

Abstract
AIM: To determine the role of leptin system in non-alcoholic fatty liver disease (NAFLD) development by delineating the changes in serum levels of leptin and soluble leptin receptor (sOB-R).
METHODS: Blood samples were collected from 30 consecutive patients with liver-biopsy-proven NAFLD and 30 patients with cholecystolithiasis (stationary phase) as controls. Serum leptin levels were determined by radioimmunoassay and concentration of sOB-R was measured by ELISA. Body mass index (BMI) was calculated for all subjects, and serum insulin, C-peptide, and lipoprotein levels were also detected.
RESULTS: Mean serum leptin level and BMI in the NAFLD group were significantly higher than in the controls (both P < 0.001), but mean sOB-R level was lower in the NAFLD group when compared to the controls. Both men and women in the NAFLD group had higher mean serum leptin levels and lower sOB-R levels than did the men and women in the control group (all P < 0.001). There was a significant negative correlation between serum leptin and sOB-R levels (r = -0.725, P < 0.001). Multivariate analysis showed that the percentage of hepatocyte steatosis, sex, BMI, and homeostasis model assessment of insulin resistance (HOMA IR) were independently related to serum leptin levels.
CONCLUSION: Elevated serum leptin seems to be a feature of steatosis, and serum leptin seems to increase as hepatocyte steatosis develops. An enhanced release of leptin is accompanied by an decrease in sOB-R concentration, which suggests higher resistance of peripheral tissues towards the action of leptin.
Keywords: Leptin, Soluble leptin receptor, Non-alcoholic fatty liver disease
INTRODUCTION
Leptin - the ob gene product - is a circulating 16-kDa peptide hormone secreted mainly by adipocytes of white fat tissue. It regulates food intake, body fat, insulin action, thermogenesis, induction of angiogenesis, and modulation of the immune system. Leptin synthesis in adipocytes is regulated by several hormones[1]. Leptin action in peripheral tissues involves interaction with specific transmembrane receptors. The leptin receptor (Ob-R), which is a member of the class-1 cytokine receptor family, may be an important determinant of leptin sensitivity. Ob-R was originally demonstrated in hypothalamic neurons, through which leptin regulates food intake and body weight[2]. Alongside several membrane-bound isoforms of Ob-R, with varying cytoplasmic length and with the same extracellular domain, a soluble form of the leptin receptor (sOb-R) can be demonstrated. sOb-R represents the main leptin-binding compound in plasma, which results in fractions of bound and free leptin in plasma[3]. The balance between the free form, the rapidly bioavailable compartment, and bound leptin regulates leptin bioavailability[4]. However, the precise pathophysiological role of sOb-R has not yet been defined.
Non-alcoholic fatty liver disease (NAFLD) is increasingly recognized as a common and potentially severe condition often associated with obesity, type 2 diabetes and hyperlipidemia[5]. However, the pathogenesis of NAFLD remains unclear. Increased synthesis of fatty acids in the liver, increased delivery of free fatty acids to the liver, and decreased β-oxidation of free fatty acids may cause the accumulation of fat in the liver[6]. Therefore, fat in hepatocytes causes cellular dysfunction and may render the liver more vulnerable to any factor that leads to inflammation. Furthermore, hepatic steatosis, the hallmark of NAFLD[7], may damage liver parenchyma.
The close relationship of leptin with adipose tissue and fat stores of the body suggests its involvement in the etiology and pathogenesis of NAFLD. Chitturi et al[8] have shown that serum leptin levels are significantly higher in subjects with non-alcoholic steatohepatitis (NASH), when compared to controls. Furthermore, they have shown that serum leptin is independently associated with the degree of hepatic steatosis but not hepatic inflammation or fibrosis. In addition to this study, there have been three other published studies that have yielded conflicting results regarding the role of leptin in the pathogenesis of NASH[9–11]. Thus, the role of leptin in NAFLD is not yet clear. Also, the relationship between sOB-R and NAFLD is an unresolved issue. Since concentrations of sOB-R are decreased in obese subjects when compared with lean controls[12], an interaction between sOB-R and NAFLD is suspected. However, currently no data on the association of sOB-R with NAFLD disease are available. Therefore, the objective of this study was to evaluate the association between serum leptin and sOB-R levels and NAFLD.
