

Abstract
The intramolecular Pd-catalyzed carboetherification of alkenes affords 2-indan-1-yltetrahydrofuran products in moderate to good yield with good to excellent levels of diastereoselectivity. The stereochemical outcome of these reactions is dependent on the structure of the Pd-catalyst. Use of PCy3 or P[(4-MeO)C6H4]3 as the ligand for Pd leads to syn-addition of the arene and the oxygen atom across the double bond, whereas use of (±)-BINAP or DPP-Benzene affords products that result from anti-addition. The catalyst-induced change in stereochemistry is likely due to a change in reaction mechanism. Evidence is presented that suggests the syn-addition products derive from an unprecedented transannular alkene insertion of an 11-membered Pd(Ar)(OR) complex. In contrast, the anti-addition products appear to arise from Wacker-type anti-oxypalladation. Studies on analogous Pd-catalyzed intramolecular carboamination reactions, which afford 2-indan-1-ylpyrrolidines that result from syn-addition, are also described.
Introduction
A large number of important biologically active molecules and natural products contain subunits comprised of heterocyclic or carbocyclic rings attached by a C-C bond between two stereogenic centers.2 Many approaches to the construction of these moieties employ strategies in which one or both stereocenters are generated prior to ring-closure.2,3,4 However, a potentially attractive and concise alternate approach would be to form both rings and both stereocenters in a single step via a stereospecific addition across a carbon-carbon double bond.5 A strategy that provides access to both possible diastereomers (syn- addition or anti-addition) from a single starting material should allow for facile preparation of analogs that could be used to optimize biological properties or probe structure/activity relationships. In addition, this strategy would permit construction of the desired product stereoisomer from the starting material with the most easily accessible alkene geometry, which could potentially decrease the length of synthetic sequences and improve overall synthetic efficiency.
We have recently described a stereoselective method for the construction of tetrahydrofurans and pyrrolidines via Pd-catalyzed intermolecular6 carboetherification7a-b or carboamination7c-d,8 reactions between aryl bromides and γ-hydroxy or -amino alkenes (eq 1). These reactions generate two bonds and up to two stereocenters in one step, and are believed to proceed via intramolecular syn-alkene insertion into a Pd(Ar)(YR) complex (e.g. 1).7b,d We reasoned that this methodology could potentially be employed for the stereoselective construction of heterocycles bearing attached carbocyclic rings (e.g. 4) by appending the aryl halide moiety to the unsaturated alcohol or amine, which would lead to the formation of two bonds, two stereocenters, and two rings in a single step (Scheme 1).9,10 However, we felt these transformations could be quite challenging, as intermolecular carboetherification reactions of internal olefins are currently limited to substrates bearing tertiary alcohol nucleophiles, and intermolecular carboamination reactions provide complex mixtures of products when acyclic internal alkenes are employed as substrates.7
Scheme 1.

 |
(1) |
In addition to the potential synthetic utility and challenges described above, these transformations also posed an interesting mechanistic question. The intramolecular reactions,6 if mechanistically analogous to the intermolecular reactions (eq 1), would involve syn-alkene insertions of eleven-membered palladacyclic intermediates such as 3 (Scheme 1, Path A). Only one previous report has described transformations that presumably involve macrocyclic palladacycles bearing both Pd-C and Pd-heteroatom bonds,11 and transannular syn-alkene insertions of macrocyclic palladacycles bearing internal olefins are unknown. Alternatively, product formation could potentially occur through other mechanistic pathways that have not been previously observed to predominate in carboamination/carboetherification processes.12 For example, a Wacker-type mechanism (Scheme 1, Path B) could generate products resulting from anti-addition across the C-C double bond (5).9,13 Although a priori we could not predict which pathway would predominate, both pathways seemed potentially viable, and we felt it might be possible to influence the mechanistic and stereochemical course of the reactions by varying catalyst structure.14,15
In this paper we describe the first examples of intramolecular carboetherification and carboamination reactions of unsaturated alcohols/amines bearing tethered aryl halides. These reactions afford 2-indan-1-yl tetrahydrofurans and pyrrolidines in good yields with stereoselectivities of up to >20:1. Products resulting from syn-addition of the arene and the alcohol/amine across the carbon-carbon double bond are formed when catalysts bearing monodentate phosphines such as PCy3 or P[(p-MeO)C6H4]3 are employed, and likely derive from unprecedented transannular alkene insertions of macrocyclic palladium(aryl)(alkoxide) or palladium(aryl)(amido) complexes (e.g. 3). In contrast, substrates bearing tethered alcohol nucleophiles provide products resulting from anti-addition when catalysts supported by chelating ligands with small bite angles such as (±)-BINAP or DPP-Benzene are used.16 The experiments described herein suggest that the change in product stereochemistry likely results from a change in reaction mechanism that is influenced by both the structure of the Pd-catalyst and the nature of the tethered heteroatom, and are the first examples of phosphine ligand-control of syn- vs. anti-oxypalladation pathways in catalytic reactions.
