

SUMMARY
We present here a rapid, highly sensitive nonradioactive assay for adenylation enzyme selectivity determination and characterization. This method measures the isotopic back exchange of unlabeled pyrophosphate into γ–18O4-labeled ATP via MALDI-TOFMS, ESI-LC/MS or ESI-LC/MS/MS and is demonstrated for both nonribosomal (TycA, ValA) and ribosomal synthetases (TrpRS, LysRS) of known specificity. This low volume (6μL) method detects as little as 0.01% (600 fmol) exchange, comparable in sensitivity to previously reported radioactive assays and readily adaptable to kinetics measurements and high throughput analysis of a wide spectrum of synthetases. Finally, a previously uncharacterized A-T didomain from anthramycin biosynthesis in the thermophile S. refuinius was demonstrated to selectively activate 4-methyl-3-hydroxyanthranilic acid at 47 °C, providing biochemical evidence for a new aromatic β–amino acid activating adenylation domain and the first functional analysis of the anthramycin biosynthetic gene cluster.
Keywords: Pyrophosphate exchange, adenylation, nrps, nonribosomal peptide synthetase, anthramycin, benzodiazepine, biosynthesis
INTRODUCTION
Synthetases play a key role in a range of cellular processes, particularly in those involving protein and peptide amide bond synthesis. In the case of ribosomal peptide synthesis, dedicated tRNA synthetases activate amino acids via adenylation, after which they are transferred by esterification to the 3′ or 2′ hydroxyl of cognate tRNA templates. In secondary metabolism, most peptide bonds are formed by nonribosomal peptide synthetases (NRPS). NRPS are multidomain systems that also transiently activate amino acids, including nonproteinogenic amino acids, via adenylation. This activation is catalyzed by adenylation (A) domains that subsequently interact with adjacent thiolation (T) domains (Figure 1A), in which pendant phosphopantetheinyl moieties covalently capture amino acid adenylates as thioesters prior to condensation (C) domain catalyzed reactions. NRPS are responsible for the biosynthesis of the peptide scaffolds of a large number of clinically significant natural product pharmaceuticals including penicillin, vancomycin and rapamycin to name a few (Fischbach and Walsh, 2006; Sieber and Marahiel, 2005).
Figure 1. ATP-PPi exchange.

A: A-domains in NRPS systems adenylate amino acids for subsequent thiolation reactions on T domains. B: The exchange reaction, performed in the absence of thiolation activity, measures equilibrium exchange of γ-18O4-ATP with 16O4-pyrophosphate. C: Time dependent formation of γ-16O4-ATP and disappearance of γ-18O4-ATP with TycA, measured by MALDI-TOFMS. Intermediary masses correspond to 18O3, 18O2, and 18O1 peaks.
The biochemical assay of decoupled synthetases poses practical challenges, as most adenylation reactions are not formally catalytic. Isolated synthetases perform half reactions (Figure 1B) for subsequent amino acid (thio)esterification that are nearly stoichiometric with regard to their respective tRNA/T domains and aminoacyl adenylates are tightly bound enzyme intermediates. Conventionally, adenylation enzyme selectivity has been assayed using the ATP-32PPi isotope exchange assay. In this method, the synthetase is incubated with excess 32PPi, amino acid and ATP and the reversible back exchange of labeled 32PPi into ATP is monitored by solid phase capture of ATP on activated charcoal followed by cintillation counting. This method, in use for more than half a century (Lee and Lipmann, 1975; Linne and Marahiel, 2004a), is highly sensitive and modern variants have recently been developed for high-throughput and kinetics applications (Otten et al., 2007). The primary drawbacks of the assay are that it requires relatively large amounts of radioactive PPi (0.2 μCi/experiment) and extensive liquid handling of highly radioactive materials. Moreover, the solid phase capture step is an indirect measure of exchange and high background signal can complicate data analysis.
