

Abstract
AIM: To evaluate the effect of rosiglitazone in a murine model of liver fibrosis induced by Schistosoma japonicum infection.
METHODS: A total of 50 mice were randomly and averagely divided into groups A, B, C, D and E. The mice in group A served as normal controls, while those in the other four groups were infected with Schistosoma japonicum to induce the model of liver fibrosis. Besides, the mice in groups C, D and E were treated with praziquantel, rosiglitazone and praziquantel plus rosiglitazone, respectively. NF-κB binding activity and expression of PPARγ-mRNA were determined by Western blot assay and real-time quantitative PCR. Radioimmunoassay technique was used to detect the serum content changes of TNF-α and IL-6. Histological specimens were stained with HE. Expression of TGF-β1, a-smooth muscle actin and type I and type III collagen was detected by immunohistochemistry and multimedia color pathographic analysis system.
RESULTS: Inflammation and fibrosis in the rosiglitazone plus praziquantel treatment group (group E) were lightest among the mice infected with Schistosoma (P < 0.05). To further explore the mechanism of rosiglitazone action, we found that rosiglitazone can significantly increase the expression of PPARγ [E: -18.212 ± (-3.909) vs B: -27.315 ± (-6.348) and C: -25.647 ± (-5.694), P < 0.05], reduce the NF-κB binding activity (E: 88.89 ± 19.34 vs B: 141.11 ± 15.37, C: 112.89 ± 20.17 and D: 108.89 ± 20.47, P < 0.05), and lower the serum level of TNF-α (E: 1.613 ± 0.420 ng/mL vs B: 2.892 ± 0.587 ng/mL, C: 2.346 ± 0.371 ng/mL and D: 2.160 ± 0.395 ng/mL, P < 0.05) and IL-6 (E: 0.106 ± 0.021 ng/mL vs B: 0.140 ± 0.031 ng/mL and C: 0.137 ± 0.027 ng/mL, P < 0.05) in mice with liver fibrosis. Rosiglitazone can also substantially reduce the hepatic expression of TGF-β1, α-SMA type I and type III collagen in mice with liver fibrosis.
CONCLUSION: The activation of PPARγ by its ligand can retard liver fibrosis and suggest the use of rosiglitazone for the treatment of liver fibrosis due to Schistosoma japonicum infection.
Keywords: Peroxisome proliferators-activated receptorγ, Rosiglitazone, Liver fibrosis, Schistosomiasis, Hepatic stellate cell
INTRODUCTION
Hepatic schistosomiasis is one of the most prevalent forms of chronic liver diseases in the world, resulting in the morbidity from infection due to its complications of liver fibrosis. However, there are few medicines or means available to control and treat fibrosis in schistosomiasis.
The key pathogenic event in liver fibrosis is the activation of hepatic stellate cells (HSC) and their transformation into myofibroblasts[1,2]. The HSC (formally called the Ito cell) is the primary cell-type in the liver responsible for excessive collagen synthesis during hepatic fibrosis[3,4]. Following liver injury, the HSC undergoes a complex transformation or activation process in which the cell changes from a quiescent, vitamin A-storing cell to an activated myofibroblast, resulting in considerable changes such as the appearance of the cytoskeletal protein smooth muscle α-actin (α-SMA), the loss in the cellular vitamin A stores[5,6], and the up-regulation of type I and III collagen genes. In addition, transforming growth factor-β (TGF-β), as the most potent fibrogenic cytokine described in HSC[7], accompanying its receptors are increased following HSC activation as well. These pathogenic alterations cause excessive depositions of extracellular matrix (ECM) proteins including three large families of protein-glycoproteins, collagens and proteoglycans, and disrupt the balance in ECM integrity to induce hepatic fibrosis[8].
Peroxisome proliferator-activated receptors (PPARs) are a family of ligand-activated nuclear transcription factors, members of the nuclear hormone receptor super-family[9]. There are 3 mammalian subtypes identified as PPAR-α, -β (or -δ), and -γ[10]; however, current studies demonstrate that only PPARγ is mainly expressed in human HSC, which contributes to the process of liver fibrogenesis, and both its translational and transcriptional levels are significantly decreased in in vitro and/or in vivo activated human HSC cells. The decreased PPARγ at the early stage of HSC activation and the amelioration of stimulated PPARγ by its ligands to some deterioration resulting from activated HSCs, suggest that PPARγ may be involved in the maintenance of a quiescent phenotype of HSC[11], although the identification of PPARγ as a novel modulator to liver fibrosis remains controversial.
PPARγ ligands, 15-deoxy-triangle up (1214) prosta-glandin J (2) (15d-PGJ (2) and rosiglitazone, significantly decreased the expression of α-SMA and proliferation in activated human HSCs induced by platelet-derived growth factor[12]. Oral administration of rosiglitazone is found able to diminish extracellular matrix deposition and HSC activation in liver fibrosis of rat models which are created by bile duct ligation[13]. They found PPARγ-specific DNA binding activities in nuclear extracts of HSCs isolated from liver fibrotic rat models are impaired significantly, although they can return, through rosiglitazone administration, to the normal levels as controls. Intriguingly, rosiglitazone induces PPARγ activation to inhibit collagen and fibronectin synthesis in human HSCs initiated by transforming growth factor (TGF)-beta1 in vitro. These findings implicate that the PPARγ activation in HSC retards fibrosis, suggesting the use of PPARγ ligands for the treatment of fibrosis following liver injury.