Results
Intramolecular Carboetherification Reactions
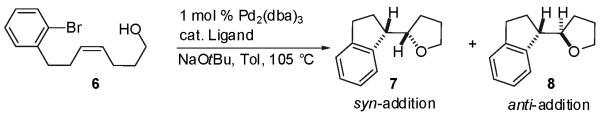
In our initial experiments we elected to explore the intramolecular carboetherification of Z-alkene 6 bearing a tethered primary alcohol group as we felt that the Z-alkene geometry combined with the high nucleophilicity of the unhindered alkoxide (generated in situ upon reaction with NaOtBu) would help to facilitate formation of the putative 11-membered palladium(aryl)(alkoxide) complex required for syn-alkoxypalladation. Our previous studies of Pd-catalyzed intermolecular carboetherification reactions demonstrated that the choice of phosphine ligand had a large impact on chemical yield of the desired tetrahydrofuran products.7a,17 Thus, our optimization studies focused on variation of this parameter while employing otherwise standard reaction conditions (toluene, NaOtBu, 105 °C).18
The alcohol substrate was prepared in three steps from commercially available materials19 and was treated with catalytic amounts of Pd2(dba)3/phosphine ligand in the presence of excess NaOtBu (2.0 equiv). As shown in Table 1, we were gratified to find that Pd-catalyzed reactions of 6 proceeded to generate the desired 2-(1-indanyl)tetrahydrofuran as a mixture of two diastereomers (7 and 8) in moderate to good yield with stereoselectivities dependent on catalyst structure. Use of a catalyst comprised of Pd2(dba)3 and P(o-tol)3 that provided optimal results in intermolecular carboetherification reactions of acyclic internal alkenes7b afforded 7 as the major diastereomer (entry 3), which derives from syn-addition of the arene and the alkoxide across the C-C double bond. The major side products observed in this reaction resulted from debromination of the starting material20 or intramolecular Heck-arylation of the alkene. After some experimentation, a mixture of Pd2(dba)3 and P[(4-MeO)C6H4]3 was found to provide product 7 in 54% isolated yield with 8:1 diastereoselectivity (entry 1). Use of this catalyst system diminished the competing Heck-arylation, although competing debromination of the substrate was still problematic. X-ray crystallographic analysis of a derivative bearing an aminobiphenyl substituent on the aromatic ring confirmed that the major diastereomer 7 possesses the (1S*,2S*)-relative stereochemistry.21
Table 1.
 | |||
|---|---|---|---|
| entry | ligand | dr(b) | Isolated Yield (7 + 8) |
| 1 | P[(p-MeO)C6H4]3 | 8:1 | 54% |
| 2 | PPh3 | 5:1 | 27% |
| 3 | P(o-tol)3 | 2:1 | 35% |
| 4 | PCy3 | 2:1 | 48% |
| 5 | DPE-Phos | 1:1 | 49% |
| 6 | DPPE | 1:2 | 46% |
| 7 | DPP-Benzene | 1:2 | 44% |
| 8 | (±)-BINAP | 1:18 | 60% |
Conditions: 1.0 equiv 6, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % ligand (monophosphines) or 2 mol % ligand (bis-phosphines), toluene (0.1 M), 105 °C, 3-8 h.
Diastereoselectivities were determined by GC and/or 1H NMR analysis of crude reaction mixtures.
Surprisingly, a complete shift in the stereochemical outcome of this reaction was observed when (±)-BINAP was employed as the ligand (entry 8). Under these conditions, the (1S*,2R*)-diastereomer (8) was formed as the major product. This substance derives from anti-addition of the arene and the alcohol across the double bond, and was obtained in 60% yield and 18:1 dr. Other chelating phosphine ligands with small bite angles (≤ 93°) such as DPPE and DPP-Benzene also provided modest selectivity for the anti-addition product.
With optimized reaction conditions in hand, we examined the intramolecular carboetherification of several different alcohol substrates with varying alkene geometries and varying degrees of alcohol substitution. As shown in Scheme 2, the transformations of substrates bearing primary alcohols were found to be stereospecific, and either diastereomer could be selectively obtained from either the E- or Z-alkene starting materials (6 or 9) with the appropriate choice of catalyst. For example, the (1S*,2R*)-2-indan-1-yl tetrahydrofuran 8 was generated in 51% yield and >20:1 dr via the Pd/PCy3-catalyzed syn-addition reaction of E-alkene substrate 9,22 and was obtained in 60% yield and 18:1 dr via the Pd/(±)-BINAP catalyzed anti-addition of Z-alkene substrate 6. Similarly, the (1S*,2S*)-isomer 7 was produced in 56% yield (15:1 dr) from 9 with dpp-benzene as ligand, and 54% yield (8:1 dr) from 6 with the ligand P[(p-MeO)C6H4]3.
Scheme 2. a.

(a) Conditions: 1.0 equiv alcohol substrate, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % ligand (monophosphines) or 2 mol % ligand (bis-phosphines), toluene (0.1 M), 105 °C, 3-8 h. Diastereoselectivities were determined by GC and/or 1H NMR analysis.
Substrates 10 and 11 bearing tethered tertiary alcohols were also selectively converted to products of either syn- or anti-addition depending on catalyst structure (Scheme 3). However, in contrast to reactions of Z-alkene primary alcohol 6, which afforded syn-addition product 7 with most catalyst systems, transformations of the analogous tertiary alcohol bearing a Z-alkene (11) gave anti-addition product 12 under most conditions examined. After some experimentation, the Pd/PMe3·HBF4 catalyzed reaction of 11 was found to provide 13,22 the product of syn-addition across the Z-alkene, in 74% yield with 9:1 dr. Use of the ligand PCy3·HBF4 for the Pd-catalyzed cyclization of 11 afforded anti-addition product 12 in 78% yield and 14:1 dr.23 Although both diastereomers 12 and 13 could be selectively obtained from either starting material (10 or 11), higher yields were obtained with the Z-alkene substrate 11.
Scheme 3.

(a) Conditions: 1.0 equiv alcohol substrate, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % ligand (monophosphines) or 2 mol % ligand (bis-phosphines), toluene (0.1 M), 105 °C, 3-8 h. Diastereoselectivities were determined by GC and/or 1H NMR analysis.
