

Abstract
Molecular profiling of human cancer is complicated by both stromal contamination and cellular heterogeneity within samples from tumor biopsies. In this study, we developed a tissue-processing protocol using mechanical dissociation and flow cytometric sorting that resulted in the respective enrichment of stromal and tumor fractions from frozen pancreatic adenocarcinoma samples. Molecular profiling of DNA from the sorted populations using high-density single nucleotide polymorphism arrays revealed widespread chromosomal loss of heterozygosity in tumor fractions but not in either the stromal fraction or unsorted tissue specimens from the same sample. Similarly, a combination of KRAS mutations and chromosomal copy number changes at key pancreatic cancer loci, such as CDK2NA and TP53, was detected in a substantial proportion of the tumor fractions but not in matched stromal fractions from the same sample. This approach to tissue processing could greatly expand the amount of archived tissue that is available for molecular profiling of human cancer and enable a more accurate diagnosis of genetic alterations in patient samples.
Pancreatic adenocarcinoma is among the 10 most common forms of cancer and is associated with particularly poor prognosis, with median survival from time of diagnosis of 3 to 6 months and an overall 5-year survival rate of less than 5%.1 A primary reason for such poor outcomes is lack of early detection, mainly because of relatively symptom-free early stages of the disease.2 As with all cancer, pancreatic adenocarcinoma is a genetic disease driven by a step-wise accumulation of somatic alterations, including point mutations, chromosome translocations, loss of heterozygosity (LOH), and changes in gene copy number, resulting in the inactivation of tumor suppressor genes and activation of proto-oncogenes.2 The role of a number of known cancer genes in the development of pancreatic adenocarcinoma has been well documented.2,3 Activating mutations of the KRAS oncogene are the most common genetic aberrations and increase in frequency with disease progression, from 30% in early stage disease to 80 to 90% of pancreatic adenocarcinomas.4,5 Cyclin-dependent kinase inhibitor 2A (CDKN2A) encodes two tumor suppressor genes, INK4a (p16) and ARF (p14) that appear to play an important role in pancreatic cancer, as loss of function of CDK2NA either by mutation, deletion or promoter hypermethylation occurs in 80 to 90% of pancreatic adenocarcinomas.2,5 The tumor suppressor gene TP53 has been shown to be either deleted or mutated in 50 to 75% of the pancreatic tumors,6,7 and although not commonly lost in pancreatic cancer, the tumor suppressor PTEN has been shown to exhibit reduced expression8 and to undergo LOH9 in pancreatic adenocarcinoma in some instances. The transforming growth factor-β pathway has also been implicated in the pathenogenesis of pancreatic cancer, primarily through loss of the transcriptional regulator SMAD4/DPC4, an event that occurs in ∼55% of pancreatic cancers and seems to be a later event in the progression of pancreatic cancer.10
In addition to the involvement of known cancer genes, pancreatic cancer is characterized by high levels of aneuploidy and extensive chromosomal instability.11 The molecular basis of such instability is not well understood but a number of studies have implicated mutations in genes associated with kinetochore structure, centrosome formation, and mitotic spindle checkpoint, in contributing to instability of chromosomes.12 The BRCA2 locus, which plays a crucial role in maintenance of genomic stability by regulating homologous recombination-linked DNA repair processes, is occasionally lost in later stage pancreatic adenocarcinomas and may impact the overall level of chromosomal instability.13
Although a number of mutational and chromosomal alterations have been characterized, molecular analyses of pancreatic cancer have been hindered by the low cancer cellularity of this neoplasm and the contaminating influence of euploid stromal cells that make up the complex anatomy and heterogeneous cellularity of pancreatic tissue.14 Indeed, the later stages of pancreatic cancer are typically characterized by a vigorous, nonneoplastic stromal response with malignant cancer cells representing only 25% of the cells in the tumor on average.15,16 The relatively low percentage of neoplastic cells may pose less of a problem for detecting mutated and dominantly acting oncogenes (eg, KRAS), because highly sensitive methods can be deployed to identify lesions in heterogeneous material with very low tumor content,17 but may be a prohibitive issue for detecting patterns of allelic loss in tumor cells. A number of methods for genome-wide screening of genetic aberrations in cancer are now available, and new innovations and methodological improvements have further enhanced the sensitivity of these technologies and have led to the identification of numerous chromosomal regions that may harbor novel genes important for pancreatic cancer pathogenesis.18 The majority of this work has been accomplished in vitro, using pancreatic cell line models, or through laborious methods such as laser capture microdissection (LCM).16 To provide a facile method to allow characterization of genomic aberrations in isolated pancreatic neoplasms, we developed a neoplastic cell enrichment protocol from snap-frozen pancreatic adenocarcinoma resection specimens. A fluorescence-activated cell sorting-based approach has previously been reported to enrich for neoplastic cells using disaggregated breast tumor samples.19 Molecular profiling of enriched neoplastic cells from whole adenocarcinoma tissue samples was used to confirm specific mutation of KRAS in neoplastic but not stromal fractions and has also revealed several regions of LOH that were undetectable in unsorted whole tissue. This method should have applications to the identification of novel genetic alterations in pancreatic cancer and molecular diagnostic applications in terms of providing a means of accurately assessing chromosomal abnormalities in pancreatic tumor samples.