MATERIALS AND METHODS
Patients
A total of 30 consecutive patients with liver-biopsy-proven NAFLD were enrolled in the study from May 2007 to September 2007. NAFLD patients were admitted or referred to our department with incidentally found liver enzyme elevations. For the diagnosis of NAFLD and to rule out other possible liver diseases, all patients underwent liver biopsy. Thirty patients with cholecystolithiasis (stationary phase) were recruited as controls. Liver biopsies were obtained during an examination that preceded laparoscopic cholecystectomy, using a 16-gauge bard core biopsy needle. Informed consent was obtained from all patients.
All subjects underwent a detailed clinical and laboratory evaluation, including liver function tests, hepatitis markers, and autoantibodies, in addition to upper abdominal ultrasonography. Alcohol consumption was absent in all subjects. Subjects with previous or current history of acute or chronic viral hepatitis; malignant disease; acute infections; pituitary, adrenal, thyroid and pancreatic disease, or evidence for any other endocrine disorder; or prolonged use of corticosteroids or sex hormones were excluded. The controls had normal liver enzymes and no clinical, laboratory or imaging evidence of liver disease.
Clinic and laboratory evaluation
Overnight (12 h) fasting samples of serum obtained after centrifugation were stored in aliquots at -70°C until assayed. Clinical and laboratory data were collected on the date the liver biopsy was performed. A complete medical history and physical examination was accomplished in all patients and controls. Body mass index (BMI) was calculated. Laboratory evaluation included liver enzymes, glucose, complete blood count, total cholesterol, triglycerides, viral serology for hepatitis B and C, and autoantibodies. C-peptide was measured by a direct, double antibody sequential radioimmunoassay (RIA) (North Bio, Beijing, China). Serum insulin was measured by enzyme immunoassay kits (North Bio). The insulin resistance (IR) index was calculated on the basis of fasting values of plasma glucose and insulin according to the homeostasis model assessment for insulin sensitivity (HOMA) model formula: HOMA IR = fasting insulin (mUI/L) × fasting glucose (mmol/L)/22.5.
Leptin levels were measured by RIA kits (North Bio). The test utilizes 125I-labelled human leptin and human leptin antiserum to determine the level of total (free of and bound to serum proteins) leptin in serum. The test was 100% specific for human leptin and the sensitivity was 0.45 ng/mL. The coefficient of variation (CV) was < 8.3% and < 6.2%, respectively, for intra- and inter-assay variations (n = 5). The total concentration of sOB-R was determined using photometric ELISA kits (BioVendor Laboratory Medicine Inc, Brno, Czech Republic), which utilizes double monoclonal antihuman leptin receptor antibodies labeled with horseradish peroxidase. This test was 100% specific for human leptin receptors and the sensitivity was 0.4 U/mL. The intra-assay (CV < 5.6%) and inter-assay precision was good (CV < 5.5%). Samples were diluted three times according to the manufacturer's recommendation, and results higher than the higher standard were run again at another dilution.
Liver histology
Liver biopsy specimens were at least 15 mm in length, and had an appropriate number of portal tracts to make a confident evaluation of histological features and diagnosis[13]. Slides were routinely stained with hematoxylin-eosin, Masson’s trichrome and special stains for iron and copper. Liver biopsies were read by a single liver pathologist who was unaware of the patients’ clinical and laboratory data. Degree of steatosis was assessed on a scale of 1-3: 1, mild (5%-33% of hepatocytes affected); 2, moderate (33%-66% of hepatocytes affected); and 3, severe (> 66% of hepatocytes affected). The grades of inflammation and stages of fibrosis were modified to two categories as follows: mild inflammation (Kleiner grades 0 and 1) and moderate-to-severe inflammation (Kleiner grades 2 and 3), mild fibrosis (stages 0-2), and advanced fibrosis (stages 3 and 4). NASH was defined as steatosis plus lobular inflammation, plus either ballooning of hepatocytes or abnormal fibrosis (stages 1-4).
