

Abstract
Fragile X Syndrome is caused by the expansion of an unstable CGG-repeat tract in the 5′-UTR of the FMR1 gene, which generally results in transcriptional silencing and consequent absence of the FMR1 protein. To date, the smallest premutation allele reported to expand to a full mutation allele in a single generation is 59 CGG repeats. Here, we report a single-generation expansion to a full mutation allele (male with ∼538 CCG repeats) from a mother who is a carrier of a premutation allele of 56 CGG repeats. Furthermore, the maternal grandfather was a carrier of a gray (or intermediate)-zone allele (45 to 54 repeats) of 52 CGG repeats. Thus, in this family, a gray-zone allele expanded to the full mutation range in two generations. Interestingly, the two AGG interruptions present in the grandfather's allele were absent in the mother's premutation allele. These observations underscore the need to consider carriers of alleles of greater than 55 CGG repeats as being at risk for transmission of a full mutation allele in a single generation, and those with even smaller alleles in the gray zone as being at risk of having grandchildren with full mutation alleles.
Fragile X Syndrome (OMIM #300624) is the most common form of inherited mental impairment and the leading known heritable form of autism.1 The disorder is caused by a trinucleotide (CGG) repeat expansion to greater than 200 repeats (full mutation) in the 5′-untranslated region of the fragile X mental retardation 1 (FMR1) gene, which generally results in transcriptional silencing and absence of the FMR1 protein.2,3,4 The fragile X syndrome phenotype includes a large spectrum of involvement including mental retardation, developmental and speech delay, physical abnormalities such as large or prominent ears, long and narrow jaw, connective tissue problems, and macro-orchidism.5 The behavioral phenotype of fragile X syndrome is also characterized by autistic symptoms in approximately 25% to 33%, including social and communication deficits, stereotypic behavior, social anxiety, withdrawal, hyperarousal, unusual responses to sensory stimuli, gaze aversion, inattention, impulsivity, and hyperactivity.1,6,7,8,9,10
Transmission of a full mutation occurs exclusively from mothers who are carriers of either a full mutation or a premutation (55 to 200 CGG repeats) FMR1 allele. For mothers who are carriers of premutation alleles, the risk of transmitting an allele in the full mutation range is a function of the repeat length. Nolin and colleagues11 reviewed the propensity for premutation-to-full mutation expansion in more than 1500 transmissions from female carriers of premutation alleles. They showed that the smallest premutation allele leading to a full mutation offspring in a single generation was 59 CGG repeats, which was observed for two female carriers. For slightly smaller alleles (45 to 54 CGG repeats; “gray zone”), the extent of repeat instability is currently unknown. However, due to the observed repeat instability for alleles in this size range,12,13,14 gray-zone alleles are potential precursors of a full mutation in subsequent generations.11
Here, we describe a family in which a 52 repeat (gray-zone) allele has undergone a two generation expansion comprising an initial (grandfather-to-mother) increase to 56 repeats (premutation) followed by an expansion (mother-to-son) to a full mutation allele of ∼538 CGG repeats. This 56 repeat allele is the smallest premutation known to give rise to a full mutation allele in a single generation, and underscores the potential for carriers of alleles at the lower end of the premutation range to have children with full mutation alleles. One potential source of repeat instability is the absence of AGG interruptions within the CGG repeat element.15,16,17 In this regard, it is noteworthy that the four (CGG) repeat expansion in the first generation is accompanied by the loss of two AGG interruptions in the mother's premutation allele.
Study Cases
Written informed consent was obtained from all subjects in accordance with approved institutional human subjects protocols (Ethical Committee, University for Research Project). The proband (42-6; Figure 1) was a 17-year-old male with severe language impairment, cognitive deficits, social deficits and autistic behavior, hyperactivity and attention deficits, and psychomotor delay. The proband underwent a thorough medical history and physical examination and presented with a number of typical features of fragile X syndrome, including large head, long face, prominent forehead, flat feet, and hyperextensible joints. His mother (42-3), did not have primary ovarian insufficiency, emotional or behavioral or neurological problems. The proband's sister, (42.7), a premutation carrier, did not present with any significant intellectual impairment, learning disabilities, or emotional/behavioral problems. The proband's grandfather (42.2), presented with symptoms consistent with fragile X-associated tremor/ataxia syndrome, which included progressive action tremor and ataxia.