To determine amelioration of PPARγ and its ligands to liver fibrosis, we studied several different treatments of ligands, rosiglitazone, with/out the additives and praziquantel in liver fibrosis model mice created by infection of schistosome.
MATERIALS AND METHODS
Animal preparation and treatment
Fifty 4-5-wk-old Kunming mice, weighing 16-22 g (obtained from the experimental animal center of Tongji Medical College, Huazhong University of Science and Technology) were used. All animals were housed in a temperature and humidity controlled environment, and all animal experiments were carried out in accordance with the Chinese Council on Animal Care Guide for the Care and Use of Laboratory Animals.
The mice were randomly divided into five groups (10 per group) as follows: group A, B, C, D and E. The mice in group A served as normal controls, while those in the other four groups were infected with 40 Schistosoma japonicum cercariae through skin to create a liver fibrosis model. The mice in groups C, D and E were treated with praziquantel, rosiglitazone, and praziquantel plus rosiglitazone, 4 wk after infection, respectively, Praziquantel (500 mg/kg) was given daily for 2 d by intragastric administration in groups C and E. Rosiglitazone (4 mg/kg) was given daily for 6 wk by intragastric administration in groups D and E. Meanwhile, the mice in the normal control group (group A) and the model control group (group B) were given normal saline daily for 6 wk. All mice were sacrificed at the end of the study period. Vein blood was centrifuged at 1500 r/min for 15 min, and the serum was stored at -20°C for use. Parts of liver tissues were fixed by 40 g/L formaldehyde, and embedded in paraffin. The remaining liver was stored at -70°C.
Histopathological examination
The sections of the liver were stained with hematoxylin-eosin (HE) staining. Criteria used in histopathological analysis strictly obeyed WHO category and nomenclature for liver fibrosis[14]. Mean degrees of liver fibrosis examined from ten randomized scopes under optical microscopy in each slice were used for statistical analysis in current study. Pathological phases are graded as “-”, “+”, “++” and “+++” respectively: “-”, no fibrosis in liver lobules marked phase “0”; “+”, limit fibrosis in lobules, phase “1”; “++”, typical fibrosis spacing around liver lobules, phase “2”; and “+++”, early cirrhosis, fibrosis enveloping liver lobules and spacing into central vein area, phase “3”.
Serum assay
Serum TNF-α and IL-6 were detected using radioim-munoassay kit (Institute of Radioimmunoassay, Science and Technology Empolder Center, General Hospital of PLA) according to the manufacturer’s instructions.
Immunohistochemistry for TGF-β, α-SMA and collagens
TGF-β, α-SMA and collagens were detected using the three step streptavidin-biotin immunoperoxidase method. Briefly, tissue sections were de-paraffinized and rehydrated, and then were heated in microwave oven for 10 min to enhance antigen retrieval. For minimizing endogenous peroxidase, activity slides were incubated with 3% H2O2. After blocking with 5% normal goat serum in 0.01% PBS, the primary antibodies (mice raised against α-SMA, rabbit raised against TGF-β, and types I and III collagens, Bosider, Wuhan, China) were applied and incubated at 37°C in a moisture chamber for 1 h. Sections were then washed with PBS 3 times. After reacted with biotinylated hircine anti-mouse (or rabbit) IgG and then avidin at 37°C for 20 min each, sections were washed with PBS 3 times. Then diaminobenzidine solution (1 mg/mL in PBS containing 0.03% hydrogen peroxide) was applied as the chromogen. Sections were counterstained with hematoxylin for 15 s before checked under microscope. As a negative control, PBS was used instead of primary antibody. The cytoplasm or membrane of the positive cell was stained brown and yellow. The sections were observed under microscope. For quantification, 10 random fields of intralobular and periportal areas were evaluated under microscope at 40 × magnification. The integral light density was determined by multimedia color pathographic analysis system.
Protein preparation and western blotting
One hundred mg of frozen tissues was homogenized and centrifuged at 5000 r/min at 4°C for 10 min. The sediment was resuspended in 200 μL of ice-cold extraction buffer A (10 mmol/L HEPES pH 7.9, 1.5 mmol/L MgCl2, 10 mmol/L KCl, 0.5 mmol/L DTT, 0.5 mmol/L phenyl-methylsulfonyl fluoride, 1 μg/mL Aprotinin), and spin down at 5000 r/min at 4°C for 10 min. The sediment was resuspended in 100 μL of extraction buffer C (25% glycerol, 420 mmol/L NaCl, 20 mmol/L HEPES pH 7.9, 1.5 mmol/L MgCl2, 1% NP40, 0.2 mmol/L EDTA, 0.5 mmol/L DTT, 0.5 mmol/L phenylmethylsulfonyl fluoride, 1 μg/mL each of aprotinin) and kept on ice for 10 min. Finally, it was transferred to a microdosis centrifuge tube and centrifuged at 15 000 r/min at 4°C for 20 min. The supernatant was stored at -70°C for use.