This study describes a nonradioactive mass spectrometry (MS) based PPi exchange assay. To validate the method, we first assayed previously characterized synthetases: TycA, responsible for L-phenylalanine activation during tyrocidine biosynthesis (Pfeifer et al., 1995), ValA, an ‘orphan’ A domain responsible for valine activation (Du and Shen, 1999) and E. coli tRNA synthetases TrpRS and LysRS (Joseph and Muench, 1971; Stern et al., 1966). Subsequently, we assayed the specificity of ORF21, a recently described uncharacterized A-T didomain of the anthramycin gene cluster (Hu et al., 2007), providing the first biochemical evidence for the anthramycin biosynthetic pathway.
RESULTS
Mass sensitive observation of PPi-exchange can hypothetically detect adenylation domain catalyzed mass shifts on either side of the exchange equation. As back exchange is favored by using PPi in excess, we introduced a heavy atom label in the starting material as γ–18O4-ATP. γ–18O4-ATP is commercially available or alternatively can be synthesized chemically (Hoard and Ott, 1965). Conditions for MS isotope exchange were based on previously reported assay methods (Linne and Marahiel, 2004a; Otten et al., 2007) and simultaneously optimized for MALDI-TOFMS and ESI-LC/MS easurement strategies. Correspondingly, adenylation domains/enzymes (200 nM) were incubated with 1 mM γ–18O4-ATP, 1 mM amino acid, 5 mM MgCl2 and 5 mM PPi for 5 – 30 minutes. For MALDITOFMS analysis, enzymatic reactions were quenched by mixing with an equal volume of 9-aminoacridine in acetone, which was found to be an optimal matrix for detection of triphosphate nucleotides (Sun et al., 2007). For ESI-LC/MS measurements, acetone quenched reactions were separated from salts and buffer using a graphitic matrix column (Hypercarb, Thermo Inc.), which reliably retains highly charged aromatic metabolites via charge quadrupole and hydrophobic interactions (Hu et al., 2007; Xing et al., 2004). The Hypercarb column was eluted with an isocratic gradient of 17.5% ACN/82.5% 20mM ammonium acetate buffer and analytes were detected in negative ion mode.
Formation of γ–16O4-ATP and consumption of γ–18O4-ATP was directly monitored by observing the 8 Da mass shift (Figure 1C) due to back exchange of unlabeled PPi. The activity of the enzyme was quantified as the integrated peak ratio of γ–16O4-ATP species to all ATP species in the reaction mixture. As the species being compared are isotopologues, peak integration and the subsequent signal ratio is quantitative in both MALDI-TOFMS and ESI-LC/MS, as there is no difference in ionization efficiency owing to differences in chemical composition. The observed mass ratios correlate directly to the fraction of ATP-PPi exchange. Using this strategy, 5 minute incubations of 6 μL reactions can be successfully analyzed in as little as 30 seconds using MALDI-TOFMS. Alternatively, and with correspondingly higher sensitivity, measurements can be made with ESI-LC/MS in 5 minutes or less.
The limit of detection (LOD) for ESI-LC/MS and MALDI-TOFMS were determined by mixing known amounts of labeled and unlabeled ATP in assay buffer and measuring integrated peak ratios (Figure S2). For ESI-LC/MS, the LOD was determined to be 3.4 μM (0.34% exchange), and for MALDI-TOFMS a higher LOD (10 μM, 1% exchange) was observed. One potential source of this difference may be ion suppression effects in MALDITOFMS by eliminating the Hypercarb sample clean-up prior to analyses. Residual γ–16O4-ATP levels found in commercial γ–18O4-ATP were estimated to be 3.4μM by ESILC/MS and fall below the threshold of detection for the MALDI-TOFMS method. Both detection methods were comparable in sensitivity to reported radioactive ATP-PPi exchange assays. Under optimized radioactive conditions, as little as 50 pmol exchange per assay (0.01%) has been detected in a 100 μL reaction (Eigner and Loftfield, 1974). Using the rapid MALDI-TOFMS method, as little as 1% (60 pmol) exchange was detectable, whereas, the full scan ESI-LC/MS method detects 0.1% (6 pmol) exchange. Further enhancement was obtained using tandem ESI-LC/MS utilizing selected reaction monitoring (SRM), in which as little as 0.01% (600 fmol) exchange was detectable.