Figure 1.
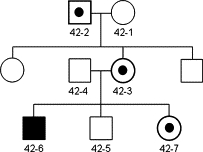
Pedigree showing the proband (42-6) family members. CGG repeat numbers were as follows: (42-2), 52 CGG repeats (gray zone) (42-3); 34, 56 CGG repeats (premutation); (42-4) 23 CGG repeats (normal); (42-6) ∼538 CGG repeats (full mutation); (42-7) 23, 91 CGG repeats (premutation).
Materials and Methods
DNA Analysis
A blood sample was obtained from each subject for the determination of CGG repeat number. Genomic DNA was isolated from peripheral blood leukocytes using the Ultraclean DNA Blood Isolation Kit (Mo Bio Laboratories, Carlsbad, CA). PCR and Southern blot analysis were performed on the proband and on all family members as described in Tassone et al,18 using either primers C and F19 or 1 and 320 and the FastStart TaqDNA Polymerase (Roche Diagnostics, Indianapolis, IN).
PCR analysis was also performed using modified primers c and f19 and the GC-Rich PCR System (Roche Diagnostics). PCR reactions, in a final volume of 30 μl, contained 0.67 mmol/L of each of the following primers: C primer, 5′-GCTCAGCTCCGTTTCGGTTTCACTTCCGG-3′; F2 primer, FAM-labeled, 5′-TTGTAGAAAGCGCCATTGGAGCCCCGCACT-3′; and 125 mmol/L each of the dNTPs. The CGG repeat region was amplified in a My Cycler system (Biorad, Hercules, CA) after DNA denaturation at 96°C for 6 minutes, followed by 30 cycles of 95°C for 1 minute, 65°C for 40 seconds, 72°C for 1 minute and 30 seconds, and finally the amplified products were extended at 72°C for 5 minutes.
PCR fragments were resolved on an ABI 3130 DNA sequencer (Applied Biosystems, Foster City, CA), in a 36 cm-capillary array, POP-7 polymer (Applied Biosystems) and 1× electrophoresis running buffer with EDTA (Applied Biosystems). A quantity of 0.5 μl of PCR product was dispensed with 0.25 μl of the size standard GeneScan−500 [ROX] (Applied Biosystems) and 10 μl of ultrapure Hi-Di Formamide (Applied Biosystems), to maintain denaturing conditions. The run temperature (60°C), run voltage (15 kV), sample injection voltage (1.2 kV), sample injection time (8 seconds), and run time (1200 seconds), were selected in the Data Collection Software, Ver. 3.0 (Applied Biosystems).
Fragment size analysis was performed with the GeneMapper Software V 3.7 (Applied Biosystems). A multiplex PCR that amplifies sex chromosome fragments and long CGG repeat tracts was also performed with the Abbott Molecular Fragile X PCR test (Abbott Laboratories, Wiesbaden, Germany) according to the manufacturer's instructions. PCR products were analyzed by agarose gel (2%) electrophoresis, and by both short and long run capillary electrophoresis. Capillary electrophoreses used an ABI 3130 DNA sequencer (Applied Biosystems) with a 36 cm-capillary array, POP-7 polymer (Applied Biosystems), and 1× electrophoresis running buffer with EDTA (Applied Biosystems), following the manufacturer's recommended electrophoresis protocol. Seven microliters of ultrapure Hi-Di Formamide (Applied Biosystems) was added to all samples. Analysis of PCR products was performed using GeneMapper Software Ver. 3.7 (Applied Biosystems); repeat number was determined following the specifications provided by the manufacturer. Finally, PCR analysis and sizing was also performed according to Tassone et al18 using the HDA-GT12 System, (Qiaxcel, Qiagen, Valencia, CA).