Statistical analysis
Hepatic steatosis was assessed as categorical (absent or present) or a continuous variable from 0%-100%. Comparisons between the NAFLD group and controls were made using Student’s t test for continuous variables and the χ2 or Fisher’s exact probability test for categorical data. All values are presented as mean ± SD. P < 0.05 was considered to be statistically significant. Multiple linear regression has been used in multivariate analysis of factors associated with leptin levels, including age, sex, steatosis, BMI, sOB-R, C-peptide and IR.
RESULTS
Comparison of patients and controls
Clinical and biochemical characteristics of the patients with NAFLD and controls are compared in Table 1. Serum leptin levels were found to be significantly higher in patients with NAFLD as compared with the controls (P < 0.001), and subjects with NAFLD had significantly lower levels of sOB-R, and higher levels of insulin and HOMA than the controls. There was no significant difference in serum total cholesterol, triglyceride, C-peptide or glucose levels in patients with NAFLD when compared with the controls.
Table 1.
Clinical and laboratory data of patients with NAFLD and controls
| NAFLD | Control | P | |
| n | 30 | 30 | |
| Gender (F/M) | 15/15 | 16/14 | 0.8 |
| Age | 41.97 ± 8.36 | 43.90 ± 8.35 | 0.374 |
| BMI (kg/m2) | 26.47 ± 3.11 | 23.97 ± 1.88 | < 0.001 |
| Total cholesterol (mmol/L) | 4.11 ± 0.85 | 3.97 ± 0.63 | 0.446 |
| Triglyceride (mmol/L) | 1.54 ± 0.62 | 1.30 ± 0.38 | 0.075 |
| C-peptide (nmol/L) | 1.73 ± 0.29 | 1.78 ± 0.34 | 0.505 |
| Glucose (mmol/L) | 5.42 ± 0.94 | 5.03 ± 0.68 | 0.072 |
| Insulin (mU/L) | 14.22 ± 2.66 | 8.64 ± 1.88 | < 0.001 |
| HOMA IR | 3.46 ± 1.04 | 1.92 ± 0.45 | < 0.001 |
| Leptin (ng/mL) | 10.90 ± 3.39 | 5.55 ± 1.96 | < 0.001 |
| sOB-R (ng/mL) | 18.00 ± 3.74 | 23.51 ± 2.62 | < 0.001 |
Gender difference in serum leptin and sOB-R levels
As expected, serum leptin levels were significantly higher in women than in men in the NAFLD group (P = 0.002). In contrast, sOB-R levels were significantly lower in women compared with men in the NAFLD group (P = 0.018). For this reason, comparisons were made between men and women separately. Both men and women in the NAFLD group had higher mean serum leptin levels and lower sOB-R levels than did the men and women in the control group (all P < 0.001, Table 2).
Table 2.
Leptin and sOB-R results according to gender
|
Male |
Female |
|||||
| NAFLD | Control | P | NAFLD | Control | P | |
| n | 15 | 14 | 15 | 16 | ||
| Leptin (ng/mL) | 9.07 ± 2.22 | 4.89 ± 1.73 | < 0.001 | 12.73 ± 3.43 | 6.13 ± 2.01 | < 0.001 |
| sOB-R (ng/mL) | 19.58 ± 4.04 | 23.92 ± 2.36 | < 0.001 | 16.41 ± 2.70 | 23.15 ± 2.86 | < 0.001 |
Histological evaluation
Upon histological staging, steatosis was mild in 17 patients, moderate to severe in 13; inflammation was mild in 16, and moderate to severe in 14; and fibrosis was absent in nine, mild in 21, and advanced in one patient. As summarized in Table 3, patients with moderate to severe steatosis had significantly higher levels of leptin than those with mild steatosis. Similarly, patients with moderate to severe steatosis were more insulin resistant as indicated by higher values of HOMA-IR. sOB-R levels were higher in the mild steatosis group than in the moderate to severe steatosis group, but the difference was not significant (P = 0.098).