Of concern was the hydrolytic 18O-lability of γ–18O4-ATP under typical assay conditions. Correspondingly, γ–18O4-ATP was incubated under assay conditions with 1) no TycA, 2) no amino acid and 3) incorrect amino acid for up to 15 hrs (Figure S3). Consistent with literature reports that 18O-substituted phosphates are relatively stable in buffered solutions (Cohn and Hu, 1978), no loss of 18O was observed at up to 15 hours in assay buffer in the absence of TycA. However, in the presence of enzyme or incorrect amino acid, slow exchange of 18O label was observed after 2 – 5 hrs. Of note, non β–γ–bridging 18O atoms exchanged more rapidly than bridging 18O atoms. Incubation of γ–18O4-ATP with TycA for 5 hours resulted in a decrease in 18O4 of 45 – 60 % whereas only a 14 – 24% increase in the -8 Da shift, corresponding to the loss of the bridging β–γ18O, was observed under the same conditions. As PPi exchange is only indicated by complete loss of bridging label, these slow shifts can be compensated for by calculating the exchange as the ratio of unlabeled ATP divided by the sum of all ATP species normalized to the theoretical equilibrium 5:1 16O/18O molar ratio. Therefore, 83.33% apparent exchange reflects 100% exchange) and % exchange = (100/0.833) • 16O/(18O + 16O).
TycA, from tyrocidine biosynthesis has been extensively studied by the conventional 32PPi exchange assay (Lee and Lipmann, 1975; Otten et al., 2007) and was selected as a model synthetase for method development of mass based PPi exchange. To demonstrate substrate selectivity, we tested the full panel of proteinogenic amino acids and Dphenylalanine, a previously identified substrate of TycA. As shown in Figure 2, L-phenylalanine and D-phenylalanine were both identified as substrates demonstrating 70 –100% exchange using both MALDI-TOFMS and ESI-LC/MS analysis. Due to the LOD for MALDI-TOFMS, no exchange was observed for all other amino acids tested. However, the signal of MALDI-TOFMS analysis could be enhanced with increased enzyme concentration and/or longer incubation times (Figure S4). Lower rates of exchange were also effectively measured by ESI-LC/MS.
Figure 2.
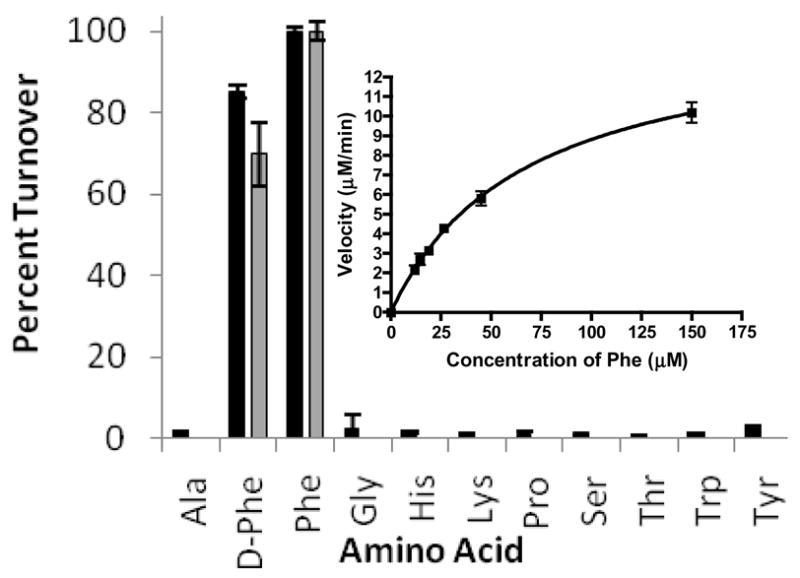
ESI-LC/MS (■) and MALDI-TOFMS (■) detection of TycA substrate activation by ATP-PPi exchange. Inlay shows concentration dependence of exchange for L-Phe.
