHC class I molecules as CEA[961](H61LFGYSWYK69) for HLA-A3 [7]. Second, modification of CEA antigens with HIV Tat, one of the well-known PTDs, could enhance the immunogenicity of CEA drastically by facilitating the delivery of the TAA into the cytosol of DCs (Fig. 2). It may allow exogenous proteins to be channelled into the MHC class I presentation pathways and may be highly effective in inducing anti-CEA immunity, regardless of the activation status of the DCs [18,39]. Indeed, we observed significantly stronger CTL responses, preferential Th1 responses, retarded tumour growth and increased survival in the mice vaccinated with DC (Tat-CEA), compared with those from mice immunized using DC (CEA).
Previously, it has been reported that Tat protein itself could enhance the maturation of DCs, thereby increasing T cell responses [41]. Furthermore, CEA has been known to inhibit DC maturation[24,25]. In our study, expression of activation markers of DCs were not significantly different between DC (CEA) and DC (LPS) (Fig. 3), suggesting that CEA did not impair DC maturation in this setting. DC (Tat-CEA) showed slightly higher expression of co-stimulatory molecules and I-Ab when compared with DC (CEA). It suggests that the enhanced immunity against tumours may be partly because of the adjuvant effect of Tat in addition to the increased antigen delivery into DCs. In our purification process, we could not remove LPS completely from the recombinant proteins (> 0·5 EU). However, after washing the DCs with PBS three times immediately prior to injection into the mice, the levels of LPS were less than 0·1 EU.
Recombinant tumour antigens purified from prokaryotes have been used widely in DC-based vaccines and have induced tumour-specific CTL responses in a number of studies [42–47]. It seems likely that there is no significant difference in the immunogenicity of a purified TAA whether it is purified from prokaryotic or eukaryotic cells [46,47]. Rather, the efficient delivery of a TAA into antigen-presenting cells (APCs) through a functional modification is probably the more important determinant to inducing effective anti-tumour immunity as shown by our current results and others [45,48].
Recently, Woo et al. immunized mice with E. coli-derived recombinant Tat-CEA proteins with CpG oligodeoxynucleotides as an adjuvant, without DCs [26]. Administration of purified recombinant Tat-CEA could induce a certain level of protective anti-CEA immune responses, although they were Th2-associated antibody responses with weak IFN-γ production. Our current results using a DC-based strategy pulsed with Tat-CEA suggests that eliciting Th1-orientated immune responses are critical for protective anti-cancer immunity in vivo.
In summary, vaccination of DCs loaded with bacteria-derived Tat-CEA induced potent CTL-mediated anti-CEA immune responses that are sufficient to protect tumour-bearing mice. While other vaccine approaches may elicit antigen-specific responses in normal human volunteers, more efficient strategies are needed to overcome the immune-suppressive tumour microenvironment in the clinical setting where CEA-specific T cell clones are generally anergized/deleted and regulatory T cells are dominant.
Acknowledgments
This work was supported by a grant from the Ministry of Health and Welfare, Republic of Korea (grant A062260) and the Korea Science and Engineering Foundation through the Pioneer Program (M10711160001-08M1116-00110).