For Southern blot analysis, 5 to 10 μg of isolated genomic DNA was digested with EcoRI and NruI. Probe hybridization used the FMR1-specific dig-labeled StB12.3. Details are as previously described.18 Analysis and measurement of trinucleotide allele size, as well as the determination of the activation ratio (fraction of normal FMR1 allele as the active allele), were determined using an α Innotech FluorChem 8800 Image Detection System (α Innotech, San Leandro, CA).18
Sequencing Analysis
The 56 CGG repeat FMR1 allele was PCR amplified using genomic DNA as a template, and primers C and F, as previously described.18 The PCR product was purified with the Qiaquick PCR Purification Kit and directly sequenced using the nested primers 1 and 3.20 PCR products were also cloned into BlpI and XhoI sites of a CMV-driven FMR1 5′ UTR-reporter construct (unpublished results). The construct was propagated in Top10 cells (Invitrogen, Carlsbad, CA), and purified using the Qiagen Plasmid miniprep kit. Clones were digested with BlpI and XhoI, and CGG-containing inserts were separated from vector by agarose electrophoresis. Four positive clones were sequenced for both the sense and antisense strands of the FMR1 5′ UTR using a CMV-forward primer (5′-CGCAAATGGGCGGTAGGCGTG-3′), and a FMR1 5′ UTR reverse primer (5′-GGAGTGGACACCTGTGGAGA-3′); the clones, 56a, 56 d, 56c and 56b, contained 53, 56, 57, and 58 CGG repeats, respectively. This slight variation in CGG repeat size appears to have occurred during the cloning process itself. Sequencing was performed using the ABI 3730 Capillary Electrophoresis Genetic Analyzer and the ABI Big Dye Terminator v3.1 Cycle Sequencing chemistry (UC David Sequencing Facility).
Results
CGG repeat analysis of the family members (Figure 1) revealed a full mutation allele of ∼538 CGG repeats (as determined by Southern blot analysis) in the proband (sample 42.6), two alleles of 34 and 56 repeats in the mother (sample 42.3), an allele of 23 CGG repeats in the father of the proband (sample 42.4), and two alleles, of 23 and 91 CGG repeats, in the sister of the proband (sample 42.7). Southern blot and PCR results are shown in Figure 2A–C. In addition, using sequencing analysis, the number of CGG repeats in the maternal grandfather (42.2) revealed a gray-zone allele of 52 CGG repeats with two AGG interruptions in a 10-9-31 pattern (Figure 3). Finally, sequencing analysis confirmed the size of the 56-CGG allele determined by CGG sizing, but also revealed that the premutation allele did not contain any AGG interruptions (Figure 3). The loss of the two AGG interruptions was observed when the 56-CGG allele (42.3) was directly sequenced from a PCR-amplified product from genomic DNA and also by sequencing of the cloned PCR fragments. Thus, the 52-CGG allele expanded to a full mutation allele of ∼538 repeats in two generations, incurring the loss of the two AGG repeats in the gray zone to premutation expansion, and the expansion from a 56 CGG repeat premutation allele to a full mutation allele in a single generation.
Figure 2.
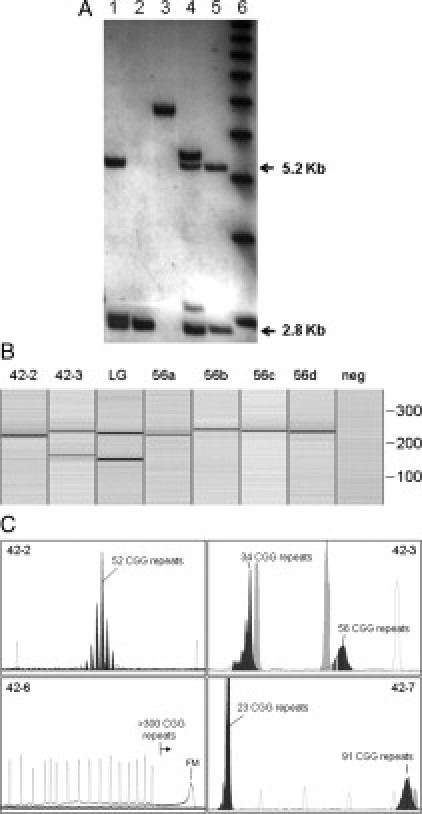
A: Southern blot analysis of genomic DNA isolated from the proband's parents; his mother (42-3) with a premutation allele, his father (42-4) with a normal allele (lanes 1 and 2, respectively). Proband, with a full mutation is shown in lane 3. His sister a premutation carrier (42-7) (lane 4). A normal female control is shown in lane 5. The normal, unmethylated band (2.8 Kb) and the normal methylated band (5.2 Kb) are indicated. A DNA 1kb ladder is shown in lane 6. B: PCR size analysis using the Qiaxcel capillary system (see: Materials and Methods) of PCR products derived from subject 42-2 (52 CGG repeats) and from subject 42-3 (34, 56 CGG repeats). PCR products derived from clones 56a (53 CGG), 56b (58 CGG), 56c (57 CGG), and 56 d (56 CGG) are also shown. PCR reactions used primers 1 and 3.20C: PCR size analysis for family members 42-2, 42-3, 42-6, and 42-7 using the ABI 3130 DNA Sequencing technology and the Abbott Molecular Fragile X PCR Test (see: Materials and Methods). In each frame, reference peaks (marker X-Rhodamine [ROX] labeled DNA ladder, size range: 50 to 1000 bp) are indicated in outline (also light gray in 42-3 and 42-7). For 42-2 and 42-3, standards were 300, 350, and 400 bp (36 to 69 CGG repeats); for 42-7, standards were 250, 300, 350, 400, 450, and 475 bp (19 to 94 CGG repeats); for 42-6, upper standard was 1000 bp (269 CGG repeats). For 42-6, the full mutation allele was sized by Southern blot.