To further validate the assay, an additional ‘orphan’ synthetase, ValA, a valine activating A-domain identified and verified by Shen and coworkers (Du and Shen, 1999), was tested. The MALDI-TOFMS assay demonstrated appreciable levels of exchange for valine in accordance with previous studies. Again, for all other amino acids tested, levels of exchange fell below the LOD for MALDI-TOFMS analysis, but low levels of exchange were easily detectable using either ESI-LC/MS analysis or longer incubation times with higher enzyme concentrations for the MALDI-TOFMS method (data not shown).
To demonstrate the applicability of the mass based exchange assay for kinetics measurements, the exchange velocities for TycA were plotted versus phenylalanine concentration as shown in Figure 2. As previously noted, decoupled A-domains are not catalytic. Therefore, concentration response curves of PPi exchange assays cannot be strictly interpreted in terms Michaelis-Menten steady-state kinetics. It should be noted that a comprehensive quantitative treatment of exchange kinetics for the purposes of deriving kinetic constants has been described (Cole and Schimmel, 1970). In any event, the apparent Michaelis-Menten kinetic parameters can be calculated for the purpose of comparison to previously reported TycA apparent parameters. ESI-LC/MS analysis yielded an apparent KM of 67 ± 2 μM and apparent kcat of 92 ± 2 min−1. These values are consistent with previously described measurements which report measurements of KM between 40 μM ((Pfeifer et al., 1995) to 13 μM (Otten et al., 2007).
In addition to the aforementioned NRPS adenylation domains, two previously characterized tRNA synthetases from E. coli, TrpRS and LysRS, were also assayed using mass based pyrophosphate exchange. As shown in Table 1, TrpRS and LysRS activate their cognate amino acids under standard assay conditions. No activation was observed for nonsubstrates (data not shown).
Table 1.
Activity of amino acid adenylating enzymes.
| Enzyme | Amino Acid | %γ–18O4-ATP exchange |
|---|---|---|
| TycA | D-Phe | 100 ±2.5 |
| L-Phe | 69.8 ±7.8 | |
| ORF21a | HA | 8.9 ± 2.3 |
| MHA | 27.3 ± 3.2 | |
| ValAa | Val | 5.5 ± 0.8 |
| TrpRSa | Trp | 9.7 ± 1.6 |
| LysRSa | Lys | 13.0 ± 2.7 |
Exchange measured by MALDI-TOFMS for a panel of amino acids. Exchange is only listed for active amino acids. All other amino acids tested fell below the threshold of detection for the MALDI-TOFMS based assay.
ORF21 from the thermophilic actinomycete Streptomyces refuinius has been proposed to encode the initiating A-T containing module of anthramycin biosynthesis (Hu et al., 2007). Previous sequence analysis and chemical complementation studies of the anthramycin pathway revealed that there are two possible substrates for ORF21 activation: 3-hydroxyanthranilic acid (HA) and 4-methyl-3-hydroxyanthranilic acid (MHA). Sequence analysis of the putative A-domain peptide sequence indicates it is highly divergent from previously studied A-domains. Analysis of residues in the substrate binding region indicate Asp-235 (GrsA numbering), essential for binding α–amino functionality, is substituted by alanine in ORF21. Furthermore, the 8 to 10 amino acid selectivity conferring code (Challis et al., 2000; Stachelhaus et al., 1999) bears no similarity to previously described A-domains including, notably, actinomycin synthase ACMS I, which has been reported to activate the MHA analog p-toluic acid in the MHA containing peptide actinomycin (Pfennig et al., 1999).