In order to examine the possibility of stereoselectively generating products with three stereocenters, the secondary alcohol 14 bearing an E-alkene was prepared and subjected to the carboetherification reaction conditions. The best results were obtained with a catalyst comprised of Pd2(dba)3/PCy3·HBF4, which provided a 40% isolated yield of 15 with 92:5:2:1 selectivity favoring syn-addition across the alkene and trans-stereochemistry around the tetrahydrofuran ring (eq 2).24 The modest yield can be attributed to the formation of large amounts of side products that derive from reduction of the aryl bromide with concomitant oxidation of the alcohol. Use of (±)-BINAP for this transformation led to complex mixtures of products; only small amounts of the desired tetrahydrofuran derivatives were detected by 1H NMR analysis of crude reaction mixtures.
 |
(2) |
The cyclization of the corresponding Z-alkene 16 proceeded in 40% yield with good selectivity (17:2:1:1 dr)25 for syn-addition/trans-thf formation (eq 3); the two most prevalent diastereomers both contained trans-tetrahydrofuran rings.24 As noted above, competing oxidation/reduction of the substrate was problematic in this system.
 |
(3) |
In contrast to the related Pd/BINAP-catalyzed transformation of the E-alkene substrate 14, treatment of 16 with catalytic Pd2(dba)3/(±)-BINAP provided 15 as the major diastereomer, which results from anti-addition with trans-thf formation, albeit in low yield (25%) (eq 4). High selectivity for anti-addition was observed (94:6), although trans/cis-selectivity was modest (ca. 2:1).24 The major side products in this transformation resulted from competing intramolecular Heck-arylation of the starting material.
 |
(4) |
Intramolecular Pd-Catalyzed Carboamination Reactions
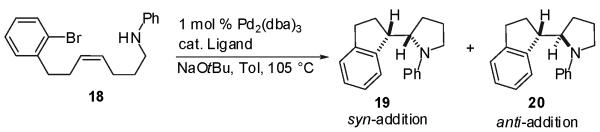
Having demonstrated the feasibility of intramolecular carboetherification reactions, we sought to determine whether or not intramolecular carboamination reactions could also be achieved. Thus, the Z-alkene substrate 18 bearing a tethered aniline moiety was prepared and treated with a catalytic amount of Pd2(dba)3 and several different phosphine ligands under reaction conditions similar to those describedabove (Table 2). Interestingly, in contrast to the results obtained in cyclizations of alcohol-containing substrate 6, all catalysts examined for the cyclization of 18 provided selectivity for formation of (1S*,2S*)-stereoisomer 19, which derives from syn-addition across the Z-alkene. As observed in the related transformations of substrates bearing alcohol nucleophiles, the major side products formed in these reactions result from debromination of the starting material or intramolecular Heck-arylation. Use of PCy3·HBF4 (entry 7) provided the optimal results for this transformation (88% yield, >20:1 dr), and effectively suppressed the formation of both side products.
Table 2.
 | |||
|---|---|---|---|
| entry | ligand | dr(b) | Isolated Yield (19 + 20) |
| 1 | (±)-BINAP | 1:1 | 46% |
| 2 | DPE-Phos | 15:1 | 49% |
| 3 | DPPE | 16:1 | 54% |
| 4 | P(o-tol)3 | 8:1 | 47% |
| 5 | Xantphos | 12:1 | 57% |
| 6 | P[(p-MeO)C6H4]3 | 13:1 | 68% |
| 7 | PCy3·HBF4 | >20:1 | 88% |
Conditions: 1.0 equiv 18, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % ligand (monophosphines) or 2 mol % ligand (bis-phosphines), toluene (0.14 M), 105 °C, 3-10 h.
Diastereoselectivities were determined by GC and/or 1H NMR analysis of crude reaction mixtures.
The intramolecular carboamination reactions also proved to be stereospecific, as E-alkene derivative 21 underwent cyclization under optimized conditions to afford (1S*,2R*)-diastereomer 20, the product of syn-addition across the E-alkene, in 71% yield with >20:1 dr (eq 5). As observed with the analogous alcohol substrates, the chemical yield obtained in the reaction of the E-alkene was slightly lower than the yield of the reaction of Z-alkene 18.
 |
(5) |
Reactions involving aniline-bearing substrates that are branched at C-1 proceeded with complete syn-selectivity. For example, treatment of 22 with catalytic Pd2(dba)3/PCy3·HBF4 afforded product 23 in 55% yield and 5:1 diastereoselectivity (eq 6). Interestingly, the major product diastereomer was found to possess 2,5-trans-disubstitution around the pyrrolidine ring; the minor diastereomer results from syn-addition with cis-pyrrolidine formation.24 In contrast, the analogous intermolecular carboamination reactions of γ-(N-arylamino)alkenes have been shown to provide generally high selectivity for the formation of 2,5-cis-disubstituted pyrrolidine products.7c The reaction of Z-alkene 24 proceeded in high yield (81%), but modest diastereoselectivity (2:1 dr) favoring product 25, which results from syn-addition across the alkene with cis-stereochemistry around the pyrrolidine ring (eq 7). As seen in the reaction of 22, both observed diastereomers resulting from the cyclization of 24 derive from syn-addition, but differ in their relative stereochemistry about the pyrrolidine ring.24
 |
(6) |
 |
(7) |
Discussion
Reactivity and Stereoselectivity
The intramolecular palladium-catalyzed carboetherification and carboamination of γ-hydroxy- or γ-aminoalkenes with tethered aryl bromides provides a new, stereoselective means to generate carbocycles bearing attached heterocycles. These reactions effect the conversion of acyclic precursors to bicyclic products with formation of two bonds, two rings, and two stereocenters in a single step. The yields and selectivities are dependent on a number of factors, but under optimal conditions the desired products are obtained in moderate to good yields with good to excellent stereocontrol.
Several trends in reactivity have been observed during the course of these experiments. From the results described above, it appears that the chemical yield is affected by the alkene geometry, the nature of the heteroatom, and the degree of substitution adjacent to the heteroatom. For example, substrates bearing Z-alkenes are usually transformed in higher chemical yield than the analogous E-alkenes. In addition, reactions involving starting materials with tethered aniline nucleophiles produce higher yields than analogous reactions of alcohol-bearing substrates (with a similar degree of substitution). Finally, in many cases substitution next to the heteroatom decreases chemical yield.