Figure 3.
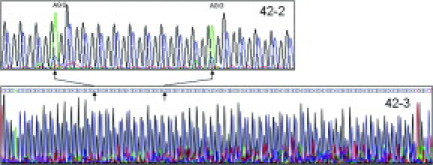
Sequencing analysis of 42-2 and 42-3. The upper electropherogram shows the sequencing analysis of subject 42-2 with an FMR1 allele comprising 52 CGG repeats and 2 AGG interruptions. The bottom electrophoregram shows the sequence of the cloned allele from subject 42-3 and the absence of AGG interruptions.
Discussion
A dynamic CGG repeat element is located within the 5′ untranslated region of the FMR1 gene. The following four allele categories have been established on the basis of repeat size by the American College of Medical Genetics21: normal (6 to 44 CGG), gray zone (45 to 54 CGG), premutation (55 to 200 CGG), and full mutation (>200 CGG). However, the boundaries among these categories are somewhat arbitrary, with functional significance depending on the outcome being considered (eg, transmission instability, gene expression levels). Indeed, while the boundary between premutation and full mutation alleles is clear, the distinctions between premutation and intermediate alleles or between intermediate and normal alleles are not well defined. Our general lack of understanding of the basic mechanisms that influence repeat instability and propensity for expansion makes it difficult to estimate the risk of expansion, particularly for gray-zone alleles.
One source of variability that may play a role in the expansion process is the presence of AGG interruptions within the CGG element. FMR1 alleles within the normal range are generally interrupted by one or more AGG trinucleotides, typically spaced at 9 to 11 repeat intervals (ie, 9−10 to 10−10). Premutation alleles become increasingly unstable in transmission by a female carrier with increasing size of the CGG repeat and the loss/absence of AGG interruptions.14,16 This association of AGG interruptions with the smaller, more stable alleles has led to the concept of an AGG “anchor”11,17,22; that is, that the AGG itself exerts a stabilizing influence on allele size in transmission. Thus, together with the size of the alleles (CGG repeat number) and the sex of the carrier, the presence of AGG interruptions appears to be an important factor influencing the stability of alleles during meiosis. Dombrowski et al23 have suggested that the loss of AGG interruptions is not the initial event leading to instability, but more likely a late event before the expansion occurs.23
The current study describes a carrier female carrier with an allele of 56 CGG repeats, lacking any AGG interruptions, which has given rise to a full mutation allele on transmission in a single generation. Before this report, the smallest allele reported to yield a full mutation in a single transmission possessed 59 CGG repeats, with the frequency of such transitions being very low (1.1%).11 As consequence, many reports have since used this value as cut off from the intermediate to the premutation range. In two additional cases, gray-zone alleles (44, 45 CGG repeats) were reported to give rise to full mutation alleles in two generations24,25; however, the premutation alleles in the intermediate generation (61, 80 CGG repeats) were both larger than the reported lower value of 59 repeats. Thus, it is evident that the lower bound for intermediate-to-premutation transition must be revised downward to at least 56 CGG repeats. Consistent with earlier observations, no AGG interruptions were found within the CGG stretch, supporting the importance in modulating the expansion process. Therefore, from the genetic counseling perspective, it is important to consider that there is a risk, albeit small, of expansion for alleles at the lower end of the premutation range, thus raising the issue of prenatal diagnosis for carriers with alleles above 55 repeats.