To provide direct biochemical evidence for substrate activation of the A-domain of ORF21, the encoding gene was cloned via PCR and LR-ligation into pETDEST-42 for overproduction as a C-terminal His6-tagged protein and purified using Ni+2-affinity chromatography (Figure S1). As summarized in Table 1, purified ORF21 activates only MHA and HA stimulating a 3-fold higher rate of exchange for MHA when the reaction mixture is incubated for 30 min at 47°C, the optimal temperature for anthramycin production. For further characterization of Orf21, apparent kinetic parameters were calculated for both MHA and HA. ESI-LC/MS analysis yielded apparent KM of 33 ± 3 μM and apparent kcat of 130 ± 7 min−1 for MHA and KM of 154 ± 9 μM and apparent kcat of 99 ± 2 min−1 for HA (Figure 3). While the exchange assay suggests MHA is a likely substrate for ORF21, further studies are required to pinpoint the timing of methylation during anthramycin biosynthesis.
Figure 3.

Above: proposed reaction catalyzed by ORF21 en route to anthramycin. Below: ORF21 substrate dependence of exchange for ORF21 double-reciprocal plot for ■ MHA and ◆ HA respectively. Data were measured in triplicate using the ESI-LC/MS method.
DISCUSSION
New strategies are continually developed for ribosomal and nonribosomal peptide synthetase analysis and characterization (Francklyn et al., 2008; Linne and Marahiel, 2004b). For example, recently described MS methods allow the determination of aminoacyl-S-enzyme intermediates on intact proteins (Dorrestein et al., 2006; Hicks et al., 2004). Other high throughput methods include fluorescence polarization assays, which utilize a competitive binding experiment with a synthetic fluorescent probe (Neres et al., 2008) and affinity capture techniques, which utilize alkyne-functional probes in ‘click’ type analysis (La Clair et al., 2004; Zou and Yin, 2008). The mass based pyrophosphate exchange assay described herein is complementary to these methods and provides an improved alternative to the conventional 32PPi exchange assay, an indispensible tool in synthetase investigation. Equilibrium exchange compared via mass isotopologue ratios is a direct measurement of exchange, eliminating artifacts that may result from radioisotope exchange methods. Stable isotopologues permit quantitative analyses in both MALDI-TOFMS and ESI-LC/MS methods and exchange kinetics parameters can be readily determined.
Several practical advantages of the mass based system with regard to the conventional assay are also evident. The use of stable isotopes circumvents the labor and regulatory expenses related to the safe handling of radioactive materials. In addition to γ–18O4-ATP being indefinitely stable at −80 °C, the low reacti on volume used in our method (6μL) permits over 3000 exchange reactions to be performed using 10 mg of γ–18O4-ATP, resulting in a materials cost of approximately 33 cents per reaction using commercially available starting materials. Lastly, the speed of the mass based assay compares favorably to conventional exchange methods. 32PPi exchange assays employ solid phase capture, centrifugation or TLC steps followed by liquid scintillation counting, typically requiring continuous monitoring of β–emission for up to 48 hours for maximal sensitivity. Conversely mass based detection can be performed in as little as 30 seconds per sample in our MALDI-TOFMS implementation, which has not yet been optimized for speed. Furthermore, the MALDIbased assay described herein requires little sample clean-up and handling prior to analysis. Future implementation with imaging MALDI-TOFMS (Cornett et al., 2007), or imaging MALDI-ion mobility-TOFMS, detection of 384-well plates could in principle accelerate the analyses to rates faster than 1 second per sample (McLean et al., 2007).
Recently, microbial genomics initiatives have identified staggering numbers of gene clusters containing cryptic putative NRPS. Given the history of NRP drugs, these synthetases are likely to encode new natural products with potential as drug leads and bioprobes. NRPS A domain substrate selectivity can be estimated, to varying degrees of accuracy, by primary sequence analysis using homology modeling approaches or neural network algorithms (Challis et al., 2000; Rausch et al., 2005; Stachelhaus et al., 1999). However, subsequent to these in silico analyses, it is often necessary to provide biochemical evidence supporting A domain selectivity. The γ–18O4-ATP-PPi exchange system provides a rapid, sensitive and reproducible means to measure adenylation domain specificity. Moreover, when combined with previously reported 96-well based liquid handling methods for evaluating A-domains in E. coli libraries (Otten et al., 2007), is readily adaptable to high through-put analysis appropriate for directed evolution studies (Fischbach et al., 2007).