References
- 1.Gold P, Freedman SO. Demonstration of tumor-specific antigens in human colonic carcinomata by immunological tolerance and absorption techniques. J Exp Med. 1965;121:439–62. doi: 10.1084/jem.121.3.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang D, Rayani S, Marshall JL. Carcinoembryonic antigen as a vaccine target. Exp Rev Vaccines. 2008;7:987–93. doi: 10.1586/14760584.7.7.987. [DOI] [PubMed] [Google Scholar]
- 3.Thompson JA, Grunert F, Zimmermann W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal. 1991;5:344–66. doi: 10.1002/jcla.1860050510. [DOI] [PubMed] [Google Scholar]
- 4.Sikorska H, Shuster J, Gold P. Clinical applications of carcinoembryonic antigen. Cancer Detect Prev. 1988;12:321–55. [PubMed] [Google Scholar]
- 5.Shively JE, Beatty JD. CEA-related antigens: molecular biology and clinical significance. Crit Rev Oncol Hematol. 1985;2:355–99. doi: 10.1016/s1040-8428(85)80008-1. [DOI] [PubMed] [Google Scholar]
- 6.Horig H, Medina FA, Conkright WA, Kaufman HL. Strategies for cancer therapy using carcinoembryonic antigen vaccines. Exp Rev Mol Med. 2000;2:1–24. doi: 10.1017/S146239940000168X. [DOI] [PubMed] [Google Scholar]
- 7.Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol. 2002;20:2197–207. doi: 10.1200/JCO.2002.08.017. [DOI] [PubMed] [Google Scholar]
- 8.Kim SG, Park MY, Kim CH, et al. Modification of CEA with both CRT and TAT PTD induces potent anti-tumor immune responses in RNA-pulsed DC vaccination. Vaccine. 2008;26:6433–40. doi: 10.1016/j.vaccine.2008.08.072. [DOI] [PubMed] [Google Scholar]
- 9.Wu Y, Wan T, Zhou X, et al. Hsp70-like protein 1 fusion protein enhances induction of carcinoembryonic antigen-specific CD8+ CTL response by dendritic cell vaccine. Cancer Res. 2005;65:4947–54. doi: 10.1158/0008-5472.CAN-04-3912. [DOI] [PubMed] [Google Scholar]
- 10.Baral RN, Saha A, Chatterjee SK, et al. G oligonucleotides enhance the immune response of anti-idiotype vaccine that mimics carcinoembryonic antigen. Cancer Immunol Immunother. 2003;52:317–27. doi: 10.1007/s00262-002-0351-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horig H, Wainstein A, Long L, et al. A new mouse model for evaluating the immunotherapy of human colorectal cancer. Cancer Res. 2001;61:8520–6. [PubMed] [Google Scholar]
- 12.Foon KA, John WJ, Chakraborty M, et al. Clinical and immune responses in resected colon cancer patients treated with anti-idiotype monoclonal antibody vaccine that mimics the carcinoembryonic antigen. J Clin Oncol. 1999;17:2889–5. doi: 10.1200/JCO.1999.17.9.2889. [DOI] [PubMed] [Google Scholar]
- 13.Bhattachary-Chatterjee M, Nath Baral R, Chatterjee SK, et al. Cancer vaccines: single-epitope anti-idiotype vaccine versus multiple-epitope antigen vaccine. Cancer Immunol Immunother. 2000;49:133–41. doi: 10.1007/s002620050612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fong L, Hou Y, Rivas A, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci USA. 2001;98:8809–14. doi: 10.1073/pnas.141226398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palucka AK, Ueno H, Fay JW, Banchereau J. Taming cancer by inducing immunity via dendritic cells. Immunol Rev. 2007;220:129–50. doi: 10.1111/j.1600-065X.2007.00575.x. [DOI] [PubMed] [Google Scholar]
- 16.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–83. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic cell immunotherapy: mapping the way. Nat Med. 2004;10:475–80. doi: 10.1038/nm1039. [DOI] [PubMed] [Google Scholar]
- 18.Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117:1195–203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarze SR, Dowdy SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci. 2000;21:45–8. doi: 10.1016/s0165-6147(99)01429-7. [DOI] [PubMed] [Google Scholar]
- 20.Shibagaki N, Udey MC. Dendritic cells transduced with protein antigens induce cytotoxic lymphocytes and elicit antitumor immunity. J Immunol. 2002;168:2393–401. doi: 10.4049/jimmunol.168.5.2393. [DOI] [PubMed] [Google Scholar]