Protein concentration was determined using the Coomassie brilliant blue method with G-250 as a standard. Nuclear or cytoplasmic proteins were electrophoresed through 12% SDS-PAGE in Tris-glycine electrophoresis buffer and transferred onto nitrocellulose membrane. The blot was pre-incubated in blocking buffer [Tris buffered saline (TBS) containing Tween and 7% non-fat dried milk powder] at room temperature for 2 h and probed with a primary antibody [rabbit anti-mouse NF-κB polyclonal antibody (Santa Cruz, CA, USA)] at 4°C overnight. After washed three times with 0.1% Tween-TBS, it was incubated with 1:2000 goat anti-rabbit conjugated with horseradish peroxidase (The Eastman Company, Beijing, China) at room temperature for 2 h. Immunoreactive bands were detected by epiluminescence and quantified in arbitrary units [optical density (A) × band area using Vilber Lourmat image analysis system].
RNA isolation, reverse transcription and quantitative PCR
Total RNA was extracted from the frozen liver tissue with Trizol reagent (GIBCO, USA). Complementary DNA (cDNA) was synthesized from total RNA using the reverse transcriptase Superscript II (GiBCO, USA) according to the manufacturer’s instructions.
Relative quantification of target gene expression was performed using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. The threshold cycle and the standard curve method were used for calculating the relative amount of the target mRNA. For PPARγ mRNA detection, the forward and reverse primers were: 5′-TTTCAAGGGTGCCAGTTTCG-3′, and 5′-TCTTTATTCATCAGGGAGGC-3′; for GAPDH, the primers were: 5′-GATGGTGAAGGTCGGTGTG-3′, and 5′-GAGGTCAATGAAGGGGTCG-3′. Real-time PCR was carried out in 50 μL of PCR reaction mixture which contained 1.5 μL of the extracted cDNA, 25 mmol/L MgCl2, 20 μmol/L each primer, and 1 μL of SYBR Green I (Biotium, USA). A “no template” control was added, which consisted of all the reagents listed above for real-time PCR, except the cDNA template was replaced with water. The following thermal profile was used: 10 min at 94°C, followed by 45 cycles of 94°C for 30 s, 53°C for 30 s, and 72°C for 30 s. Dissociation curve was run to prove the purity of the product.
Data analysis: The relative PPARγ mRNA expression levels were calculated by the ΔΔCt (threshold cycle) method in relation to PPARγ expression in the liver tissues. ΔΔCt = ΔCt (specimen) - ΔCt (GAPDH), ΔCt = Ct (negative control) - Ct (specimen). ΔCt is the relative gene expression (Ct-the number of fractional cycle at which the reporter fluorescence was generated by cleavage of the probe passes a fixed threshold above baseline). The relative difference in expression of the gene of interest and of the internal reference gene is represented by ΔCt. Changes of gene expression in relation to the calibrator are represented by 2ΔΔCt [relative quantification (RQ)].
Statistical analysis
Means of triplicates were used for statistical analysis by one-way analysis of variance (ANOVA) with post hoc Tukey test for pairwise group comparisons (SPSS 15, SPSS Inc, Chicago, IL, USA). The level of statistical significance was set at 0.05 (two-sided).
RESULTS
Histopathological analysis
Compared with normal mice, slice of the liver from fibrosis model (group B) showed typical damage in the liver lobules: the segregation of liver by collagen fibers, necrosis lesions in the granulomas and inflammatory cells extensively in the periphery of granulomas (Figure 1A). Compared with group B, the decreases were not significant in the fibroplasias, hepatocellular necrosis and inflammatory cell infiltration in the liver slice from groups C and D (fibrosis models treated only with praziquantel or rosiglitazone respectively) (Figure 1B). However, the severity of hepatocellular necrosis and fibroplasias was significantly decreased in the liver of mice treated with praziquantel plus rosiglitazone (group E) compared with group B (Figure 1C). Group E mice also showed thin fibroseptal and decreased inflammatory cell infiltrations in the livers, demonstrating a markedly improved or normal architecture of hepatic lobule.
Figure 1.