The stereochemical outcome of these reactions is influenced by both the nature of the heteroatom and the catalyst. All substrates containing nitrogen nucleophiles that were examined in these studies are selectively converted to products resulting from syn-addition across the double bond. Although the catalyst structure has an effect on the diastereoselectivity of the pyrrolidine-forming reactions, with diastereomeric ratios varying from 1:1 to >20:1, no catalyst examined led to selective formation of the anti-addition product. In contrast, primary and tertiary alcohol substrates were transformed to products of syn-addition when catalysts bearing electron-rich monodentate ligands such as PCy3, PMe3, or P[(MeO)C6H4]3 were employed, but reactions of these starting materials gave products of anti-addition when chelating phosphines with small bite angles (e.g. BINAP, DPP-Benzene, or DPPE; bite angles ≤ 93°) were used as supporting ligands. Notably, in carboetherification reactions of primary and tertiary alcohols either product diastereomer could be obtained selectively from either starting alkene stereoisomer.
Reactions involving α-substituted alcohols catalyzed by Pd/PCy3 or P[(MeO)C6H4]3 proceed with good to excellent levels of selectivity for the formation of one of four possible diastereomeric products; the major diastereomer results from syn-addition with trans-thf formation. However, use of Pd/BINAP with these substrates leads to low yields and modest selectivities. The analogous Pd/PCy3 catalyzed reactions of α-branched anilines proceed in good yields with very high syn-selectivity, but give mixtures of cis- and trans-pyrrolidine products.
Mechanism of Intramolecular Carboetherification Reactions that Provide syn-Addition Products
In our previous studies on intermolecular Pd-catalyzed carboetherification and carboamination reactions we obtained results that were most consistent with a mechanistic pathway involving syn-insertion of the alkene moiety into the Pd-heteroatom bond of intermediate Pd(Ar)(OR) or Pd(Ar)(NRR’) complexes (e.g. 1).7b,d Other mechanistic scenarios such as product formation via a Wacker-type anti-addition or via intermolecular carbopalladation followed by C-O or C-N bond-forming reductive elimination were ruled out on the basis of the observed product stereochemistry, the side products formed in the reactions, and the high regioselectivity for formation of 5-membered ring products. The fact that syn-addition products are also obtained in the intramolecular reactions suggests that a similar mechanism may operate. However, this pathway would necessitate the formation of 11-membered ring intermediates (e.g. 3), which may be entropically unfavorable, and would require transannular alkene insertion of a macrocyclic palladacycle bearing an internal alkene, which is unprecedented.26 Thus, two plausible mechanisms for the formation of the observed syn-addition products of intramolecular reactions merit consideration.

As shown in Scheme 4, oxidative addition of the aryl bromide 26 to the LnPd(0) species generated in situ from Pd2(dba)3 and a monodentate phosphine ligand, followed by coordination of the tethered alkene would afford the 16-electron intermediate 27, a species common to both mechanistic scenarios. This intermediate could potentially be converted to the observed products via a Heck-type 5-exo carbopalladation26a and deprotonation by NaOtBu to provide 28 (Path A), which could undergo nucleophilic displacement of the bromide by the tethered alkoxide to afford 29.27 An unprecedented sp3-carbon-oxygen bond-forming reductive elimination from Pd(II)-complex 29 (with retention of configuration) would generate the observed product 30.28,29 However, the fact that the cyclizations of secondary alcohol substrates 14 and 16 proceed with good to excellent stereoselectivity for trans-tetrahydrofuran formation and syn-addition suggests that this pathway is much less likely than the mechanism shown in Path B (see below).30 If the reactions proceed via the mechanism outlined in Path A, the 5-exo carbopalladation event would determine both the syn/anti-stereochemistry as well as the stereochemistry around the thf-ring. If the stereochemistry-determining event occurs via intramolecular carbopalladation of intermediate 27 with no communication between the alcohol and the metal, it is unlikely that high selectivity for trans-thf formation would be observed.31 In addition, Heck-type side products that would result from slow reductive elimination following the 5-exo carbopalladation are not formed in significant amounts in optimized reactions that lead to syn-addition.
Scheme 4.

In contrast, the mechanistic pathway outlined in Scheme 4, Path B, which is analogous to that proposed for the intermolecular reactions,7b,d can account for both high syn-selectivity, and high selectivity for trans-thf formation. In this scenario, the 16-electron intermediate 27 could undergo deprotonation followed by associative ligand substitution32 of the alkoxide for the bromide27 to afford eleven-membered palladacycle 31 in which the alkyl group is oriented in a pseudoequatorial position.33 Formation of the eleven-membered palladacycle 31 is presumably facilitated by complexation of the alkene to the metal, which allows for Pd-O bond formation via an effectively smaller ring.30 Transannular insertion of the alkene into the palladium-oxygen bond would generate 32, which could undergo C-C bond-forming reductive elimination to form the observed product 30. Pseudoequatorial orientation of the R-substituent in the transition state for alkene insertion would lead to the observed products bearing trans-2,5-disubstituted tetrahydrofuran groups, and should be lower in energy than the related transition state with axial orientation of the R-group due to developing transannular interactions. The side products that comprise the remainder of the mass balance in these reactions result from oxidation of the alcohol with concomitant reduction of the aryl bromide, and are consistent with competing β-hydride elimination of 31 prior to alkene insertion. Although the experiments described in this paper cannot rule out product formation via alkene insertion into the Pd-C bond of macrocycle 31, literature precedent suggests that alkene insertion into the metal-heteroatom bond of late-metal M(R)(OR) complexes is usually more facilethan insertion into the M-C bond.7,34,35
Mechanism of Intramolecular Carboetherification Reactions that Provide anti-Addition Products
In contrast to all previously described intermolecular carboetherification reactions of internal alkenes with aryl bromides,7a-b,36,37 which provide syn-addition products, the intramolecular carboetherifications provide products resulting from anti-addition when chelating ligands with small bite angles (e.g. BINAP or DPP-Benzene) are employed. Formation of anti-addition products via syn-insertion followed by reversible β-hydride elimination/reinsertion/σ-bond rotation processes that are occasionally observed in the intermolecular transformations7b does not appear to be likely in the intramolecular reactions (Scheme 5), as this pathway would require a 7-endo-hydridopalladation of an aryl(hydrido)palladium alkene complex (33), which is energetically unfavorable.38 In addition, side products that would result from reductive elimination of 33 prior to 7-endo-hydridopalladation are not observed; the major side products in reactions that afford anti-addition products derive from intramolecular Heck-type reactions (see below).