Table 3.
Leptin and sOB-R results in relatin to degree of steatosis
|
Steatosis (n = 30) |
|||
| Mild | Moderate to severe | P | |
| n | 17 | 13 | |
| Gender (F/M) | 9/8 | 6/7 | 0.7 |
| Leptin (ng/mL) | 9.42 ± 2.76 | 12.84 ± 3.23 | 0.004 |
| sOB-R (ng/mL) | 18.99 ± 2.95 | 16.70 ± 4.36 | 0.098 |
| HOMA IR | 2.98 ± 0.86 | 4.20 ± 0.83 | 0.001 |
Correlation between serum leptin and sOB-R
There was a significant negative correlation between serum leptin and sOB-R levels (r = -0.725, P < 0.001; Figure 1).
Figure 1.
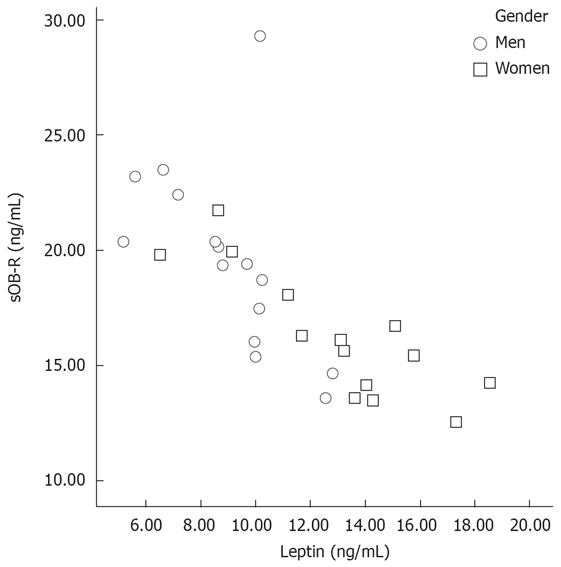
Correlation between serum leptin and sOB-R.
Relationship of serum leptin and sOB-R levels with BMI
The proportion of obese patients was rather low in the study population. Therefore, to determine the relationship between obesity and leptin more clearly, the NAFLD patients were divided into two groups according to mean BMI (26.5) as a cut-off point. Thus, patients with a BMI of 26.5 were defined as overweight. There were 12 patients with a BMI ≥ 26.5, and there were 18 patients with a BMI < 26.5. The serum leptin levels and HOMA IR values were significantly higher in the overweight group than in the normal weight group (P < 0.001 and P = 0.01, respectively). In addition, the sOB-R levels were significantly lower in the overweight group than in the normal weight group (P < 0.001,Table 4). Age, sex, BMI, HOMA IR, C-peptide and steatosis were tested in an overall multiple linear regression model, and the percentage of hepatocyte steatosis, sex, and BMI were independently related to serum leptin levels (Table 5).
Table 4.
The results of BMI, leptin and sOB-R tests in NASH patients with normal weight and overweight
| Overweight | Normal weight | ||
| (BMI ≥ 26.5) | (BMI < 26.5) | P | |
| n | 12 | 18 | |
| Female/Male | 8/4 | 9/9 | 0.5 |
| Leptin (ng/mL) | 13.73 ± 2.96 | 9.02 ± 2.14 | < 0.001 |
| sOB-R (ng/mL) | 15.16 ± 2.10 | 19.89 ± 3.40 | < 0.001 |
| HOMA IR | 4.06 ± 1.06 | 3.14 ± 0.86 | 0.01 |
Table 5.