EXPERIMENTAL PROCEDURES
ATP-PPi Exchange Assay Conditions
In order to avoid precipitation of magnesium pyrophosphate, assay components were divided into stock solutions comprising 1) 3 mM amino acids containing 15 mM PPi in 20 mM Tris pH 7.5, 2) 3 mM γ–18O4-ATP containing 15mM MgCl2 in 20 mM Tris pH 7.5 and 3) 600 nM enzyme in 20 mM Tris pH 7.5 containing 5% glycerol and 1mM DTT. Exchange reactions containing 2 μL of each component were initiated by the addition of enzyme solution. 6μL reactions therefore contained final concentrations of 5 mM MgCl2, 5 mM PPi, 1 mM γ-18O4-ATP, 1mM amino acid and 20 mM Tris-HCl pH 7.5. After an incubation period (30 min at 25 °C for TycA, ValA, TrpRS and LysRS or 47 °C for ORF21), the reactions were stopped by the addition of 6 μL 9-aminoacridine in acetone (10 mg/mL) for MALDI-TOFMS analysis or 6 μL acetone for ESI-LC/MS analysis. For enhanced MALDI-TOFMS signal, 1μM TycA was incubated for 2 hours. Detailed mass spectrometric parameters and HPLC/MS conditions are described in detail in supplementary data.
Data Analysis
The equilibrium molar ratio of unlabeled PPi to γ–18O4-ATP under assay conditions is 5:1. Therefore 83.33% apparent exchange corresponds to 100% exchange. Percent exchange was determined by comparison of the ratio of γ–16O4-ATP to the sum of all ATP species normalized with this modifier: % exchange = (100/0.833) • 16O/(18O + 16O). Monoisotopic peak areas were determined using manufacturer’s software. For MALDI-TOFMS analysis, the ratio of the area of γ–16O4-ATP (m/z 506) to the area of total ATP including unlabeled, partially labeled, fully labeled and monosodium-coordinated ions (m/z 506, 508, 510, 512, 514, 528, 530, 532, 534, 536) was calculated. For ESI-LC/MS analysis, the ratio of the area of γ–16O4-ATP to the area of total ATP including unlabeled, partially labeled, fully labeled, monosodium-coordinated and sodium acetate adduct ions was similarly calculated. For selected reaction monitoring, the ratio of the area of γ–16O4-ATP, taken as the area of the product ion (m/z 408), to the area of total ATP including unlabeled, partially labeled, and fully labeled ions, taken as the sum of the area of their respective product ions ((514 → 408, 410, 412, 414, 416), (512 → 408, 410, 412, 414), (510 → 408, 410, 414), (508 → 408. 410) and (506 → 408)), was calculated.