Scheme 5.

Instead, it appears likely that the change in observed product stereochemistry arises from a fundamental change in the mechanism through which the products are formed. This change can be attributed to the notion that tightly chelating ligands are expected to inhibit the associative ligand substitution process required for palladium-alkoxide formation.32 As outlined in Scheme 6, Path A, oxidative addition of the aryl bromide 26 to a (L-L)Pd(0) complex followed by alkene coordination would generate 18-electron complex 34. This coordinatively saturated complex could not be converted to a palladium(aryl)(alkoxide) analogous to 31 via an associative ligand substitution process, and it is unlikely that the 11-membered palladacycle 31 would be generated in the absence of alkene coordination, as 11-membered ring formation is entropically unfavorable. Thus, conversion of 34 to 31 is likely to be relatively slow.39 Instead, a mechanistic pathway involving a Wacker-type anti-alkoxypalladation may be more accessible in this system than in other carboetherification processes. The anti-alkoxypalladation of 34 via an ordered transition state such as 35 could generate 36, which would provide anti-addition product 37 upon C-C bond-forming reductive elimination. The stereochemistry around the tetrahydrofuran ring would again be dictated by nonbonding interactions in the transition state, with a preference for pseudoequatorial orientation of substituents. However, prior studies have shown that differences in transition state energies for pseudoaxial vs. pseudoequatorial orientation of the α-substituents or the alkene moiety in related Wacker-type cyclizations may be relatively small, as the preference for trans-thf formation is often modest in the absence of additional substituents.9,37
Scheme 6.

Alternatively, the products of anti-addition could derive from Heck-type carbopalladation of 34 to generate 38, followed by C-O bond-forming reductive elimination through an SN2 mechanism, which would lead to inversion of the C2 stereocenter to provide 37. Although we cannot rule out product formation via this mechanism, we currently favor the hypothesis involving a Wacker-type pathway, as the sp3C-O bond-forming reductive elimination required for product formation via Path B is not known to occur from Pd(II) complexes,28,29 and would require SN2 substitution to occur at a hindered secondary carbon atom flanked by an adjacent secondary carbon stereocenter. The fact that Heck-type side products (e.g 39) are generated in these transformations suggests that complex 38 is kinetically accessible, but mechanistic pathway B cannot account for control of relative stereochemistry around the tetrahydrofuran ring. The observed selectivity for formation of trans-disubstituted tetrahydrofuran products in the Pd/BINAP catalyzed reaction of 16 is more consistent with an ordered transition state similar to 35. Thus, although 38 may be accessible under the reaction conditions, it is not necessarily an intermediate along the pathway between 34 and 37.
Mechanism and Stereochemistry of Intramolecular Carboamination Reactions
The intramolecular carboamination reactions described above provide products that derive from syn-addition of the amine and the arene across the carbon-carbon double bond. It is likely that these transformations proceed via a mechanism similar to that outlined above (Scheme 4) for syn-addition reactions of alcohol substrates, which involves an transannular alkene insertion of an 11-membered palladium(aryl)(amido) intermediate through transition state 40 or 41. However, in reactions of substrates bearing tethered anilines with α-stereocenters, the factors leading to a preference for either trans-pyrrolidine stereochemistry (E-alkenes) or cis-pyrrolidine stereochemistry (Z-alkenes) are more complicated than in the related transformations of secondary alcohols, and the origin of the observed stereochemistry is not entirely clear. One possible explanation of these results is that pseudoequatorial orientation of the C1-R-group (40) would minimize developing transannular interactions in the transition state,40 but could lead to developing A1,3-strain between the Nsp2-aryl group and the pseudoequatorial R-substituent.41 Alternatively, pseudoaxial orientation of the R-group (41) may minimize allylic strain at the expense of increased unfavorable transannular interactions. The results of the experiments shown in eq 6-7 are consistent with preferred pseudoequatorial orientation of the substituent when the substrate contains the E-alkene geometry, which suggests the energetic effects of the transannular interactions outweigh the effects of A(1,3)-strain for this system. In contrast, there appears to be a slight preference for pseudoaxial orientation with the Z-alkene geometry, which implies the transannular interactions may be lessened relative to the degree of allylic strain in this case.42

In contrast to Pd/BINAP or Pd/DPP-Benzene catalyzed reactions of alcohol substrates, which provide good selectivity for products of anti-addition, use of BINAP or DPP-Benzene with substrates bearing aniline nucleophiles results in low syn/anti stereoselectivity. The differences in the stereochemical outcome of these transformations may be due to the effect of heteroatom nucleophilicity (anionic alkoxide vs. neutral amine) on the rate of anti-heteropalladation. In the presence of NaOtBu there is likely a significant equilibrium concentration of highly nucleophilic alkoxides that result from deprotonation of the substrate alcohol.43 In contrast, deprotonation of the less acidic aniline nucleophile would occur to a lesser extent, hence the concentration of anilide anion is likely to be low, which may result in relatively slower rates of Wacker-type addition of the nitrogen nucleophiles. The fact that yields of anti-addition products decrease (under identical conditions) with increasing steric bulk of the alcohol nucleophile is consistent with this notion.23,44
Conclusions
In conclusion, we have demonstrated the feasibility of a new strategy for the stereoselective construction of products bearing two attached rings via Pd-catalyzed carboetherification and carboamination reactions. Depending on the structure of the catalyst and the substrate, selectivity for either syn-addition or anti-addition is observed. The products of syn-addition appear to derive from unprecedented transannular alkene insertions of unusual 11-membered Pd(Ar)(YR) intermediates. In contrast, the products of anti-addition are consistent with reaction via a different mechanistic pathway that may involve Wacker-type anti-heteropalladation of the alkene. These studies represent the first examples of phosphine ligand-control of syn-insertion vs. anti-addition pathways in catalytic reactions involving olefin oxypalladation, which allows for stereoselective construction of either of two possible product diastereomers from a given alcohol substrate. Further studies on the scope and synthetic applications of this new methodology are currently underway.