- 21.Wang HY, Fu T, Wang G, et al. Induction of CD4(+) T cell-dependent antitumor immunity by TAT-mediated tumor antigen delivery into dendritic cells. J Clin Invest. 2002;109:1463–70. doi: 10.1172/JCI15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibagaki N, Udey MC. Dendritic cells transduced with TAT protein transduction domain-containing tyrosinase-related protein 2 vaccinate against murine melanoma. Eur J Immunol. 2003;33:850–60. doi: 10.1002/eji.200323709. [DOI] [PubMed] [Google Scholar]
- 23.Viehl CT, Tanaka Y, Chen T, et al. Tat mammaglobin fusion protein transduced dendritic cells stimulate mammaglobin-specific CD4 and CD8 T cells. Breast Cancer Res Treat. 2005;91:271–8. doi: 10.1007/s10549-005-0450-4. [DOI] [PubMed] [Google Scholar]
- 24.Nonaka M, Ma BY, Murai R, et al. Glycosylation-dependent interactions of C-type lectin DC-SIGN with colorectal tumor-associated Lewis glycans impair the function and differentiation of monocyte-derived dendritic cells. J Immunol. 2008;180:3347–56. doi: 10.4049/jimmunol.180.5.3347. [DOI] [PubMed] [Google Scholar]
- 25.van Gisbergen KP, Aarnoudse CA, Meijer GA, Geijtenbeek TB, van Kooyk Y. Dendritic cells recognize tumor-specific glycosylation of carcinoembryonic antigen on colorectal cancer cells through dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin. Cancer Res. 2005;65:5935–44. doi: 10.1158/0008-5472.CAN-04-4140. [DOI] [PubMed] [Google Scholar]
- 26.Woo SJ, Kim CH, Park MY, et al. Co-administration of carcinoembryonic antigen and HIV TAT fusion protein with CpG-oligodeoxynucleotide induces potent antitumor immunity. Cancer Sci. 2008;99:1034–9. doi: 10.1111/j.1349-7006.2008.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robbins PF, Kantor JA, Salgaller M, Hand PH, Fernsten PD, Schlom J. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res. 1991;51:3657–62. [PubMed] [Google Scholar]
- 28.Usharauli D, Perez-Diez A, Matzinger P. The JAM test and its daughter P-JAM: simple tests of DNA fragmentation to measure cell death and stasis. Nat Protoc. 2006;1:672–82. doi: 10.1038/nprot.2006.107. [DOI] [PubMed] [Google Scholar]
- 29.Lopez JA, Hart DN. Current issues in dendritic cell cancer immunotherapy. Curr Opin Mol Ther. 2002;4:54–63. [PubMed] [Google Scholar]
- 30.Nockel J, van den Engel NK, Winter H, Hatz RA, Zimmermann W, Kammerer R. Characterization of gastric adenocarcinoma cell lines established from CEA424/SV40 T antigen-transgenic mice with or without a human CEA transgene. BMC Cancer. 2006;6:57. doi: 10.1186/1471-2407-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sarkar K, Bose A, Chakraborty K, et al. Neem leaf glycoprotein helps to generate carcinoembryonic antigen specific anti-tumor immune responses utilizing macrophage-mediated antigen presentation. Vaccine. 2008;26:4352–62. doi: 10.1016/j.vaccine.2008.06.048. [DOI] [PubMed] [Google Scholar]
- 32.Kavanagh B, Ko A, Venook A, et al. Vaccination of metastatic colorectal cancer patients with matured dendritic cells loaded with multiple major histocompatibility complex class I peptides. J Immunother. 2007;30:762–72. doi: 10.1097/CJI.0b013e318133451c. [DOI] [PubMed] [Google Scholar]
- 33.Koido S, Hara E, Homma S, et al. Synergistic induction of antigen-specific CTL by fusions of TLR-stimulated dendritic cells and heat-stressed tumor cells. J Immunol. 2007;179:4874–83. doi: 10.4049/jimmunol.179.7.4874. [DOI] [PubMed] [Google Scholar]
- 34.Ojima T, Iwahashi M, Nakamura M, et al. Successful cancer vaccine therapy for carcinoembryonic antigen (CEA)-expressing colon cancer using genetically modified dendritic cells that express CEA and T helper-type 1 cytokines in CEA transgenic mice. Int J Cancer. 2007;120:585–93. doi: 10.1002/ijc.22298. [DOI] [PubMed] [Google Scholar]