Materials and Methods
Snap-frozen surgically resected samples from patients diagnosed with pancreatic adenocarcinoma were obtained from a commercial vendor and archived in the Genentech Tumor Bank. All samples were subjected to review by a board-certified pathologist (D.A.E.) to confirm diagnosis and assess overall tumor content (Table 1). Fourteen of the sixteen samples were confirmed as adenocarcinoma or ductal adenocarcinoma, although one sample was found to exhibit neuroendocrine differentiation and one sample found to exhibit primarily inflammation and fibrosis with no evident neoplastic cells (Table 1). Before sorting, tissue samples were slowly thawed on ice and then cut into ∼5-mm3 pieces with tweezers and scalpel in a Petri dish on ice. Cut pieces were loaded with 500 ml of phosphate-buffered saline (PBS) into a sterile Medicon chamber, a component of the MediMachine (BD Biosciences, San Jose, CA). Tissue was disaggregated within the chamber by microblades spinning at constant 100 rpm. The optimal time for disaggregation of pancreatic tissue was determined to be 3 minutes. Minced tissue was then aspirated out of the chamber and forced into single cell suspension using a 70-μm syringe filter (BD Biosciences). Cell suspensions were slide-mounted via cytospin to evaluate cellular integrity after tissue dissociation.
Table 1.
Characteristics of Pancreatic Cancer Samples Used in This Study
| Sample number | Profiled on SNP arrays? | Tissue | Tissue diagnosis | Tumor content (%) | Patient diagnosis | Stage | Age (years) | Sex |
|---|---|---|---|---|---|---|---|---|
| HF-18411 | Yes | Pancreas | Adenocarcinoma | 20 | Pancreatic cancer | T4N1M0 (IVA) | 48 | Female |
| HF-18996 | Yes | Pancreas | Adenocarcinoma | 30 | Pancreatic cancer | T2N0M0 | 49 | Female |
| HF-18998 | Yes | Pancreas | Adenocarcinoma | 30 | Pancreatic cancer | T2N0M0 | 50 | Male |
| HF-19000 | Yes | Pancreas | Adenocarcinoma | 10 | Pancreatic cancer | T2N0M0 | 59 | Female |
| HF-16979 | No | Pancreas | Adenocarcinoma ductal | 20 | Pancreatic cancer | pT3pN1pMX (IIB) | 60 | Female |
| HF-18764 | No | Pancreas | Adenocarcinoma | 30 | Pancreatic cancer | T3N0M0 (II) | ||
| HF-16976 | No | Pancreas | Adenocarcinoma ductal | 20 | Pancreatic cancer | pT3pN1bpMX (III) | 64 | Male |
| HF-2035 | No | Pancreas | Adenocarcinoma | 75 | Adenocarcinoma | 72 | Female | |
| HF-18409 | No | Pancreas | Adenocarcinoma | 30 | Pancreatic cancer | T3N0M0 (II) | 63 | Female |
| HF-16359 | No | Pancreas | Adenocarcinoma ductal | 10 | Pancreatic cancer | T 3N0M0R1 (II) | 66 | Female |
| HF-16349 | No | Pancreas | Adenocarcinoma ductal | 10 | Pancreatic cancer | T3N1M0R 0 (III) | 50 | Male |
| HF-16345 | Yes | Pancreas | Adenocarcinoma ductal | 20 | Pancreatic cancer | T4N 1bM0R0 (IVA) | 75 | Female |
| HF-16975 | No | Pancreas | Adenocarcinoma ductal | 10 | Pancreatic cancer | pT3pN1pMX (IIB) | 69 | Female |
| HF-16980 | Yes | Pancreas | Adenocarcinoma ductal | 10 | Pancreatic cancer | pT3pN1pMX (IIB) | 68 | Female |
| HF-17928 | No | Pancreas | Carcinoma, neuroendocrine | 90 | Pancreatic cancer | T3N0M0 (II) | 79 | Male |
| HF-17700 | No | Pancreas | Inflammation and fibrosis | <5% | Pancreatic cancer | T4N0M0 (IVa) | Male |
Cytokeratin expression is a common marker of adenocarcinomas and so a pan-cytokeratin antibody was chosen to define tumor cell populations. In preparation for flow cytometric sorting, cell suspensions were permeabilized by 0.2% Triton X in PBS, and then fixed with 80% ethanol (−20°C). Cells were stained with fluorescein isothiocyanate-conjugated C-11, a pan-cytokeratin mAb (AbCam, Cambridge, MA), then counterstained with DNA-binding 4,6-diamidino-2-phenylindole (DAPI) (Roche, Indianapolis, IN). Sorting of tumor cells [high-fluorescein isothiocyanate (+), high-DAPI (+)] from somatic cells [fluorescein isothiocyanate (−), DAPI (−)] was conducted on a MoFlo cell sorter system (DAKO, Carpinteria, CA).