Supplementary Material
Acknowledgment
The authors thank the NIH-NIGMS (GM-076150) and the University of Michigan for financial support of this work. JPW acknowledges the Camille and Henry Dreyfus Foundation for a New Faculty Award, Research Corporation for an Innovation Award, and Eli Lilly for a Grantee Award. The authors also thank Amgen, 3M, and Eli Lilly for additional unrestricted support.
References and Notes
- 1.X-ray structure analysis was performed by Jeff W. Kampf.
- 2.For examples of heterocycles bearing attached heterocycles, see:Bermejo A, Figadere B, Zafra-Polo M-C, Barrachina I, Estornell E, Cortes D. Nat. Prod. Rep. 2005;22:269–303. doi: 10.1039/b500186m.Faul MM, Huff BE. Chem. Rev. 2000;100:2407–2474. doi: 10.1021/cr940210s.Hoppe R, Scharf H-D. Synthesis. 1995:1447–1464.Koert U. Angew. Chem., Int. Ed. 1995;34:298–300.
- 3.For examples of heterocycles bearing attached carbocycles, see:Ukiya M, Akihisa T, Yasukawa K, Kasahara Y, Kimura Y, Koike K, Nikaido T, Takido M. J. Agric. Food Chem. 2001;49:3187–3197. doi: 10.1021/jf010164e.Spinella A, Mollo E, Trivellone E, Cimino G. Tetrahedron. 1997;53:16891–16896.Rasmusson GH, Reynolds GF, Steinberg NG, Walton E, Patel GF, Liang T, Cascieri MA, Cheung AH, Brooks JR, Berman C. J. Med. Chem. 1986;29:2298–2315. doi: 10.1021/jm00161a028.Arthur HR, Ko PDS. Phytochemistry. 1974;13:2551–2557.
- 4.For representative examples of other strategies that generate attached rings via C-C bond-forming reactions between cyclic precursors, see:Lemieux RM, Meyers AI. J. Am. Chem. Soc. 1998;120:5453–5457.Lebsack AD, Overman LE, Valentekovich RJ. J. Am. Chem. Soc. 2001;123:4851–4852. doi: 10.1021/ja015802o.
- 5.a) For attached-ring synthesis via pincaol-terminated Prins cyclizations, which generate two stereocenters and one ring in a single step see:Overman LE, Pennington LD. Can. J. Chem. 2000;78:732–738.b) For attached-ring synthesis via intramolecular 1,3-dipolar cycloaddition followed by Mo-catalyzed ring cleavage, see:Tranmer GK, Tam W. Org. Lett. 2002;4:4101–4104. doi: 10.1021/ol026846k.c) For attached ring synthesis via Mn(III)-promoted oxidative cyclization reactions of malonates bearing tethered unsaturated alcohols, see:Hulcoop DG, Sheldrake HM, Burton JW. Org. Biomol. Chem. 2004;2:965–967. doi: 10.1039/b402411g.
- 6.Both the intermolecular and intramolecular carboetherification/carboamination reactions have an intramolecular component, as the heteroatom is tethered to the alkene in both cases. In this manuscript, the term “intermolecular carboetherification/carboamination” is used to describe the reaction between an aryl bromide and a γ-unsaturated alcohol/amine that are not tethered, whereas the term “intramolecular carboetherification/carboamination” describes reactions in which the γ-unsaturated alcohol/amine are tethered to the aryl bromide.
- 7.a) Wolfe JP, Rossi MA. J. Am. Chem. Soc. 2004;126:1620–1621. doi: 10.1021/ja0394838. [DOI] [PubMed] [Google Scholar]; b) Hay MB, Wolfe JP. J. Am. Chem. Soc. 2005;127:16468–16476. doi: 10.1021/ja054754v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ney JE, Wolfe JP. Angew. Chem., Int. Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; d) Ney JE, Wolfe JP. J. Am. Chem. Soc. 2005;127:8644–8651. doi: 10.1021/ja0430346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For recent examples of stoichiometric Cu-promoted carboamination reactions, see:Sherman ES, Chemler SR, Tan TB, Gerlits O. Org. Lett. 2004;6:1573–1575. doi: 10.1021/ol049702+.
- 9.Balme has described elegant studies on related intramolecular reactions of alkenes bearing a bromoaryl group tethered to one sp2carbon and a malonate derivative tethered to the other. These reactions proceed via Wacker-type trans-carbopalladation of intermediate six-membered arylpalladium alkene complexes to afford products resulting from anti-addition across the C-C double bond. See:Bruyère D, Bouyssi D, Balme G. Tetrahedron. 2004;60:4007–4017.
- 10.For intramolecular anti-addition reactions of alkynes bearing an alkyl(bromoaryl) group tethered to one end and an alkyl carboxylic acid to the other see:Cavicchioli M, Bouyssi D, Gore J, Balme G. Tetrahedron Lett. 1996;37:1429–1432.Bouyssi D, Balme G. Synlett. 2001:1191–1193.
- 11.For examples of Pd-catalyzed N-arylation reactions that likely proceed through macrocyclic palladium(aryl)(amido) complexes, see:Beletskaya IP, Bessmertnykh AG, Averin AD, Denat F, Guilard R. Eur. J. Org. Chem. 2005:281–305.and references cited therein.
- 12.In our previous studies we have noted that the large majority of products formed in intermolecular carboetherification and carboamination reactions of internal alkenes derive from syn-insertion of the alkene into the Pd-heteroatom bond of intermediate Pd(Ar)(OR) or Pd(Ar)(NRR’) complexes. See references 4b and 4d.
- 13.For examples of intramolecular anti-carboacetoxylation reactions of alkynes, which proceed through a Wacker-type mechanism, see:Bouyssi D, Balme G. Synlett. 2001:1191–1193.