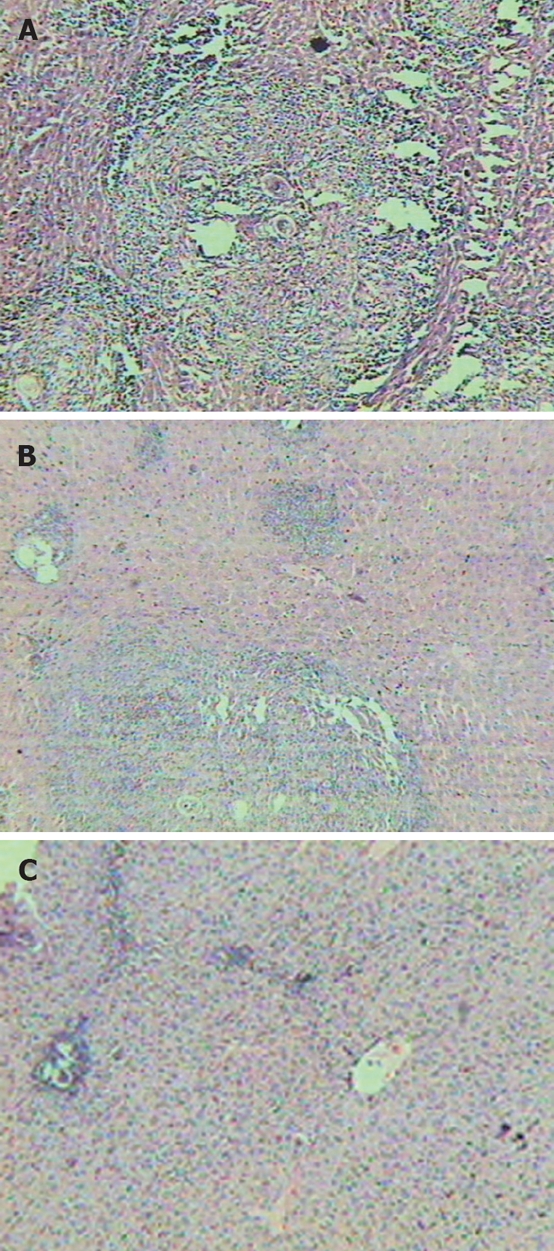
Liver histopathology of mice (HE staining, × 10). A: Represents the model group; B: Represents the group treated with praziquantel; C: Represents the group treated with rosiglitazone plus praziquantel.
The pathological phase quantities of groups B, C, D and E were 2.31 ± 0.63, 1.49 ± 0.77, 1.38 ± 0.60 and 0.78 ± 0.53, respectively. Fibrosis in the rosiglitazone plus praziquantel treatment group (group E) was lightest among those with Schistosoma infection (P < 0.05).
Immunohistochemical findings
TGF-β1-positive cells were concentrated in the portal venule pericytes with perisinusoidal distribution in the normal mouse liver. Similarly distributed and decreased TGF-β1-positive cells were found in group E mice (treated with praziquantel plus rosiglitazone). However, there was a significantly large number of TGF-β1-positive cells in groups B, C and D, which distributed mainly at the fibro-septa band, the area of necrosis and inflammatory cell infiltration, few hepatic cells and lipid-containing vacuoles.
There were few expressions of α-SMA positive cells in venule pericytes and periphery of granulomas in the mouse livers of normal control and group E. Nevertheless, the positive cells of α-SMA were substantially distributed at venule pericytes, inflammatory cell infiltration, periphery of granulomas and fibrous proliferation in groups B, C and D.
Moreover, few expressions of type I & III collagen were only found in the central venule pericytes and the linkage region of lobules from the mouse liver of group A (normal control), group D or E. In contrast, there was a significant larger number of positive collagen cells distributed not only in these areas but also in fibrous proliferation and Disec cavities of mouse livers from groups B (fibrosis model) and C treated with praziquantel alone.
TGF-β1, α-SMA, type I & III collagen positive cells were quantified with multimedia color pathographic analysis. Quantities of TGF-β1 and type I collagen in the mouse livers of praziquantel treatment group (group C) were reduced much more than in group B (P < 0.05). No significant difference was found in the volumes of α-SMA and type III collagen in the mouse livers of groups C and group B (P > 0.05). Additionally, volumes of α-SMA and type III collagen in the mouse livers of rosiglitazone treatment group (group D) decreased dramatically compared with groups B and C (P < 0.05). TGF-β1 and type I collagen in mouse livers of group D decreased more significantly than in group B (P < 0.05). Notably, volumes of TGF-β1, α-SMA, type I & III collagen in the mouse livers of group E (praziquantel plus rosiglitazone treatment) decreased substantially compared to groups B and C as well (P < 0.05). All data are summarized in Table 1.
Table 1.
Quantitative analysis of TGF-β1, α-SMA, type I and type III collagen in mouse livers (mean ± SD)
| Groups | n | TGF-β1 | α-SMA | Type I collagen | Type IIIcollagen |
| A | 10 | 0.356 ± 0.145 | 0.602 ± 0.116 | 0.303 ± 0.117 | 0.317 ± 0.133 |
| B | 10 | 0.829 ± 0.154a | 0.915 ± 0.172a | 0.654 ± 0.186a | 0.735 ± 0.192a |
| C | 10 | 0.655 ± 0.117ac | 0.902 ± 0.155a | 0.505 ± 0.103ac | 0.701 ± 0.174a |
| D | 10 | 0.603 ± 0.126ac | 0.732 ± 0.109ace | 0.477 ± 0.132ac | 0.508 ± 0.127ace |
| E | 10 | 0.459 ± 0.107aceg | 0.689 ± 0.132ce | 0.382 ± 0.125ceg | 0.436 ± 0.112ace |
Compared with group A,
P < 0.05; Compared with group B,
P < 0.05; Compared with group C,
P < 0.05; Compared with group D,
P < 0.05.