Finally, the grandfather of the proband, carrying a gray-zone allele of 52 CGG repeats, presented with the two principle core features of fragile X-associated tremor/ataxia syndrome, action tremor and ataxia. To our knowledge, this is first reported case of (probable) fragile X-associated tremor/ataxia syndrome arising from a gray-zone allele. In this regard, however, there is now preliminary evidence of an association between gray-zone alleles and parkinsonism,26 suggesting that subtle forms of central nervous system dysregulation may be occurring even for alleles in the gray zone range.
Acknowledgements
We thank the patients and their relatives for their participation in this study. This work is dedicated to the memory of Matteo.
Footnotes
Supported by the National Institutes of Child Health and Development Grant R01 HD02274 (F.T.) and by the Instituto de Salud Carlos III (G03/098 and PI05098) (I.F.).
Contributor Information
Isabel Fernandez-Carvajal, Email: metabol@ped.uva.es.
Flora Tassone, Email: ftassone@ucdavis.edu.
References
- 1.Hagerman RJ, Hagerman PJ. The fragile X premutation: into the phenotypic fold. Curr Opin Genet Dev. 2002;12:278–283. doi: 10.1016/s0959-437x(02)00299-x. [DOI] [PubMed] [Google Scholar]
- 2.Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 3.Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, Nelson DL. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 4.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen GB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 5.Hagerman RJ, Hagerman PJ, editors. Fragile X Syndrome: Diagnosis, Treatment, and Research. 3rd ed. The Johns Hopkins University Press; Baltimore: 2002. [Google Scholar]
- 6.Demark JL, Feldman MA, Holden JJ. Behavioral relationship between autism and fragile X syndrome. Am J Ment Retard. 2003;108:314–326. doi: 10.1352/0895-8017(2003)108<314:BRBAAF>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 7.Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB, Jr, Roberts J, Mirrett P. Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006;140A:1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- 8.Kaufmann WE, Cortell R, Kau AS, Bukelis I, Tierney E, Gray RM, Cox C, Capone GT, Stanard P. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am J Med Genet A. 2004;129A:225–234. doi: 10.1002/ajmg.a.30229. [DOI] [PubMed] [Google Scholar]
- 9.Reiss AL, Dant CC. The behavioral neurogenetics of fragile X syndrome: analyzing gene-brain-behavior relationships in child developmental psychopathologies. Dev Psychopathol. 2003;15:927–968. doi: 10.1017/s0954579403000464. [DOI] [PubMed] [Google Scholar]
- 10.Rogers SJ, Wehner DE, Hagerman R. The behavioral phenotype in fragile X: symptoms of autism in very young children with fragile X syndrome, idiopathic autism, and other developmental disorders. J Dev Behav Pediatr. 2001;22:409–417. doi: 10.1097/00004703-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 11.Nolin SL, Brown WT, Glicksman A, Houck GE, Jr, Gargano AD, Sullivan A, Biancalana V, Brondum-Nielsen K, Hjalgrim H, Holinski-Feder E, Kooy F, Longshore J, Macpherson J, Mandel JL, Matthijs G, Rousseau F, Steinbach P, Vaisanen ML, von Koskull H, Sherman SL. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003;72:454–464. doi: 10.1086/367713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crawford DC, Schwartz CE, Meadows KL, Newman JL, Taft LF, Gunter C, Brown WT, Carpenter NJ, Howard-Peebles PN, Monaghan KG, Nolin SL, Reiss AL, Feldman GL, Rohlfs EM, Warren ST, Sherman SL. Survey of the fragile X syndrome CGG repeat and the short-tandem-repeat and single-nucleotide-polymorphism haplotypes in an African American population. Am J Hum Genet. 2000;66:480–493. doi: 10.1086/302762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crawford DC, Zhang F, Wilson B, Warren ST, Sherman SL. Fragile X CGG repeat structures among African-Americans: identification of a novel factor responsible for repeat instability. Hum Mol Genet. 2000;9:1759–1769. doi: 10.1093/hmg/9.12.1759. [DOI] [PubMed] [Google Scholar]