Genomic DNA (gDNA) was extracted from sorted fractions by the DNEasy kit (Qiagen, Valencia, CA). Amplification of exons 2 to 3 of KRAS was accomplished using nested primers (KRAS exon 2, 10: sense 5′-GCAGTCAACTGGAATTTTCATG-3′, antisense 5′-ACTCATGAAAATGGTCAGAGAAACC-3′, 20: sense 5′-TGTAAAACGACGGCCAGTGTACTGGTGGAGTATTTGATAGTGT-3′, antisense 5′-CAGGAAACAGCTATGACCTGGTCAGAGAAACCTTTATCTG-3′, KRAS exon 3, 10: sense 5′-AACGCATCGATAGCTCTGCCC-3′, antisense 5′-AGGAAGGAAGGGGTTCCCCGTC-3′, 20: sense 5′-TGTAAAACGACGGCCAGTTCGATAGCTCTGCCCTCTGCG-3′, antisense 5′-CAGGAAACAGCTATGACCGGGACCCCTAATTCATTCACT-3′). Amplicons were sequenced in sense and antisense directions using dye-terminator chemistry (Applied Biosystems, Foster City, CA). Subsequent rounds of independent PCR and sequencing reactions confirmed detected KRAS mutations.
The GeneChip mapping assay (Affymetrix, Santa Clara, CA) was performed according to the manufacturer's protocol by the Genentech Microarray Facility on genomic DNA from unsorted tissue, postsort tumor tissue, and postsort stromal tissue from five tumor specimens. Restriction enzyme-digested genomic DNA is PCR-amplified in triplicate, fragmented, and then fluorescent-labeled before loading on Affymetrix Human Map100k array chips. Single nucleotide polymorphism (SNP) data were analyzed using the Affymetrix Chromosome Copy Number Analysis Tool (CNAT) and The Loss of Heterozygosity (LOH) Algorithm in Genotyping Console 2.0, and copy number data were processed using the Affymetrix GSA_CN algorithm. Data were visualized using Spotfire Software (TIBCO, Palo Alto, CA). All SNP array data from pancreatic tumors described in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE14579.
LCM and DNA Extraction
Snap-frozen pancreatic tumor samples were embedded in the cryoprotectant OCT and serial 7-μm sections were cut and mounted on microscope slides. The sections were maintained at −80°C until used for LCM. At the time of processing, frozen sections immediately fixed in 75% ethanol for 30 seconds, nuclease-free water for 30 seconds, stained with HistoGene stain (MDS Analytical Technologies, Sunnyvale, CA) for 20 seconds, washed in nuclease-free water for 30 seconds, followed by sequential dehydration in 70, 95, and 100% ethanol, and a final 5-minute dehydration in xylene. Once air-dried, neoplastic cells and normal cells were identified by a pathologist and microdissected using the Veritas microdissection instrument (MDS Analytical Technologies, Sunnyvale, CA) on to separate CapSure Macro LCM caps using instrument settings recommended by the manufacturer. After microdissection, each cap was incubated overnight with 50 μl of proteinase K using the PicoPure DNA Extraction Kit (MDS Analytical Technologies). After proteinase K inactivation by heating at 95°C for 10 minutes, samples were collected and directly used for PCR and sequencing.
Results
Selection of Tumors and Sorting
Histopathological review and analysis of neoplastic cell content was conducted on a large number of samples representing a diverse group of solid tumor malignancies (Figure 1A). Overall, the pancreatic adenocarcinomas had the lowest percent tumor cell content and thus were selected for tumor enrichment sorting studies. Figure 1 shows examples of samples with high (Figure 1B) and low (Figure 1C) tumor cell content. The majority of pancreatic cancer samples used in this study had an overall neoplastic cell content of less than 30% (Table 1).
Figure 1.
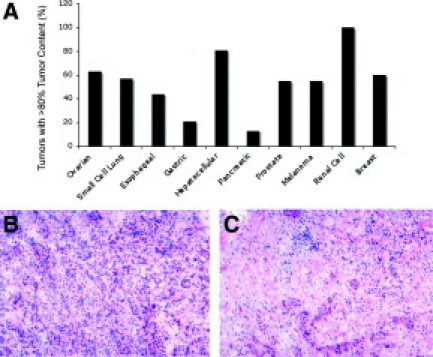
Analysis of tumor content in a range of solid tumor malignancies. A: Histopathological review was conducted on at least 10 samples from a range of tumor types and the percentage of tumors with at least 80% tumor cell content is shown. Gastric and pancreatic cancers were found to have substantially lower tumor content than other tumor types. H&E-stained sections showing relatively high (B) and low (C) tumor cell content sections. Samples similar to that shown in C were used for sorting experiments described in this study.
A key step to sorting tumor from stromal tissue was the selection of a pan-cytokeratin marker specifically expressed in tumor cells but not in surrounding tissues. The C-11 antibody has been shown to have broad specificity for keratins 7, 8, and 18 and hence was ideal for this purpose because keratin 7 is the mostly commonly expressed cytokeratin in pancreatic cancer.20 Immunofluorescence and histopathological analyses were used to show that the pan-cytokeratin antibody C-11 specifically labels neoplastic pancreatic cells but not cells in the surrounding stroma (Figure 2A).
Figure 2.
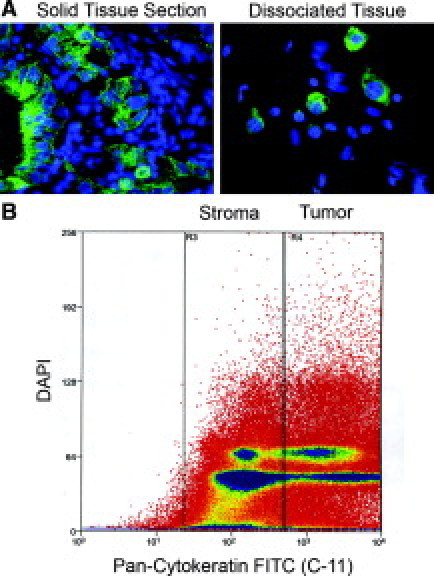
Fluorescence-activated cell sorting tumor sorting data. A: Example of a solid tumor section stained with DAPI (blue) and anti-pan-cytokeratin antibody (C-11) (green) in the left panel and after mechanical dissociation in the right panel. B: Representative flow cytometric sorting data from a pancreatic adenocarcinoma sample. The y axis indicates DAPI staining intensity and the x axis anti-pan-cytokeratin staining. Boxes indicate subpopulations enriched for stromal or tumor cells, as indicated.