Supplementary Material
Acknowledgments
Funding was provided by the National Institutes for Health 1R01GM077189-3 (B.O.B.), the Vanderbilt University College of Arts and Sciences, and the Vanderbilt Institute of Chemical Biology (J.A.M.). We wish to acknowledge Professor Ben Shen (University of Wisconsin) for the gift of the ValA vector, Professor Torsten Stachelhaus (AureoGen Biosciences) for the gift of the TycA vector, Professor Anthony Forster (Vanderbilt University) for the gift of TrpRS and LysRS, and Ms. Randi Gant-Branum and Mr. Michal Kliman for instrumental assistance (Vanderbilt University).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Challis GL, Ravel J, Townsend CA. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chemistry and Biology. 2000;7:211–224. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- Clark RL, Neidhardt FC. Roles of the two lysyl-tRNA synthetases of Escherichia coli: analysis of nucleotide sequences and mutant behavior. Journal of bacteriology. 1990;172:3237–3243. doi: 10.1128/jb.172.6.3237-3243.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn M, Hu A. Isotopic (18O) shift in 31P nuclear magnetic resonance applied to a study of enzyme-catalyzed phosphate--phosphate exchange and phosphate (oxygen)--water exchange reactions. Proceedings of the National Academy of Sciences of the United States of America. 1978;75:200–203. doi: 10.1073/pnas.75.1.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole FX, Schimmel PR. On the rate law and mechanism of the adenosine triphosphate--pyrophosphate isotope exchange reaction of amino acyl transfer ribonucleic acid synthetases. Biochemistry. 1970;9:480–489. doi: 10.1021/bi00805a005. [DOI] [PubMed] [Google Scholar]
- Cornett DS, Reyzer ML, Chaurand P, Caprioli RM. MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nature methods. 2007;4:828–833. doi: 10.1038/nmeth1094. [DOI] [PubMed] [Google Scholar]
- Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL. Facile detection of acyl and peptidyl intermediates on thiotemplate carrier domains via phosphopantetheinyl elimination reactions during tandem mass spectrometry. Biochemistry. 2006;45:12756–12766. doi: 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du LC, Shen B. Identification and characterization of a type II peptidyl carrier protein from the bleomycin producer Streptomyces verticillus ATCC 15003. Chemistry & Biology. 1999;6:507–517. doi: 10.1016/S1074-5521(99)80083-0. [DOI] [PubMed] [Google Scholar]
- Eigner EA, Loftfield RB. Kinetic techniques for the investigation of amino acid: tRNA ligases (aminoacyl-tRNA synthetases, amino acid activating enzymes) Methods in enzymology. 1974;29:601–619. doi: 10.1016/0076-6879(74)29053-0. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, Lai JR, Roche ED, Walsh CT, Liu DR. Directed evolution can rapidly improve the activity of chimeric assembly-line enzymes. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:11951–11956. doi: 10.1073/pnas.0705348104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- Francklyn CS, First EA, Perona JJ, Hou YM. Methods for kinetic and thermodynamic analysis of aminoacyl-tRNA synthetases. Methods (San Diego, Calif) 2008;44:100–118. doi: 10.1016/j.ymeth.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CV, Yanofsky C. Cloning and characterization of the gene for Escherichia coli tryptophanyl-transfer ribonucleic acid synthetase. Journal of bacteriology. 1981;148:941–949. doi: 10.1128/jb.148.3.941-949.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks LM, O'Connor SE, Mazur MT, Walsh CT, Kelleher NL. Mass spectrometric interrogation of thioester-bound intermediates in the initial stages of epothilone biosynthesis. Chemistry & Biology. 2004;11:327–335. doi: 10.1016/j.chembiol.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Hoard DE, Ott DG. Conversion of Mono- and Oligodeoxyribonucleotides to 5-Triphosphates. Journal of the American Chemical Society. 1965;87:1785–1788. doi: 10.1021/ja01086a031. [DOI] [PubMed] [Google Scholar]