Serum assays
The serum level of TNF-α was markedly lower in group E (1.613 ± 0.420 ng/mL) than that in groups B (2.892 ± 0.587 ng/mL), C (2.346 ± 0.371 ng/mL) and D (2.160 ± 0.395 ng/mL) (P < 0.05). However, the serum level of IL-6 was markedly higher in groups B (0.140 ± 0.031 ng/mL), and/or C (0.137 ± 0.027 ng/mL) than that in groups D (0.108 ± 0.021 ng/mL) and/or E (0.106 ± 0.021 ng/mL) (P < 0.05) as summarized in Table 2.
Table 2.
Serum level of TNF-α and IL-6 in mice (mean ± SD)
| Groups | n | TNF-α | IL-6 |
| A | 10 | 1.530 ± 0.380 | 0.094 ± 0.026 |
| B | 10 | 2.892 ± 0.587a | 0.140 ± 0.031a |
| C | 10 | 2.346 ± 0.371ac | 0.137 ± 0.027a |
| D | 10 | 2.160 ± 0.395ac | 0.108 ± 0.021ce |
| E | 10 | 1.613 ± 0.420ceg | 0.106 ± 0.021ce |
Compared with group A,
P < 0.05; Compared with group B,
P < 0.05; Compared with group C,
P < 0.05; Compared with group D,
P < 0.05.
NF-κB binding activity
Less NF-κB activity was found in groups A, C, D or E than in group B (without any treatment) (P < 0.05). Moreover, the activity of NF-κB decreased much more in group A and E than in either group C or D. The activity of NF-κB in groups C and D (112.89 ± 20.17 and 108.89 ± 20.47) was higher than in groups A and E (78.89 ± 18.12, 88.89 ± 19.34, P < 0.05), but lower than in group B (141.11 ± 15.37, P < 0.05). There was no significant difference between groups C and D (P > 0.05) as seen in Table 3 and Figure 2.
Table 3.
NF-κB binding activity and the expression of PPARγ-mRNA in mouse livers (mean ± SD)
| Groups | n | NF-κB | PPAR-γ mRNA |
| A | 10 | 78.89 ± 18.12 | -16.557 ± (-3.022) |
| B | 10 | 141.11 ± 15.37a | -27.315 ± (-6.348)a |
| C | 10 | 112.89 ± 20.17ac | -25.647 ± (-5.694)a |
| D | 10 | 108.89 ± 20.47ac | -18.217 ± (-4.498)ce |
| E | 10 | 88.89 ± 19.34ceg | -18.212 ± (-3.909)ce |
Compared with group A,
P < 0.05; Compared with group B,
P < 0.05; Compared with group C,
P < 0.05; Compared with group D,
P < 0.05.
Figure 2.
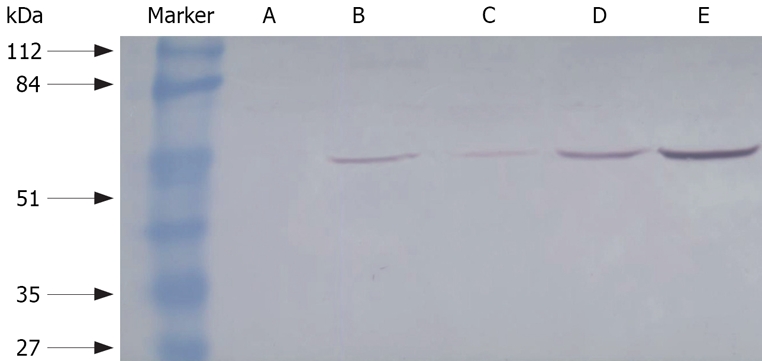
NF-κB binding activity in mouse livers with Western blot assay. A: Normal group; B: Rosiglitazone treatment; C: Praziquantel plus rosiglitazone treatment; D: Praziquantel treatment; E: Model group.
Expression of PPARγ-mRNA
Levels of PPARγ mRNAs were markedly higher in groups A [-16.557 ± (-3.022)], D [-18.217 ± (-4.498)] and E [-18.212 ± (-3.909)] than that in either group B [-27.315 ± (-6.348)] or C [-25.647 ± (-5.694), P < 0.05] as summarized in Table 3.
DISCUSSION
PPARγ is a ligand-activated nuclear transcription factor which belongs to the nuclear hormone receptor super-family. Normally, PPARγ expresses in quiescent human liver HSC, but significantly lowers both its translational and transcriptional activities after HSC activation in culture[15,16]. Some recent findings support a role of PPARγ in the development of liver fibrosis and suggest that PPARγ ligands can be therapeutically used to inhibit HSC activation and the progression to liver fibrosis[17–19].