- 35.Saha A, Chatterjee SK, Foon KA, et al. Dendritic cells pulsed with an anti-idiotype antibody mimicking carcinoembryonic antigen (CEA) can reverse immunological tolerance to CEA and induce antitumor immunity in CEA transgenic mice. Cancer Res. 2004;64:4995–5003. doi: 10.1158/0008-5472.CAN-04-0626. [DOI] [PubMed] [Google Scholar]
- 36.Babatz J, Rollig C, Lobel B, et al. Induction of cellular immune responses against carcinoembryonic antigen in patients with metastatic tumors after vaccination with altered peptide ligand-loaded dendritic cells. Cancer Immunol Immunother. 2006;55:268–76. doi: 10.1007/s00262-005-0021-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ueda Y, Itoh T, Nukaya I, et al. Dendritic cell-based immunotherapy of cancer with carcinoembryonic antigen-derived, HLA-A24-restricted CTL epitope: clinical outcomes of 18 patients with metastatic gastrointestinal or lung adenocarcinomas. Int J Oncol. 2004;24:909–17. [PubMed] [Google Scholar]
- 38.Tamir A, Basagila E, Kagahzian A, et al. Induction of tumor-specific T-cell responses by vaccination with tumor lysate-loaded dendritic cells in colorectal cancer patients with carcinoembryonic-antigen positive tumors. Cancer Immunol Immunother. 2007;56:2003–16. doi: 10.1007/s00262-007-0299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morse MA, Lyerly HK, Gilboa E, Thomas E, Nair SK. Optimization of the sequence of antigen loading and CD40-ligand-induced maturation of dendritic cells. Cancer Res. 1998;58:2965–8. [PubMed] [Google Scholar]
- 40.Ullenhag GJ, Frodin JE, Jeddi-Tehrani M, et al. Durable carcinoembryonic antigen (CEA)-specific humoral and cellular immune responses in colorectal carcinoma patients vaccinated with recombinant CEA and granulocyte/macrophage colony-stimulating factor. Clin Cancer Res. 2004;10:3273–81. doi: 10.1158/1078-0432.CCR-03-0706. [DOI] [PubMed] [Google Scholar]
- 41.Fanales-Belasio E, Moretti S, Nappi F, et al. Native HIV-1 Tat protein targets monocyte-derived dendritic cells and enhances their maturation, function, and antigen-specific T cell responses. J Immunol. 2002;168:197–206. doi: 10.4049/jimmunol.168.1.197. [DOI] [PubMed] [Google Scholar]
- 42.Santin AD, Bellone S, Palmieri M, et al. Induction of tumor-specific cytotoxicity in tumor infiltrating lymphocytes by HPV16 and HPV18 E7-pulsed autologous dendritic cells in patients with cancer of the uterine cervix. Gynecol Oncol. 2003;89:271–80. doi: 10.1016/s0090-8258(03)00083-0. [DOI] [PubMed] [Google Scholar]
- 43.Paglia P, Chiodoni C, Rodolfo M, Colombo MP. Murine dendritic cells loaded in vitro with soluble protein prime cytotoxic T lymphocytes against tumor antigen in vivo. J Exp Med. 1996;183:317–22. doi: 10.1084/jem.183.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morse MA, Clay TM, Colling K, et al. HER2 dendritic cell vaccines. Clin Breast Cancer. 2003;3(Suppl. 4):S164–72. doi: 10.3816/cbc.2003.s.007. [DOI] [PubMed] [Google Scholar]
- 45.Cho HI, Kim EK, Park SY, Lee SK, Hong YK, Kim TG. Enhanced induction of anti-tumor immunity in human and mouse by dendritic cells pulsed with recombinant TAT fused human survivin protein. Cancer Lett. 2007;258:189–98. doi: 10.1016/j.canlet.2007.08.023. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Zan Y, Shan M, et al. Effects of heat shock protein gp96 on human dendritic cell maturation and CTL expansion. Biochem Biophys Res Commun. 2006;344:581–7. doi: 10.1016/j.bbrc.2006.03.171. [DOI] [PubMed] [Google Scholar]
- 47.Frankenburg S, Elias O, Gelbart Y, et al. Recombinant hydrophilic human gp100: uptake by dendritic cells and stimulation of autologous CD8+ lymphocytes from melanoma patients. Immunol Lett. 2004;94:253–9. doi: 10.1016/j.imlet.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 48.Oizumi S, Strbo N, Pahwa S, Deyev V, Podack ER. Molecular and cellular requirements for enhanced antigen cross-presentation to CD8 cytotoxic T lymphocytes. J Immunol. 2007;179:2310–7. doi: 10.4049/jimmunol.179.4.2310. [DOI] [PubMed] [Google Scholar]