- 14.Zhong N, Ju W, Pietrofesa J, Wang D, Dobkin C, Brown WT. Fragile X “gray zone” alleles: aGG patterns, expansion risks, and associated haplotypes. Am J Med Genet. 1996;64:261–265. doi: 10.1002/(SICI)1096-8628(19960809)64:2<261::AID-AJMG5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 15.Eichler EE, Holden JJ, Popovich BW, Reiss AL, Snow K, Thibodeau SN, Richards CS, Ward PA, Nelson DL. Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat Genet. 1994;8:88–94. doi: 10.1038/ng0994-88. [DOI] [PubMed] [Google Scholar]
- 16.Kunst CB, Warren ST. Cryptic and polar variation of the fragile X repeat could result in predisposing normal alleles. Cell. 1994;77:853–861. doi: 10.1016/0092-8674(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 17.Zhong N, Yang W, Dobkin C, Brown WT. Fragile X gene instability: anchoring AGGs and linked microsatellites. Am J Hum Genet. 1995;57:351–361. [PMC free article] [PubMed] [Google Scholar]
- 18.Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008;10:43–49. doi: 10.2353/jmoldx.2008.070073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu YH, Kuhl DP, Pizzuti A, Pieretti M, Sutcliffe JS, Richards S, Verkerk AJ, Holden JJ, Fenwick RG, Jr, Warren ST, Oostra BA, Nelson DL, Caskey CT. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 20.Brown WT, Houck GE, Jr, Jeziorowska A, Levinson FN, Ding X, Dobkin C, Zhong N, Henderson J, Brooks SS, Jenkins EC. Rapid fragile X carrier screening and prenatal diagnosis using a nonradioactive PCR test. JAMA. 1993;270:1569–1575. [PubMed] [Google Scholar]
- 21.Maddalena A, Richards CS, McGinniss MJ, Brothman A, Desnick RJ, Grier RE, Hirsch B, Jacky P, McDowell GA, Popovich B, Watson M, Wolff DJ. Technical standards and guidelines for fragile X: the first of a series of disease-specific supplements to the Standards and Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics. Quality Assurance Subcommittee of the Laboratory Practice Committee. Genet Med. 2001;3:200–205. doi: 10.1097/00125817-200105000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin P, Warren ST. Understanding the molecular basis of fragile X syndrome. Hum Mol Genet. 2000;9:901–908. doi: 10.1093/hmg/9.6.901. [DOI] [PubMed] [Google Scholar]
- 23.Dombrowski C, Levesque S, Morel ML, Rouillard P, Morgan K, Rousseau F. Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of fragile X syndrome alleles. Hum Mol Genet. 2002;11:371–378. doi: 10.1093/hmg/11.4.371. [DOI] [PubMed] [Google Scholar]
- 24.Terracciano A, Pomponi MG, Marino GM, Chiurazzi P, Rinaldi MM, Dobosz M, Neri G. Expansion to full mutation of a FMR1 intermediate allele over two generations. Eur J Hum Genet. 2004;12:333–336. doi: 10.1038/sj.ejhg.5201154. [DOI] [PubMed] [Google Scholar]
- 25.Zuniga A, Juan J, Mila M, Guerrero A. Expansion of an intermediate allele of the FMR1 gene in only two generations. Clin Genet. 2005;68:471–473. doi: 10.1111/j.1399-0004.2005.00514.x. [DOI] [PubMed] [Google Scholar]
- 26.Hall DA, Howard K, Hagerman R, Leehey MA. Parkinsonism in FMR1 premutation carriers may be indistinguishable from Parkinson Disease. Parkinsonism Relat Disord. 2009;15:156–159. doi: 10.1016/j.parkreldis.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]