A mouse model of liver fibrosis through Japonicum cercariae infection was generated, and then treated with PPARγ ligand, rosiglitazone and its additive, and praziquantel to diminish liver fibrosis in the current study. Our results demonstrated that the level of hepatic PPARγ mRNA markedly decreased in the model mice and praziquantel treated group as compared with the normal mice. However, rosiglitazone significantly increased the expression of PPARγ and decreased HSC activation and liver fibrosis progression.
Our mouse model successfully showed the key pathogenic events in liver fibrosis: the activation of HSCs and transformation of HSCs into myofibroblasts. Consequently, livers of model mice exhibited cellular changes including the appearance of cytoskeletal protein α-SMA and substantial expression of TGF-β, both fibrogenic cytokines which are activated accompanying liver HSC activation[20,21].
Based on the results from histopathological and immunohistochemical analyses, current fibrosis model clearly confirmed that an imbalance between the synthesis and degradation of ECM in chronic liver injury is the indispensable process developing into liver fibrosis. Although there are three large families of ECM proteins, glycoproteins, collagens and proteoglycans[22,23], excessive depositions of types I and III collagens are mainly pathologic characteristics of liver fibrosis due to Schistosoma japonicum infection. Our results further demonstrated that rosiglitazone alone or plus praziquantel can reduce the inflammation and liver fibrosis with Schistosoma infection by reducing the hepatic expression of TGF-β1, α-SMA, types I & III collagens. These results also suggest that PPARγ ligand, rosiglitazone can impede liver fibrosis after Schistosoma infection.
To seek possible regulatory mechanism in liver fibrosis, we further measured some cell factors such as TNF-α and IL-6, and the nuclear factors NF-κB in our model mice with/out treatment. TNF-α, a pro-inflammatory cytokine, plays a key role in a wide variety of physiological processes, including inflammation, proliferation and programmed cell death[24] which lead to regeneration of ECM and fibrogenesis[25]. Our results showed that the serum level of TNF-α and IL-6 increased in liver tissues. These data also provided a further evidence for the role of these cytokines in inducing hepatic fibrosis. Rosiglitazone alone or rosiglitazone plus praziquantel significantly reduced the serum level of TNF-α and IL-6, suggesting that rosiglitazone could be an alternative treatment for liver fibrosis after liver injury or infection.
NF-κB, a heterodimer, is a functional protein which is regulated by interaction with a family of regulatory proteins, the inhibitor of nuclear factor κB (IκB) proteins may be stimulated by many factors, such as TNF-α and lipopolysaccharide, via the phosphorylation and degradation of IκB. Activated NF-κB then transports into the cell nucleus and combines with gene promoters to induce the transcription of many cytokines and adhesion molecules[26,27]. Physical stress, oxidative stress, and exposure to certain chemicals can also activate NF-κB, suggesting its critical functions in mediating stress responses as well[28]. Recent studies show that the activation of NF-κB has a great impact on the pathogenesis of liver fibrosis through regulating hepatocyte, HSC and Kupffer cells[29,30]. Some studies demonstrate that NF-κB is also associated with the development of the activated phenotype of HSC and promotes survival of activated HSC[31] by protecting activated HSCs against TNF-induced apoptosis[32]. Notably, NF-κB binding activity increased in our mouse fibrosis model, while rosiglitazone significantly decreased the activity of, suggesting further that rosiglitazone could partially inhibit liver fibrosis by down-regulating NF-κB as well.
As indicated, our study showed a significant reduction of liver fibrosis following the treatment with rosiglitazone, a PPAR-γ ligand, in a murine model of hepatic fibrosis induced by Schistosoma japonicum. The effect of this compound on preventing liver fibrosis may be through down-regulation of liver TGF-β1 and collagens, and reduction of inflammatory mediators (IL-6, TNF-α, NF-κB). It suggested the use of rosiglitazone for the treatment of liver fibrosis.
Interestingly, our results showed that praziquantel alone reduced markedly the serum level of TNF-α, NF-κB binding activity, and volumes of TGF-β1 and type I collagen in liver fibrosis due to Schistosoma infection. These data may suggest an antifibrotic effect of praziquantel in the early stage of liver fibrosis. Praziquantel might be able to block liver fibrosis through killing parasite to alleviate liver inflammation. Further studies on mechanisms of rosiglitazone and praziquantel during liver injury or infection may shed lights on developing therapeutic methods in clinical practice.
COMMENTS
Background
Hepatic schistosomiasis is one of the most prevalent forms of chronic liver diseases in the world, resulting in the morbidity due to its complications of liver fibrosis from infection. However, there are currently few medicines or means available to control and treat fibrosis in schistosomiasis. Many studies demonstrate that PPARγ is mainly expressed in human HSC and contributes to the process of liver fibrogenesis, but the identification of PPARγ as a novel modulator to liver fibrosis remains controversial.