Samples were then mechanically dissociated as described in the Materials and Methods and immunofluorescence was used to confirm the presence of intact cells with DAPI-positive nuclei as well as cytokeratin-positive cytoplasmic staining (Figure 2A). Dissociated samples were fixed, stained, and then flow cytometrically sorted into high and low cytokeratin (CK)-staining populations as shown in Figure 2B with the idea that the high CK population comprises neoplasm-enriched tissue and the low CK population comprises stromal-enriched tissue.
Validation of the Sorting Protocol
We next performed a variety of genomic analyses to demonstrate that the sorting protocol was having the desired effect in terms of enriching the population of neoplastic cells relative to stromal cells. First, we profiled genomic DNA from unsorted starting tumor material as well as fractions enriched for tumor or stromal cells on Affymetrix high-density SNP arrays. The rationale was to determine whether copy number alterations (ie, chromosomal gains or losses) or copy neutral LOH that are not apparent in unsorted tumors are detectable in the sorted neoplastic cell fraction. LOH is a common mechanism that contributes to tumorigenesis, primarily by inactivation of tumor suppressor genes, and may be assessed using SNP arrays by identification of stretches of homozygous SNPs unlikely to arise strictly by chance.21 LOH is difficult to evaluate in tumors in the absence of patient-matched nontumor DNA because stromal DNA compromises genotyping accuracy. The LOH scores we used reflect the lengths of contiguous regions of homozygosity and the data are represented in the form of a heatmap with yellow regions indicating contiguous runs of homozygous SNPs. Genome-wide analysis showed multiple contiguous regions of chromosomal LOH across the genome that was detected specifically in the sorted neoplastic fraction but not in the sorted stromal fraction or the starting unsorted tissue (Figure 3). Analysis of specific chromosomal regions clearly showed that LOH in the chromosomal regions spanning the tumor suppressors PTEN (Figure 4, top) and CDK2NA (Figure 4, bottom) can be detected specifically in the tumor fraction but not in the starting tumor material or in the stromal fraction. Recent studies have also shown that high-density SNP arrays can be used to detect genome-wide DNA copy number changes in human cancers,22 including pancreatic ductal adenocarcinomas,23 suggesting that these arrays have utility in identifying recurrently amplified or deleted chromosomal regions that may harbor oncogenes or tumor suppressor genes, respectively. We compared genome-wide copy number gains and losses in sorted stromal and neoplastic fractions and found that chromosomal gains and losses were substantially more apparent in DNA from tumor fractions. Genome-wide analysis showed evidence of chromosomal loss of key tumor suppressors such as CDK2NA, TP53, and SMAD4 with substantial enrichment in neoplastic compared with stromal fractions (Figure 5A). In addition, deletion of a number of other chromosomal regions reported to be altered in pancreatic adenocarcinoma, such as loss of 6q and 4q and gain of 8q and 3q,2 were also apparent predominantly in the neoplastic fraction (Figure 5A). Analysis of smaller chromosomal regions (Figure 5, B and C) identified elevated copy numbers of chromosomal regions harboring the microphthalmia-associated transcription factor (MITF) and cyclin E1 (CCNE1)24 that were apparent in neoplastic but not stromal fractions. Because MITF amplification has not been reported in pancreatic cancer previously, we sought independent confirmation of copy number gains at the MITF locus by examination of pancreatic cell line DNA on Affymetrix SNP arrays because cell lines offer a source of neoplastic DNA free of stromal contamination. We found that a number of pancreatic cell lines showed between 2.5 and 3 copies of MITF in this analysis (see Supplemental Figure S1 at http://jmd.amjpathol.org). Although somewhat lower in magnitude and frequency than we and others observe in melanoma cell lines (see Supplemental Figure S1 at http://jmd.amjpathol.org),24 this finding does support the data obtained from the sorted tumor cells that copy number alterations at the MITF locus may occur in pancreatic adenocarcinomas. Together these results suggest that the sorting protocol can reveal copy number alterations and copy neutral LOH that are not apparent in starting tumor material or surrounding stromal material.
Figure 3.