Multiple linear regression analysis (R2 = 0.895) with serum leptin levels as a dependent variable in patients with NAFLD
| Variable | Coefficient | SE | t | P |
| Age | -0.040 | -0.099 | -1.371 | 0.183 |
| Sex | 2.128 | 0.319 | 4.295 | < 0.001 |
| BMI (kg/m2) | 0.529 | 0.485 | 5.838 | < 0.001 |
| C-peptide (nmol/L) | -0.866 | -0.075 | -1.072 | 0.295 |
| HOMA IR | 0.744 | 0.227 | 2.399 | 0.025 |
| Steatosis (%) | 1.329 | 0.247 | 2.675 | 0.014 |
DISCUSSION
The findings of the present study show that the adipocyte-derived hormone leptin is involved in metabolic abnormalities that lead to steatosis in NAFLD patients. Significantly high levels of serum leptin in NAFLD patients were found. In contrast to increased leptin level, the concentration of sOB-R significantly decreased in patients with NAFLD as compared to controls. As expected, the serum leptin level was significantly higher in women. Such a gender difference in serum leptin levels has been reported previously[14]. Leptin is exclusively expressed in adipose tissue and secreted from white adipose cells[15]. The difference is, therefore, in part, due to the higher percentage body fat in women. In addition, our study showed that serum leptin levels were strongly correlated with BMI or fat mass, consistent with a number of previous studies[16–18]. Also, the above results suggest a significant difference between normal and overweight NAFLD patients with regard to serum leptin levels.
The antisteatotic effects of leptin, which can curtail intrahepatic lipid accumulation (lipotoxicity), have been demonstrated in rodents. Thus, we considered whether leptin could play a similar role in NAFLD. Steatosis is a feature of clinical states characterized by defective leptin signalling. Steatosis is therefore often present in obese individuals[19] who are generally leptin resistant, and also in patients with congenital generalized lipodystrophy, a syndrome of leptin deficiency[20]. One of the important results of the study was that higher serum leptin levels were detected in patients with moderate to severe steatosis, compared with mild steatosis. The finding was inconsistent with a simple antisteatotic effect of leptin, which assumes that serum leptin values correlate with leptin biological activity in hepatocytes. There are two most likely explanations for the above finding. Firstly, leptin is inextricably related to IR. It has been suggested that leptin may contribute to hepatic steatosis by promoting IR and by altering insulin signaling in hepatocytes, so as to promote increased intracellular fatty acids[21]. Moreover, at a later stage, leptin may cause hepatic steatosis to turn into steatohepatitis by amplifying selected proinflammatory responses[21]. This explanation is mainly based on the effect of insulin on hepatic fat metabolism. In this study, an elevation of serum insulin level and HOMA IR in NAFLD patients, in comparison to the controls, was detected, which supports the latter suggestion. Secondly, those patients who develop NAFLD differ from obese individuals who do not develop NASH by having a state of peripheral (hepatic) leptin resistance (inadequate leptin signaling or/and decreased levels of sOB-R).
The biological relationship between leptin and its soluble receptor has previously been well established in animal models, as well as in various other clinical conditions, such as obesity, puberty[22], pregnancy[23] and inflammation[24]. However, to the best of our knowledge, the serum level of sOB-R has not previously been described in NAFLD patients. In this study, a strong negative correlation was found between serum levels of leptin and sOB-R in patients with NAFLD.