Research frontiers
Current findings further support the role of PPARγ in the development of liver fibrosis and suggest that PPARγ ligands can be therapeutically used to inhibit HSC activation and the progression of liver fibrosis.
Innovations and breakthroughs
This is a first report about PPARγ and liver fibrosis due to Schistosoma infection. This study evaluates the relationship of PPARγ ligand and hepatic fibrosis due to Schistosoma infection.
Applications
This study provides a perspective for a new therapeutic approach to prevent liver fibrosis following Schistosoma japonicum infection.
Peer review
The study reported a significant reduction of liver fibrosis following the treatment with rosiglitazone, a PPAR-γ ligand, in a murine model of hepatic fibrosis induced by Schistosoma japonicum. The effect of this compound on preventing liver fibrosis may be through down-regulation of liver TGF-β1 and collagens, and reduction of inflammatory mediators (IL-6, TNF-α, NF-κB). The study is interesting and may herald a new therapeutic approach to prevent liver fibrosis following Schistosoma japonicum infection.
Peer reviewer: Ana Cristina Simões e Silva, Professor, Pediatrics Department, Federal University of Minas Gerais Institution, Avenida Professor Alfredo Balena, 190, Belo Horizonte 30130-100, Brazil
S- Editor Li DL L- Editor Ma JY E- Editor Yin DH
References
- 1.Bartley PB, Ramm GA, Jones MK, Ruddell RG, Li Y, McManus DP. A contributory role for activated hepatic stellate cells in the dynamics of Schistosoma japonicum egg-induced fibrosis. Int J Parasitol. 2006;36:993–1001. doi: 10.1016/j.ijpara.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 2.Gutierrez-Ruiz MC, Gomez-Quiroz LE. Liver fibrosis: searching for cell model answers. Liver Int. 2007;27:434–439. doi: 10.1111/j.1478-3231.2007.01469.x. [DOI] [PubMed] [Google Scholar]
- 3.Parsons CJ, Takashima M, Rippe RA. Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol Hepatol. 2007;22 Suppl 1:S79–S84. doi: 10.1111/j.1440-1746.2006.04659.x. [DOI] [PubMed] [Google Scholar]
- 4.Galli A, Svegliati-Baroni G, Ceni E, Milani S, Ridolfi F, Salzano R, Tarocchi M, Grappone C, Pellegrini G, Benedetti A, et al. Oxidative stress stimulates proliferation and invasiveness of hepatic stellate cells via a MMP2-mediated mechanism. Hepatology. 2005;41:1074–1084. doi: 10.1002/hep.20683. [DOI] [PubMed] [Google Scholar]
- 5.Zhang XL, Liu JM, Yang CC, Zheng YL, Liu L, Wang ZK, Jiang HQ. Dynamic expression of extracellular signal-regulated kinase in rat liver tissue during hepatic fibrogenesis. World J Gastroenterol. 2006;12:6376–6381. doi: 10.3748/wjg.v12.i39.6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reeves HL, Friedman SL. Activation of hepatic stellate cells-a key issue in liver fibrosis. Front Biosci. 2002;7:d808–d826. doi: 10.2741/reeves. [DOI] [PubMed] [Google Scholar]
- 7.Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131:1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- 8.Yang C, Zeisberg M, Mosterman B, Sudhakar A, Yerramalla U, Holthaus K, Xu L, Eng F, Afdhal N, Kalluri R. Liver fibrosis: insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology. 2003;124:147–159. doi: 10.1053/gast.2003.50012. [DOI] [PubMed] [Google Scholar]
- 9.Shearer BG, Hoekstra WJ. Recent advances in peroxisome proliferator-activated receptor science. Curr Med Chem. 2003;10:267–280. doi: 10.2174/0929867033368295. [DOI] [PubMed] [Google Scholar]
- 10.Berkenstam A, Gustafsson JA. Nuclear receptors and their relevance to diseases related to lipid metabolism. Curr Opin Pharmacol. 2005;5:171–176. doi: 10.1016/j.coph.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Marra F, Efsen E, Romanelli RG, Caligiuri A, Pastacaldi S, Batignani G, Bonacchi A, Caporale R, Laffi G, Pinzani M, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466–478. doi: 10.1053/gast.2000.9365. [DOI] [PubMed] [Google Scholar]
- 12.Galli A, Crabb D, Price D, Ceni E, Salzano R, Surrenti C, Casini A. Peroxisome proliferator-activated receptor gamma transcriptional regulation is involved in platelet-derived growth factor-induced proliferation of human hepatic stellate cells. Hepatology. 2000;31:101–108. doi: 10.1002/hep.510310117. [DOI] [PubMed] [Google Scholar]
- 13.Galli A, Crabb DW, Ceni E, Salzano R, Mello T, Svegliati-Baroni G, Ridolfi F, Trozzi L, Surrenti C, Casini A. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology. 2002;122:1924–1940. doi: 10.1053/gast.2002.33666. [DOI] [PubMed] [Google Scholar]