- Hu Y, Phelan V, Ntai I, Farnet CM, Zazopoulos E, Bachmann BO. Benzodiazepine biosynthesis in Streptomyces refuineus. Chemistry & Biology. 2007;14:691–701. doi: 10.1016/j.chembiol.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Joseph DR, Muench KH. Tryptophanyl transfer ribonucleic acid synthetase of Escherichia coli. I. Purification of the enzyme and of tryptrophan transfer ribonucleic acid. Journal of Biological Chemistry. 1971;246:7602–7609. [PubMed] [Google Scholar]
- La Clair JJ, Foley TL, Schegg TR, Regan CM, Burkart MD. Manipulation of carrier proteins in antibiotic biosynthesis. Chem Biol. 2004;11:195–201. doi: 10.1016/j.chembiol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Lee SG, Lipmann F. Tyrocidine synthetase system. Methods in enzymology. 1975;43:585–602. doi: 10.1016/0076-6879(75)43121-4. [DOI] [PubMed] [Google Scholar]
- Linne U, Marahiel MA. Reactions catalyzed by mature and recombinant nonribosomal peptide synthetases. Protein Engineering. 2004a;388:293–315. doi: 10.1016/S0076-6879(04)88024-8. [DOI] [PubMed] [Google Scholar]
- Linne U, Marahiel MA. Reactions catalyzed by mature and recombinant nonribosomal peptide synthetases. Methods in enzymology. 2004b;388:293–315. doi: 10.1016/S0076-6879(04)88024-8. [DOI] [PubMed] [Google Scholar]
- McLean JA, Ridenour WB, Caprioli RM. Profiling and imaging of tissues by imaging ion mobility-mass spectrometry. Journal of Mass Spectrometry. 2007;42:1099–1105. doi: 10.1002/jms.1254. [DOI] [PubMed] [Google Scholar]
- Neres J, Wilson DJ, Celia L, Beck BJ, Aldrich CC. Aryl acid adenylating enzymes involved in siderophore biosynthesis: fluorescence polarization assay, ligand specificity, and discovery of non-nucleoside inhibitors via high-throughput screening. Biochemistry. 2008;47:11735–11749. doi: 10.1021/bi801625b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten LG, Schaffer ML, Villiers BR, Stachelhaus T, Hollfelder F. An optimized ATP/PP(i)-exchange assay in 96-well format for screening of adenylation domains for applications in combinatorial biosynthesis. Biotechnol J. 2007;2:232–240. doi: 10.1002/biot.200600220. [DOI] [PubMed] [Google Scholar]
- Pfeifer E, Pavela-Vrancic M, von Dohren H, Kleinkauf H. Characterization of tyrocidine synthetase 1 (TY1): requirement of posttranslational modification for peptide biosynthesis. Biochemistry. 1995;34:7450–7459. doi: 10.1021/bi00022a019. [DOI] [PubMed] [Google Scholar]
- Pfennig F, Schauwecker F, Keller U. Molecular characterization of the genes of actinomycin synthetase I and of a 4-methyl-3-hydroxyanthranilic acid carrier protein involved in the assembly of the acylpeptide chain of actinomycin in Streptomyces. The Journal of biological chemistry. 1999;274:12508–12516. doi: 10.1074/jbc.274.18.12508. [DOI] [PubMed] [Google Scholar]
- Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH. Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs) Nucleic acids research. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber SA, Marahiel MA. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- Stachelhaus T, Mootz HD, Marahiel MA. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chemistry and Biology. 1999;6:493–505. doi: 10.1016/S1074-5521(99)80082-9. [DOI] [PubMed] [Google Scholar]
- Stern R, DeLuca M, Mehler AH, McElroy WD. Role of sulfhydryl groups in activating enzymes. Properties of Escherichia coli lysine-transfer ribonucleic acid synthetase. Biochemistry. 1966;5:126–130. [PubMed] [Google Scholar]
- Sun G, Yang K, Zhao ZD, Guan SP, Han XL, Gross RW. Shotgun metabolomics approach for the analysis of negatively charged water-soluble cellular metabolites from mouse heart tissue. Analytical Chemistry. 2007;79:6629–6640. doi: 10.1021/ac070843+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Apedo A, Tymiak A, Zhao N. Liquid chromatographic analysis of nucleosides and their mono-, di- and triphosphates using porous graphitic carbon stationary phase coupled with electrospray mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:1599–1606. doi: 10.1002/rcm.1524. [DOI] [PubMed] [Google Scholar]
- Zou Y, Yin J. Alkyne-functionalized chemical probes for assaying the substrate specificities of the adenylation domains in nonribosomal Peptide synthetases. Chembiochem. 2008;9:2804–2810. doi: 10.1002/cbic.200800480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.