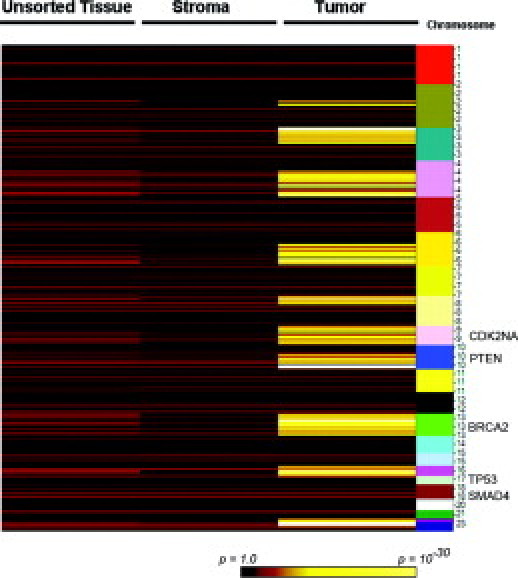
Genome-wide LOH analysis from a single pancreatic adenocarcinoma sample. Heatmap showing regions of normal heterozygosity (red, black) or regions of LOH (yellow) as determined by SNP array analysis. Colors represent the probability that a stretch of homozygous SNPs could arise by chance as indicated in the key. Columns represent data from genomic DNA from unsorted tissue and sorted stromal and tumor (ie, neoplastic) fractions and chromosomal location is indicated to the right. The approximate locations of key tumor suppressor loci in pancreatic cancer are indicated to the right. Widespread LOH is observed specifically in the tumor fraction but not unsorted or stromal fractions.
Figure 4.
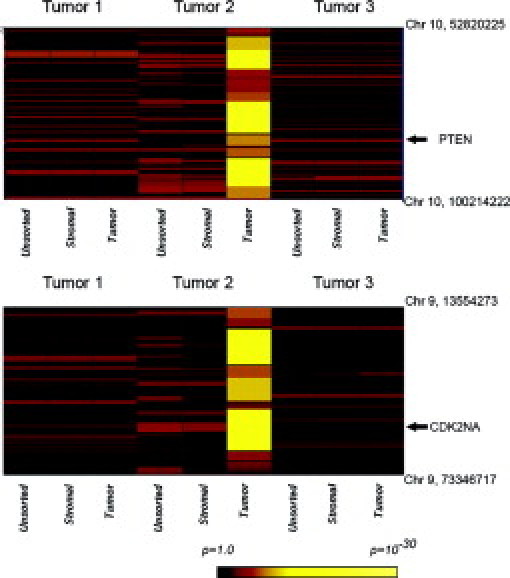
Specific examples of LOH in pancreatic adenocarcinoma samples. Top: Chromosomal region of chromosome 10 that harbors the PTEN tumor gene (coordinates indicated to the right) in unsorted and sorted fractions from three separate tumors. Heatmap colors represent the probability that a stretch of homozygous SNPs could arise by chance as indicated in the key. Bottom: Region of chromosome 9 that harbors the CDK2NA locus. Tumor 2 shows LOH at the PTEN and CDK2NA loci specifically in the neoplastic fraction but not in unsorted tissue or the stromal fraction.
Figure 5.
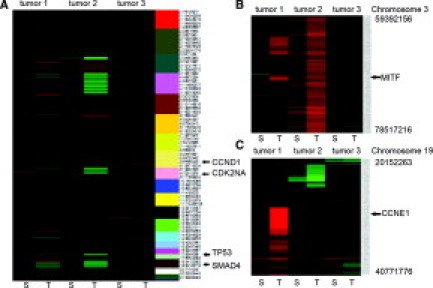
Copy number alterations in sorted tumor and stromal fractions from pancreatic adenocarcinomas. A: Genome-wide patterns of chromosomal gains (red) and losses (green) in sorted stromal (S) and tumor (T) fractions from three tumor samples. The key at the bottom indicates copy number in the heatmap chromosomal location is indicated to the right of the heatmap. The location of several important tumor suppressors in pancreatic cancer are indicated by arrows. B: Analysis of the region on chromosome 3 harboring the MITF transcription factor, with focal amplification apparent in tumor fraction 1 and broader amplification of the region apparent in tumor fraction 2. Chromosomal coordinates are given to the right of the figure. C: Analysis of the region on chromosome 19 harboring CCNE1, with high-level amplification apparent specifically in tumor fraction 1.
Because the KRAS oncogene is frequently mutated in pancreatic adenocarcinoma, we also sought to determine whether our sorting protocol allowed detection of KRAS mutations in sorted neoplastic cells but not stromal cells. KRAS exon 2 mutations were found in 8 of 16 samples screened, and were exclusively detected in neoplastic fractions (Figure 6, Table 2). Samples that were negative for KRAS mutations were clearly wild type at the locus with clear sequence reads and not scored as negative because of a failure of the sequencing reaction (data not shown). A KRAS exon 3 mutation was detected in both neoplastic and stromal fractions of one sample, although apparent enrichment of mutant DNA was observed in the tumor fraction (Figure 6, Table 2). To confirm these results with a more established method, we performed LCM to isolate both neoplastic and normal cells from four of the samples that had been analyzed using our sorting protocol (Table 2). Notably we found the same KRAS alteration in DNA from the captured neoplastic cell population but not the captured stromal cell population in three of the four samples (Table 2), suggesting reasonably good agreement between genetic analysis on material obtained by these two different methods. The failure to detect a KRAS mutation in the LCM-captured neoplastic tissue from sample HF-19000 might potentially be because of the fact that only a subsection of the sample was captured and analyzed. Overall these results suggest that the tumor sorting protocol is successfully enriching for neoplastic cells, because for the most part mutations detected in the neoplastic fraction are not apparent from analysis of the stromal fraction.
Figure 6.