- 14.Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The morphology of cirrhosis. Recommendations on definition, nomenclature, and classification by a working group sponsored by the World Health Organization. J Clin Pathol. 1978;31:395–414. doi: 10.1136/jcp.31.5.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang L, Chan CC, Kwon OS, Liu S, McGhee J, Stimpson SA, Chen LZ, Harrington WW, Symonds WT, Rockey DC. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G902–G911. doi: 10.1152/ajpgi.00124.2006. [DOI] [PubMed] [Google Scholar]
- 16.Guo YT, Leng XS, Li T, Peng JR, Song SH, Xiong LF, Qin ZZ. Effect of ligand of peroxisome proliferator-activated receptor gamma on the biological characters of hepatic stellate cells. World J Gastroenterol. 2005;11:4735–4739. doi: 10.3748/wjg.v11.i30.4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawaguchi K, Sakaida I, Tsuchiya M, Omori K, Takami T, Okita K. Pioglitazone prevents hepatic steatosis, fibrosis, and enzyme-altered lesions in rat liver cirrhosis induced by a choline-deficient L-amino acid-defined diet. Biochem Biophys Res Commun. 2004;315:187–195. doi: 10.1016/j.bbrc.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 18.Marra F, DeFranco R, Robino G, Novo E, Efsen E, Pastacaldi S, Zamara E, Vercelli A, Lottini B, Spirli C, et al. Thiazolidinedione treatment inhibits bile duct proliferation and fibrosis in a rat model of chronic cholestasis. World J Gastroenterol. 2005;11:4931–4938. doi: 10.3748/wjg.v11.i32.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 2006;350:385–391. doi: 10.1016/j.bbrc.2006.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang W, Yang CQ, Liu WB, Wang YQ, He BM, Wang JY. Blockage of transforming growth factor beta receptors prevents progression of pig serum-induced rat liver fibrosis. World J Gastroenterol. 2004;10:1634–1638. doi: 10.3748/wjg.v10.i11.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kershenobich Stalnikowitz D, Weissbrod AB. Liver fibrosis and inflammation. A review. Ann Hepatol. 2003;2:159–163. [PubMed] [Google Scholar]
- 22.Tsukada S, Parsons CJ, Rippe RA. Mechanisms of liver fibrosis. Clin Chim Acta. 2006;364:33–60. doi: 10.1016/j.cca.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 23.Benyon RC, Arthur MJ. Extracellular matrix degradation and the role of hepatic stellate cells. Semin Liver Dis. 2001;21:373–384. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 24.Theiss AL, Simmons JG, Jobin C, Lund PK. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. J Biol Chem. 2005;280:36099–36109. doi: 10.1074/jbc.M505291200. [DOI] [PubMed] [Google Scholar]
- 25.Simeonova PP, Gallucci RM, Hulderman T, Wilson R, Kommineni C, Rao M, Luster MI. The role of tumor necrosis factor-alpha in liver toxicity, inflammation, and fibrosis induced by carbon tetrachloride. Toxicol Appl Pharmacol. 2001;177:112–120. doi: 10.1006/taap.2001.9304. [DOI] [PubMed] [Google Scholar]
- 26.Cogswell PC, Kashatus DF, Keifer JA, Guttridge DC, Reuther JY, Bristow C, Roy S, Nicholson DW, Baldwin AS Jr. NF-kappa B and I kappa B alpha are found in the mitochondria. Evidence for regulation of mitochondrial gene expression by NF-kappa B. J Biol Chem. 2003;278:2963–2968. doi: 10.1074/jbc.M209995200. [DOI] [PubMed] [Google Scholar]
- 27.Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Stark GR. NFkappaB-dependent signaling pathways. Exp Hematol. 2002;30:285–296. doi: 10.1016/s0301-472x(02)00777-4. [DOI] [PubMed] [Google Scholar]
- 29.Mann DA, Smart DE. Transcriptional regulation of hepatic stellate cell activation. Gut. 2002;50:891–896. doi: 10.1136/gut.50.6.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smart DE, Vincent KJ, Arthur MJ, Eickelberg O, Castellazzi M, Mann J, Mann DA. JunD regulates transcription of the tissue inhibitor of metalloproteinases-1 and interleukin-6 genes in activated hepatic stellate cells. J Biol Chem. 2001;276:24414–24421. doi: 10.1074/jbc.M101840200. [DOI] [PubMed] [Google Scholar]
- 31.Muhlbauer M, Weiss TS, Thasler WE, Gelbmann CM, Schnabl B, Scholmerich J, Hellerbrand C. LPS-mediated NFkappaB activation varies between activated human hepatic stellate cells from different donors. Biochem Biophys Res Commun. 2004;325:191–197. doi: 10.1016/j.bbrc.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 32.Elsharkawy AM, Oakley F, Mann DA. The role and regulation of hepatic stellate cell apoptosis in reversal of liver fibrosis. Apoptosis. 2005;10:927–939. doi: 10.1007/s10495-005-1055-4. [DOI] [PubMed] [Google Scholar]