- 14.a) Through a series of deuterium labeling studies, Hayashi has demonstrated that the Pd(II)-catalyzed oxidative cyclization of an o-allylphenol derivative proceeds via anti-alkoxypalladation in the presence of LiCl, and via syn-alkoxypalladation in the absence of LiCl. However, one of the stereocenters formed in these transformations is destroyed by the β-hydride elimination step that terminates the catalytic cycle. Thus, in the absence of labeled substrates both transformations would provide identical products. See:Hayashi T, Yamasaki K, Mimura M, Uozumi Y. J. Am. Chem. Soc. 2004;126:3036–3037. doi: 10.1021/ja031946m.b) Stoltz has recently described experiments analogous to Hayashi’s deuterium labeling studies that provide further evidence for an accessible syn-oxypalladation pathway in Wacker-type cyclizations of unsaturated alcohol derivatives. See:Trend RM, Ramtohul YK, Stoltz BM. J. Am. Chem. Soc. 2005;127:17778–17788. doi: 10.1021/ja055534k.c) For additional studies on the effect of chloride ion concentration on the mechanistic/stereochemical pathway of the Wacker-oxidation, see:Hamed O, Thompson C, Henry PM. J. Org. Chem. 1997;62:7082–7083. doi: 10.1021/jo971051q.and references cited therein.
- 15.Previously described Wacker-type cyclizations of alkenes bearing tethered heteroatoms that afford tetrahydrofuran or pyrrolidine products generally proceed via a Pd(II)-Pd(0) catalytic cycle. The mechanism of these transformations involves complexation of the alkene to Pd(II) followed by nucleophilic attack of the tethered heteroatom and β-hydride elimination to generate the heterocyclic product. The resulting Pd(H)(X) complex undergoes reductive elimination of HX to provide a Pd(0) complex, which is then reoxidized to Pd(II) by an added oxidant. Catalysts employed for these reactions generally contain halide, carboxylate, or amine ligands rather than phosphine ligands. For further details, see: a) reference 14;Semmelhack MF, Bodurow C. J. Am. Chem. Soc. 1984;106:1496–1498.Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J. Org. Chem. 1997;62:2113–2122. doi: 10.1021/jo961988b.
- 16.(±)-BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; Dpp-benzene = 1,2-bis(diphenylphosphino)benzene.
- 17.Hay MB, Hardin AR, Wolfe JP. J. Org. Chem. 2005;70:3099–3107. doi: 10.1021/jo050022+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Further optimization experiments revealed that use of bases such as triethylamine and potassium carbonate did not afford the desired cyclization products.
- 19.Complete details on the synthesis of all substrates is provided in the Supporting Information.
- 20.Aldehyde products derived from oxidation of the primary alcohol moiety were not observed, but it is likely that these side products are not stable under the reaction conditions.
- 21.See the Supporting Information for complete details of stereochemical assignments.
- 22.Basic trialkylphosphine ligands such as tricyclohexylphosphine and trimethylphosphine were employed in the form of their air-stable tetrafluoroborate salts. These ligands (and all other reagents employed in these reactions) were weighed and handled in air. See:Netherton MR, Fu GC. Org. Lett. 2001;3:4295–4298. doi: 10.1021/ol016971g.
- 23.The Pd2(dba)3/BINAP catalyst system also provided the anti-addition product as the major diastereomer, albeit in low yield. Use of BINAP as ligand for the cyclization of 9 gave 35% yield (3:1 dr) of 7, and the cyclization of 11 proceeded to afford 24% yield (>20:1 dr) of 12 under similar conditions.
- 24.Only the major product diastereomer is shown in eq 2-4 and 6-7. The structures of the other minor diastereomers are shown in the Supporting Information.
- 25.The products were isolated in 40% yield as a 17:2:1:1 mixture of diastereomers. However, analysis of the crude reaction mixture indicated that the diastereomers were formed in a 9:2:1:1 ratio, suggesting that partial resolution of the diastereomers occurred upon chromatographic purification.
- 26.For examples of transannular alkene insertion reactions of intermediates bearing exocyclic alkylpalladium moieties and/or exocyclic alkenes, see:Link JT. Org. React. 2002;60:157–534.(b) For representative examples, see:Hulin B, Newton LS, Cabral S, Walker AJ, Bordner J. Org. Lett. 2004;6:4343–4345. doi: 10.1021/ol048140r.Fox ME, Li C, Marino JP, Jr., Overman LE. J. Am. Chem. Soc. 1999;121:5467–5480.
- 27.For studies on the formation of Pd(Ar)(OR) and Pd(Ar)(NR2) complexes, see:Yamashita M, Cuevas Vicario JV, Hartwig JF. J. Am. Chem. Soc. 2003;125:16347–16360. doi: 10.1021/ja037425g.and references cited therein;Widenhoefer RA, Buchwald SL. J. Am. Chem. Soc. 1998;120:6504–6511.and references cited therein.
- 28.No examples of sp3 C-O bond-forming reductive elimination from Pd(II) complexes have been previously described. One transformation that may involve sp3 C-N bond-forming reductive elimination from a Pd(II) complex with no syn-β-hydrogen atoms has been described by Stahl. See:Brice JL, Harang JE, Timokhin VI, Anastasi NR, Stahl SS. J. Am. Chem. Soc. 2005;127:2868–2869. doi: 10.1021/ja0433020.For examples of related sp3-carbon-heteroatom bond forming reductive elimination from Pd(IV), Pt(IV), and Ni(III), see:Lin BL, Clough CR, Hillhouse GL. J. Am. Chem. Soc. 2002;124:2890–2891. doi: 10.1021/ja017652n.and references cited therein;Desai LV, Hull KL, Sanford MS. J. Am. Chem. Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c.Williams BS, Goldberg KI. J. Am. Chem. Soc. 2001;123:2576–2587. doi: 10.1021/ja003366k.Stahl SS, Labinger JA, Bercaw JE. Angew. Chem., Int. Ed. 1998;37:2180–2192. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2180::AID-ANIE2180>3.0.CO;2-A.