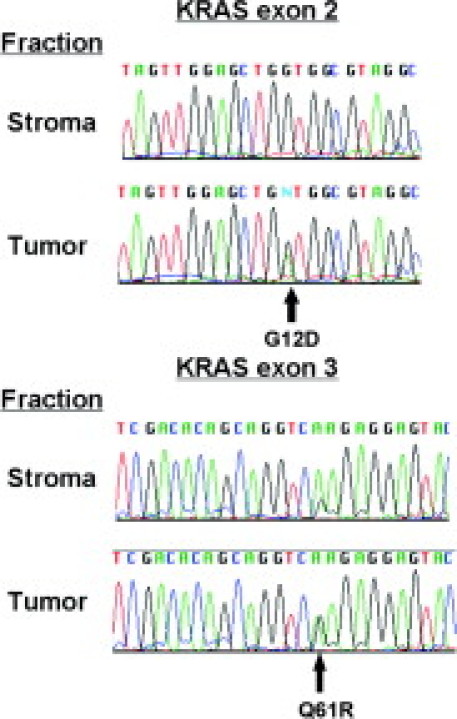
Sequence analysis of the KRAS locus in sorted pancreatic adenocarcinoma tissue. Upper sequence traces show analysis of a region of exon 2 in KRAS with nucleotide change predicted to cause a G12D amino acid substitution apparent in the neoplastic fraction but not the stromal fraction. Lower sequence traces show analysis of a region of exon 3 in KRAS with nucleotide change predicted to cause a Q61R amino acid substitution apparent in the tumor and stromal fractions but substantially enriched in the tumor fraction.
Table 2.
KRAS Mutation Detection in Sorted Tumor Samples
| Sample number | Fraction* | KRAS status, FACS† | KRAS status, LCM† | Tissue diagnosis |
|---|---|---|---|---|
| HF-18411 | T | G12D | G12D | Adenocarcinoma |
| S | WT | WT | ||
| HF-18996 | T | G12D | G12D | Adenocarcinoma |
| S | WT | WT | ||
| HF-18998 | T | G12D | G12D | Adenocarcinoma |
| S | WT | WT | ||
| HF-19000 | T | G12R | WT | Adenocarcinoma |
| S | WT | WT | ||
| HF-16979 | T | G12R | ND | Adenocarcinoma ductal |
| S | WT | ND | ||
| HF-18764 | T | G12R | ND | Adenocarcinoma |
| S | WT | ND | ||
| HF-16976 | T | G12V | ND | Adenocarcinoma ductal |
| S | WT | ND | ||
| HF-2035 | T | Q61R | ND | Adenocarcinoma |
| S | Q61R | ND | ||
| HF-18409 | T | WT | ND | Adenocarcinoma |
| S | WT | ND | ||
| HF-16359 | T | WT | ND | Adenocarcinoma ductal |
| S | WT | ND | ||
| HF-16349 | T | WT | ND | Adenocarcinoma ductal |
| S | WT | ND | ||
| HF-16345 | T | WT | ND | Adenocarcinoma ductal |
| S | WT | ND | ||
| HF-16975 | T | WT | ND | Adenocarcinoma ductal |
| S | WT | ND | ||
| HF-16980 | T | WT | ND | Adenocarcinoma ductal |
| S | WT | ND | ||
| HF-17928 | T | WT | ND | Carcinoma (neuroendocrine) |
| S | WT | ND | ||
| HF-17700 | T | WT | ND | Inflammation |
| S | WT | ND |
CK+ neoplastic tumor fraction (T) or CK− stromal fraction (S).
FACS tumor sorting protocol.
LCM, laser capture microdissection protocol.
Discussion
Tumor cell sorting enabled identification and detailed characterization of genetic changes in pancreatic adenocarcinoma tumors, some of which were difficult or impossible to detect in the original heterogeneous tumor environment. In particular, the method revealed widespread LOH in pancreatic neoplastic fractions that was not apparent in unsorted tissue or sorted stromal material. Whereas previous efforts to separate neoplastic cells from stromal contaminants have required methods such as LCM that require specialized pathology training to identify relevant cell populations,25 the fluorescence-activated cell sorting protocol we describe here is robust, rapid, and provides ample material for genomic characterization.
One somewhat discordant result of our findings compared with previous studies is that we identified a relatively low percentage of KRAS mutations in the samples in our study. Whereas previous reports have identified KRAS mutation frequencies well in excess of 80% of pancreatic adenocarcinomas,4,5 we found mutations in only 8 of the 16 samples we analyzed (50%). There are several possible explanations for this observation. First, as noted in the Results and Table 1, 2 of the 16 samples are not bona fide adenocarcinomas and thus might not be expected to have KRAS mutations. Excluding those samples we identified mutations in 8 of 14 or 57% of samples. This is still low compared with most estimates in the literature, but we note that a January 15, 2009 search of the Sanger COSMIC mutation database http://www.sanger.ac.uk/genetics/CGP/cosmic/) shows a mutation rate of 71% when the search is restricted to pancreatic ductal adenocarcinoma (n = 3404 samples). Thus one interpretation would be that our relatively small sample size has led to sampling error and an underestimate of the true frequency of KRAS mutations. It is also possible that the failure to detect KRAS mutations in some of the samples may be because of the presence of significant amounts of contaminating nonneoplastic epithelial cells in the cytokeratin-positive fraction, because we are sorting based solely on cytokeratin expression and not aneuploidy or some other specific marker of neoplastic cells. Still, the fact that we can identify alterations specifically in the neoplastic fraction but not stromal fraction, and that we have corroborated this finding by using LCM, supports the utility of the fluorescence-activated cell sorting tumor sorting protocol in enrichment of neoplastic tissue for genetic analyses.