The low sOB-R levels in NAFLD patients may be part of a feedback mechanism aimed at reducing the increase in leptin. High free leptin levels were observed in morbidly obese subjects, probably caused by high leptin release by adipocytes due to abundant food intake. Interestingly, during food restriction, circulating leptin levels in lean and obese subjects drop dramatically within 1 d after onset of starvation[25], which indicates that leptin release is strongly reduced. On the other hand, plasma sOB-R might function like the soluble IL-6 receptor for the cytokine IL-6 (also a member of the gp-130 receptor family) and enhance leptin signaling. An increase in the bioavailability of leptin in ob/ob mice by overexpression of sOB-R leads to an improved weight-reducing effect of exogenous leptin[26]. It has been suggested that decreased serum levels of sOB-R result in adequate leptin signaling in NAFLD. Furthermore, the origin of sOB-R in plasma is also unclear at present. It may originate from alternative splicing of the OB-R or from full-length functional OB-Rs released by enzymatic cleavage. In line with this, sOB-R levels in plasma may reflect the amount of OB-R expressed by tissues. The finding of decreased serum levels of sOB-R in NAFLD suggests an increased resistance of peripheral tissues to the action of leptin, due to decreased expression and secretion of the OB-R. This might be in agreement with the proposed leptin resistance in morbidly obese subjects[27–29].
In conclusion, this study shows that increased serum concentrations of leptin are involved in NAFLD. Enhanced release of leptin is accompanied by a decrease in sOB-R concentration, which suggests higher resistance of peripheral tissues towards the action of leptin. However, the precise pathophysiological role of leptin and sOB-R has not yet been defined. We suggest that further studies should focus on local expression of leptin and its receptor in tissues of non-adipose origin, since the expression of leptin has been shown to be non-specific for adipocytes.
COMMENTS
Background
The exact pathogenesis in non-alcoholic fatty liver disease (NAFLD) is poorly understood, but some studies have analyzed the role of a variety of soluble peptides and other mediators. The close relationship of leptin with adipose tissue and fat stores of the body suggests its possible involvement in the etiology and pathogenesis of NAFLD.
Research frontiers
There have been several studies on the association between leptin and NAFLD, which have yielded conflicting results regarding the role of leptin in the pathogenesis of NASH. Since concentrations of the sOB-R are decreased in obese subjects when compared with lean controls, interaction between sOB-R and NAFLD is suspected. However, currently no data on the association of sOB-R with NAFLD disease are available.
Innovations and breakthroughs
The study showed elevated levels of leptin with a corresponding decrease in the concentration of sOB-R in patients with NAFLD. Although the results of laboratory experiments are not readily applicable to the clinical situation, these findings offer further insight into the role of leptin in the pathogenesis of NAFLD.
Applications
The results from this study confirm higher resistance of liver towards the action of leptin. This encourages us to study promising new drugs for treating NAFLD to improve leptin resistance in future clinical trials.
Peer review
This study aimed to investigate the role of leptin system in NAFLD development by delineating the changes in serum levels of leptin and sOB-R. The authors found a higher resistance of peripheral tissues towards the action of leptin. This is an interesting subject.
Acknowledgments
The authors thank Dr. Luo for useful comments and expert technical assistance.