- 29.Although related sp2C-O bond forming reductive elimination is well established, reductive elimination in aryl sp2-systems has been demonstrated to proceed via an addition/elimination mechanism similar to nucleophilic aromatic substitution, which is not an accessible pathway for reductive elimination to form sp3C-O bonds. For details, see reference 27b.
-
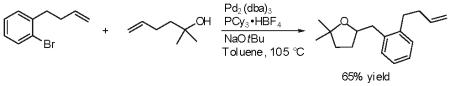
30.Palladium alkoxide formation and alkene 5-exo-insertion into a Pd-O bond appears to be kinetically faster than 5-exo carbopalladation under our optimal reaction conditions. As shown below, treatment of 1-bromo-2-(but-3-enyl)benzene with 2-methylhex-5-en-2-ol with a catalytic amount of Pd2(dba)3/PCy3 in the presence of NaOtBu afforded only the 2-benzyltetrahydrofuran derivative. Products derived from 5-exo-Heck cyclization of the aryl bromide were not observed.
- 31.A mechanism involving fast and reversible carbopalladation followed by selective Pd-heteroatom bond-formation of one possible diastereomeric intermediate could also account for the high diastereoselectivity. However, reversible carbopalladation reactions have only been observed in complexes lacking β-hydrogen atoms; carbopalladation is believed to be irreversible in most other systems. The fact that oxidized/reduced side products are observed instead of Heck-type side products also suggests this pathway is less plausible than Path B. For lead references on reversible carbopalladation, see:Campora J, Gutierrez-Puebla E, Lopez JA, Monge A, Palma P, del Rio D, Carmona E. Angew. Chem., Int. Ed. 2001;40:3641–3644. doi: 10.1002/1521-3773(20011001)40:19<3641::aid-anie3641>3.0.co;2-9.Catellani M, Frignani F, Rangoni A. Angew. Chem., Int. Ed. 1997;36:119–122.
- 32.The vast majority of ligand substitutions at 16-electron Pd(II) complexes proceed via an associative mechanism. The very rare examples of dissociative ligand substitution at 16-electron Pd(II) complexes involve the extremely bulky, monodentate, and unusually labile ligands tris(2,4,6-trifluoromethylphenyl)phosphine and P(o-tol)3. See:Bartolome C, Espinet P, Martin-Alvarez JM, Villafane F. Eur. J. Inorg. Chem. 2004:2326–2337.Louie J, Hartwig JF. J. Am. Chem. Soc. 1995;117:11598–11599.
- 33.Although formation of eleven membered rings is usually entropically unfavorable, formation of the eleven-membered palladacycle 31 is presumably facilitated by complexation of the alkene to the metal, which brings the heteroatom closer to the complex.
- 34.a) Villanueva LA, Abboud KA, Boncella JM. Organometallics. 1992;11:2963–2965. [Google Scholar]; b) Bryndza HE. Organometallics. 1985;4:406–408. [Google Scholar]
- 35.For other recent examples of syn-alkene insertion into late transition metal-heteroatom bonds, see:Helaja J, Göttlich R. J. Chem. Soc., Chem. Commun. 2002:720–721. doi: 10.1039/b201209j.Tsutsui H, Narasaka K. Chem. Lett. 1999:45–46.c) reference 28a.
- 36.Yeh M-CP, Tsao W-C, Tu L-H. Organometallics. 2005;24:5909–5915. [Google Scholar]
- 37.Wacker-type carbonylative carboetherifications described by Semmelhack afford products resulting from anti-addition of an oxygen atom and an ester group across an alkene. See reference 15b.
- 38.7-Endo carbopalladation reactions are extremely rare, and 7-endo hydridopalladation reactions are unknown. For lead references, see:Gibson SE, Guillo N, Middleton RJ, Thuilliez A, Tozer MJ. J. Chem. Soc., Perkin Trans. 1. 1997:447–456.and references cited therein.
- 39.Conversion of 34 to 31 could occur via dissociation of one arm of the chelating ligand followed by associative substitution of alkoxide for bromide, or by dissociation of the bromide followed by associative ligand substitution. Either of these pathways is likely to be higher in energy than the analogous conversion of 27 to 31.
- 40.Dosen-Micovic L, Lorenc L, Mihailovic ML. Tetrahedron. 1990;46:3659–3666. [Google Scholar]
- 41.(a) Hoffmann RW. Chem. Rev. 1989;89:1841–1860. [Google Scholar]; (b) Hart DJ. J. Am. Chem. Soc. 1980;102:397–398. [Google Scholar]; (c) Williams RM, Sinclair PJ, Zhai D, Chen D. J. Am. Chem. Soc. 1988;110:1547–1557. [Google Scholar]
- 42.As noted above, our previously described intermolecular carboamination reactions of terminal alkenes have proceeded with high stereoselectivity for the formation of trans-2,5-disubstituted N-arylpyrrolidines, which is also consistent with favorable pseudoaxial orientation of a C-1 substituent in the absence of medium-ring transannular interactions. However, the effects of E/Z-alkene geometry on the ratio of trans/cis-pyrrolidine pyrrolidine formation has not yet been examined in intermolecular transformations. For further details, see reference 7c.
- 43.The addition of base has been observed to dramatically increase the rate and/or yield of oxidative Wacker-cyclizations of o-allylphenol derivatives. See:Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew. Chem., Int. Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196.Larock RC, Wei L, Hightower TR. Synlett. 1998:522–524.
- 44.The rate and/or yield of Wacker-cyclization reactions that afford tetrahydrofuran products have also been observed to decrease with increasing steric bulk at the C1-position. See:Meulemans TM, Kiers NH, Feringa BL, van Leeuwen PWNM. Tetrahedron Lett. 1994;35:455–458.Hosokawa T, Hirata M, Murahashi S. -i., Sonoda A. Tetrahedron Lett. 1976;17:1821–1824.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.