We also note that the tumor sorting protocol should have utility in the identification of novel targets and genetic alterations that characterize pancreatic adenocarcinoma that have been obscured by the significant heterogeneity common to this tumor type. Cyclin E1 (CCNE1) is an oncogene that is expressed in the G1 phase of the cell cycle and is involved in initiating DNA synthesis via activation of cyclin-dependent kinases.26 Although CCNE1 has been shown to be overexpressed in a number of tumor types and in two pancreatic cell lines,27,28 amplification has not been reported in pancreatic tumor tissue. We have shown here that amplification of CCNE1 can be detected specifically in sorted tumor material pancreatic adenocarcinoma and suggest that additional confirmatory studies in larger data sets are warranted. Similarly, the microphthalmia-associated transcription factor (MITF) has been shown to be amplified specifically in melanoma and to constitute a lineage-specific oncogene because it can transform primary human melanocytes when transfected in conjunction with BRAF.24 Our data suggest that MITF copy number gains occur in pancreatic tumors and cell lines and thus may not be unique to the biology of melanoma disease progression.
Understanding the pattern of loss of tumor suppressor genes and activation of oncogenes in a particular patient's tumor may also have utility in understanding patient prognosis or selecting targeted therapies in the future. Indeed, previous studies have shown that overall LOH at 15 loci indicate aggressive tumor biology and correlate strongly with survival in resected pancreatic carcinomas,9 and the methods described here offer a facile method for obtaining material for such analyses. In addition, identification of LOH or oncogenic mutations at specific loci may be of key importance in the emerging field of personalized medicine.29 For instance, loss of PTEN function in breast cancer models has recently been shown to predict preclinical response to PI3 kinase inhibitors,30 whereas KRAS mutations have been shown to predict preclinical response to selective inhibitors of MEK.31 Thus the ability to use heterogeneous patient tumor samples to accurately assess chromosomal alterations in PTEN or other tumor suppressor genes, or activating mutations in key oncogenes, may become crucial as novel targeted therapies are clinically validated and associated companion diagnostic tests are used to select patients for therapy. The tumor sorting protocol described herein may facilitate more accurate diagnosis and personalized therapy for patients with pancreatic cancer by providing a clearer picture of the alterations in a given tumor.
Acknowledgements
We thank Fred de Sauvage and the Genentech Cancer Genome Project for access to samples; Zora Modrusan, Somesekar Seshagiri, and Guy Cavet for assistance with molecular characterization and analysis; and Peter Haverty for discussions and assistance in uploading SNP array data to GEO.
Footnotes
Disclosure: All of the authors are current or former (D.A.E.) employees of Genentech, Inc.
Supplemental material for this article can be found on http://jmd.amjpathol.org
Supplementary data
References
- 1.Jemal A, Clegg LX, Ward E, Ries LA, Wu X, Jamison PM, Wingo PA, Howe HL, Anderson RN, Edwards BK. Annual report to the nation on the status of cancer, 1975–2001, with a special feature regarding survival. Cancer. 2004;101:3–27. doi: 10.1002/cncr.20288. [DOI] [PubMed] [Google Scholar]
- 2.Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 3.Baumgart M, Heinmoller E, Horstmann O, Becker H, Ghadimi BM. The genetic basis of sporadic pancreatic cancer. Cell Oncol. 2005;27:3–13. doi: 10.1155/2005/632939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klimstra DS, Longnecker DS. K-ras mutations in pancreatic ductal proliferative lesions. Am J Pathol. 1994;145:1547–1550. [PMC free article] [PubMed] [Google Scholar]
- 5.Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M, Goodman SN, Sohn TA, Hruban RH, Yeo CJ, Kern SE. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997;57:1731–1734. [PubMed] [Google Scholar]
- 6.Coppola D, Lu L, Fruehauf JP, Kyshtoobayeva A, Karl RC, Nicosia SV, Yeatman TJ. Analysis of p53, p21WAF1, and TGF-beta1 in human ductal adenocarcinoma of the pancreas: TGF-beta1 protein expression predicts longer survival. Am J Clin Pathol. 1998;110:16–23. doi: 10.1093/ajcp/110.1.16. [DOI] [PubMed] [Google Scholar]
- 7.Scarpa A, Capelli P, Mukai K, Zamboni G, Oda T, Iacono C, Hirohashi S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol. 1993;142:1534–1543. [PMC free article] [PubMed] [Google Scholar]
- 8.Ebert MP, Fei G, Schandl L, Mawrin C, Dietzmann K, Herrera P, Friess H, Gress TM, Malfertheiner P. Reduced PTEN expression in the pancreas overexpressing transforming growth factor-beta 1. Br J Cancer. 2002;86:257–262. doi: 10.1038/sj.bjc.6600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franko J, Krasinskas AM, Nikiforova MN, Zarnescu NO, Lee KK, Hughes SJ, Bartlett DL, Zeh Iii HJ, Moser AJ. Loss of heterozygosity predicts poor survival after resection of pancreatic adenocarcinoma. J Gastrointest Surg. 2008;12:1664–1672. doi: 10.1007/s11605-008-0577-9. [DOI] [PubMed] [Google Scholar]