Supported by Clinical Key Project of Health Bureau of Wuhan
Peer reviewer: Philip Abraham, Professor, Consultant Gastroenterologist & Hepatologist, PD Hinduja National Hospital & Medical Research Centre, Veer Savarkar Marg, Mahim, Mumbai 400 016, India
S- Editor Zhong XY L- Editor Kerr C E- Editor Ma WH
References
- 1.Stenvinkel P, Lonnqvist F, Schalling M. Molecular studies of leptin: implications for renal disease. Nephrol Dial Transplant. 1999;14:1103–1112. doi: 10.1093/ndt/14.5.1103. [DOI] [PubMed] [Google Scholar]
- 2.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 3.Lammert A, Kiess W, Bottner A, Glasow A, Kratzsch J. Soluble leptin receptor represents the main leptin binding activity in human blood. Biochem Biophys Res Commun. 2001;283:982–988. doi: 10.1006/bbrc.2001.4885. [DOI] [PubMed] [Google Scholar]
- 4.Lahlou N, Clement K, Carel JC, Vaisse C, Lotton C, Le Bihan Y, Basdevant A, Lebouc Y, Froguel P, Roger M, et al. Soluble leptin receptor in serum of subjects with complete resistance to leptin: relation to fat mass. Diabetes. 2000;49:1347–1352. doi: 10.2337/diabetes.49.8.1347. [DOI] [PubMed] [Google Scholar]
- 5.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 6.Sheth SG, Gordon FD, Chopra S. Nonalcoholic steatohepatitis. Ann Intern Med. 1997;126:137–145. doi: 10.7326/0003-4819-126-2-199701150-00008. [DOI] [PubMed] [Google Scholar]
- 7.Day CP, James OF. Hepatic steatosis: innocent bystander or guilty party? Hepatology. 1998;27:1463–1466. doi: 10.1002/hep.510270601. [DOI] [PubMed] [Google Scholar]
- 8.Chitturi S, Farrell G, Frost L, Kriketos A, Lin R, Fung C, Liddle C, Samarasinghe D, George J. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology. 2002;36:403–409. doi: 10.1053/jhep.2002.34738. [DOI] [PubMed] [Google Scholar]
- 9.Uygun A, Kadayifci A, Yesilova Z, Erdil A, Yaman H, Saka M, Deveci MS, Bagci S, Gulsen M, Karaeren N, et al. Serum leptin levels in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2000;95:3584–3589. doi: 10.1111/j.1572-0241.2000.03297.x. [DOI] [PubMed] [Google Scholar]
- 10.Giannini E, Botta F, Cataldi A, Tenconi GL, Ceppa P, Barreca T, Testa R. Leptin levels in nonalcoholic steatohepatitis and chronic hepatitis C. Hepatogastroenterology. 1999;46:2422–2425. [PubMed] [Google Scholar]
- 11.Nakao K, Nakata K, Ohtsubo N, Maeda M, Moriuchi T, Ichikawa T, Hamasaki K, Kato Y, Eguchi K, Yukawa K, et al. Association between nonalcoholic fatty liver, markers of obesity, and serum leptin level in young adults. Am J Gastroenterol. 2002;97:1796–1801. doi: 10.1111/j.1572-0241.2002.05846.x. [DOI] [PubMed] [Google Scholar]
- 12.van Dielen FM, van ,t Veer C, Buurman WA, Greve JW. Leptin and soluble leptin receptor levels in obese and weight-losing individuals. J Clin Endocrinol Metab. 2002;87:1708–1716. doi: 10.1210/jcem.87.4.8381. [DOI] [PubMed] [Google Scholar]
- 13.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 14.Couillard C, Mauriege P, Prud’homme D, Nadeau A, Tremblay A, Bouchard C, Despres JP. Plasma leptin concentrations: gender differences and associations with metabolic risk factors for cardiovascular disease. Diabetologia. 1997;40:1178–1184. doi: 10.1007/s001250050804. [DOI] [PubMed] [Google Scholar]
- 15.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 16.Ankarberg-Lindgren C, Dahlgren J, Carlsson B, Rosberg S, Carlsson L, Wikland KA, Norjavaara E. Leptin levels show diurnal variation throughout puberty in healthy children, and follow a gender-specific pattern. Eur J Endocrinol. 2001;145:43–51. doi: 10.1530/eje.0.1450043. [DOI] [PubMed] [Google Scholar]
- 17.Ahmed ML, Ong KK, Watts AP, Morrell DJ, Preece MA, Dunger DB. Elevated leptin levels are associated with excess gains in fat mass in girls, but not boys, with type 1 diabetes: longitudinal study during adolescence. J Clin Endocrinol Metab. 2001;86:1188–1193. doi: 10.1210/jcem.86.3.7320. [DOI] [PubMed] [Google Scholar]
- 18.Horlick MB, Rosenbaum M, Nicolson M, Levine LS, Fedun B, Wang J, Pierson RN Jr, Leibel RL. Effect of puberty on the relationship between circulating leptin and body composition. J Clin Endocrinol Metab. 2000;85:2509–2518. doi: 10.1210/jcem.85.7.6689. [DOI] [PubMed] [Google Scholar]
- 19.James OF, Day CP. Non-alcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J Hepatol. 1998;29:495–501. doi: 10.1016/s0168-8278(98)80073-1. [DOI] [PubMed] [Google Scholar]
- 20.Chandalia M, Garg A, Vuitch F, Nizzi F. Postmortem findings in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 1995;80:3077–3081. doi: 10.1210/jcem.80.10.7559900. [DOI] [PubMed] [Google Scholar]
- 21.Kaplan LM. Leptin, obesity, and liver disease. Gastroenterology. 1998;115:997–1001. doi: 10.1016/s0016-5085(98)70272-0. [DOI] [PubMed] [Google Scholar]
- 22.Quinton ND, Smith RF, Clayton PE, Gill MS, Shalet S, Justice SK, Simon SA, Walters S, Postel-Vinay MC, Blakemore AI, et al. Leptin binding activity changes with age: the link between leptin and puberty. J Clin Endocrinol Metab. 1999;84:2336–2341. doi: 10.1210/jcem.84.7.5834. [DOI] [PubMed] [Google Scholar]
- 23.Lewandowski K, Horn R, O‘Callaghan CJ, Dunlop D, Medley GF, O‘Hare P, Brabant G. Free leptin, bound leptin, and soluble leptin receptor in normal and diabetic pregnancies. J Clin Endocrinol Metab. 1999;84:300–306. doi: 10.1210/jcem.84.1.5401. [DOI] [PubMed] [Google Scholar]
- 24.Voegeling S, Fantuzzi G. Regulation of free and bound leptin and soluble leptin receptors during inflammation in mice. Cytokine. 2001;14:97–103. doi: 10.1006/cyto.2001.0859. [DOI] [PubMed] [Google Scholar]
- 25.Kolaczynski JW, Considine RV, Ohannesian J, Marco C, Opentanova I, Nyce MR, Myint M, Caro JF. Responses of leptin to short-term fasting and refeeding in humans: a link with ketogenesis but not ketones themselves. Diabetes. 1996;45:1511–1515. doi: 10.2337/diab.45.11.1511. [DOI] [PubMed] [Google Scholar]
- 26.Pelleymounter MA, Cullen MJ, Healy D, Hecht R, Winters D, McCaleb M. Efficacy of exogenous recombinant murine leptin in lean and obese 10- to 12-mo-old female CD-1 mice. Am J Physiol. 1998;275:R950–R959. doi: 10.1152/ajpregu.1998.275.4.R950. [DOI] [PubMed] [Google Scholar]
- 27.Arch JR, Stock MJ, Trayhurn P. Leptin resistance in obese humans: does it exist and what does it mean? Int J Obes Relat Metab Disord. 1998;22:1159–1163. doi: 10.1038/sj.ijo.0800779. [DOI] [PubMed] [Google Scholar]
- 28.Zimmet P, Boyko EJ, Collier GR, de Courten M. Etiology of the metabolic syndrome: potential role of insulin resistance, leptin resistance, and other players. Ann N Y Acad Sci. 1999;892:25–44. doi: 10.1111/j.1749-6632.1999.tb07783.x. [DOI] [PubMed] [Google Scholar]
- 29.Martin RL, Perez E, He YJ, Dawson R Jr, Millard WJ. Leptin resistance is associated with hypothalamic leptin receptor mRNA and protein downregulation. Metabolism. 2000;49:1479–1484. doi: 10.1053/meta.2000.17695. [DOI] [PubMed] [Google Scholar]