- 10.Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH, Kern SE. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 11.Nowak NJ, Gaile D, Conroy JM, McQuaid D, Cowell J, Carter R, Goggins MG, Hruban RH, Maitra A. Genome-wide aberrations in pancreatic adenocarcinoma. Cancer Genet Cytogenet. 2005;161:36–50. doi: 10.1016/j.cancergencyto.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 13.Naderi A, Couch FJ. BRCA2 and pancreatic cancer. Int J Gastrointest Cancer. 2002;31:99–106. doi: 10.1385/IJGC:31:1-3:99. [DOI] [PubMed] [Google Scholar]
- 14.Moore PS, Beghelli S, Zamboni G, Scarpa A. Genetic abnormalities in pancreatic cancer. Mol Cancer. 2003;2:7. doi: 10.1186/1476-4598-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seymour AB, Hruban RH, Redston M, Caldas C, Powell SM, Kinzler KW, Yeo CJ, Kern SE. Allelotype of pancreatic adenocarcinoma. Cancer Res. 1994;54:2761–2764. [PubMed] [Google Scholar]
- 16.Shekouh AR, Thompson CC, Prime W, Campbell F, Hamlett J, Herrington CS, Lemoine NR, Crnogorac-Jurcevic T, Buechler MW, Friess H, Neoptolemos JP, Pennington SR, Costello E. Application of laser capture microdissection combined with two-dimensional electrophoresis for the discovery of differentially regulated proteins in pancreatic ductal adenocarcinoma. Proteomics. 2003;3:1988–2001. doi: 10.1002/pmic.200300466. [DOI] [PubMed] [Google Scholar]
- 17.Bjorheim J, Minarik M, Gaudernack G, Ekstrom PO. Mutation detection in KRAS exon 1 by constant denaturant capillary electrophoresis in 96 parallel capillaries. Anal Biochem. 2002;304:200–205. doi: 10.1006/abio.2002.5629. [DOI] [PubMed] [Google Scholar]
- 18.Karhu R, Mahlamaki E, Kallioniemi A. Pancreatic adenocarcinoma—genetic portrait from chromosomes to microarrays. Genes Chromosom Cancer. 2006;45:721–730. doi: 10.1002/gcc.20337. [DOI] [PubMed] [Google Scholar]
- 19.Schubert EL, Hsu L, Cousens LA, Glogovac J, Self S, Reid BJ, Rabinovitch PS, Porter PL. Single nucleotide polymorphism array analysis of flow-sorted epithelial cells from frozen versus fixed tissues for whole genome analysis of allelic loss in breast cancer. Am J Pathol. 2002;160:73–79. doi: 10.1016/S0002-9440(10)64351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartek J, Vojtesek B, Staskova Z, Bartkova J, Kerekes Z, Rejthar A, Kovarik J. A series of 14 new monoclonal antibodies to keratins: characterization and value in diagnostic histopathology. J Pathol. 1991;164:215–224. doi: 10.1002/path.1711640306. [DOI] [PubMed] [Google Scholar]
- 21.Huang J, Wei W, Zhang J, Liu G, Bignell GR, Stratton MR, Futreal PA, Wooster R, Jones KW, Shapero MH. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum Genom. 2004;1:287–299. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao X, Li C, Paez JG, Chin K, Janne PA, Chen TH, Girard L, Minna J, Christiani D, Leo C, Gray JW, Sellers WR, Meyerson M. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–3071. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
- 23.Harada T, Chelala C, Bhakta V, Chaplin T, Caulee K, Baril P, Young BD, Lemoine NR. Genome-wide DNA copy number analysis in pancreatic cancer using high-density single nucleotide polymorphism arrays. Oncogene. 2008;27:1951–1960. doi: 10.1038/sj.onc.1210832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, Golub TR, Rimm DL, Meyerson ML, Fisher DE, Sellers WR. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436:117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- 25.Abe T, Fukushima N, Brune K, Boehm C, Sato N, Matsubayashi H, Canto M, Petersen GM, Hruban RH, Goggins M. Genome-wide allelotypes of familial pancreatic adenocarcinomas and familial and sporadic intraductal papillary mucinous neoplasms. Clin Cancer Res. 2007;13:6019–6025. doi: 10.1158/1078-0432.CCR-07-0471. [DOI] [PubMed] [Google Scholar]
- 26.Howe JA, Newport JW. A developmental timer regulates degradation of cyclin E1 at the midblastula transition during Xenopus embryogenesis. Proc Natl Acad Sci USA. 1996;93:2060–2064. doi: 10.1073/pnas.93.5.2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, Hempen PM, Hilgers W, Yeo CJ, Hruban RH, Kern SE. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol. 2003;163:1255–1260. doi: 10.1016/S0002-9440(10)63485-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sutherland RL, Musgrove EA. Cyclins and breast cancer. J Mammary Gland Biol Neoplasia. 2004;9:95–104. doi: 10.1023/B:JOMG.0000023591.45568.77. [DOI] [PubMed] [Google Scholar]
- 29.Ross JS, Greene B. Targeted therapy in oncology: the agony and ecstasy of personalized medicine. Expert Rev Anticancer Ther. 2001;1:321–322. doi: 10.1586/14737140.1.3.321. [DOI] [PubMed] [Google Scholar]
- 30.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, Horlings H, Berns K, Hung MC, van de Vijver MJ, Valero V, Gray JW, Bernards R, Mills GB, Hennessy BT. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, Rosen N. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.