

Abstract
The heterologous expression and activation of the human P2Y2 nucleotide receptor (P2Y2R) in human 1321N1 astrocytoma cells stimulates α-secretase-dependent cleavage of the amyloid precursor protein (APP), causing extracellular release of the non-amyloidogenic protein sAPPα. To determine whether a similar response occurs in a neuronal cell, we analyzed whether P2Y2R-mediated production of sAPPα occurs in rat primary cortical neurons (rPCNs). In rPCNs, P2Y2R mRNA and receptor activity were virtually absent in quiescent cells, whereas overnight treatment with the pro-inflammatory cytokine interleukin-1β (IL-1β) upregulated both P2Y2R mRNA expression and receptor activity by four-fold. The upregulation of the P2Y2R was abrogated by pre-incubation with Bay 11-7085, an IκB-α phosphorylation inhibitor, which suggests that P2Y2R mRNA transcript levels are regulated through NF-κB signaling. Furthermore, the P2Y2R agonist UTP enhanced the release of sAPPα in rPCNs treated with IL-1β or transfected with P2Y2R cDNA. UTP-induced release of sAPPα from rPCNs was completely inhibited by pretreatment of the cells with the metalloproteinase inhibitor TAPI-2 or the phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002, and was partially inhibited by the MAPK/ERK kinase (MEK) inhibitor U0126 and the protein kinase C (PKC) inhibitor GF109203. These data suggest that P2Y2R-mediated release of sAPPα from cortical neurons is directly dependent on ADAM10/17 and PI3K activity, whereas ERK1/2 and PI3K activity may indirectly regulate APP processing. These results demonstrate that elevated levels of pro-inflammatory cytokines associated with neurodegenerative diseases, such as IL-1β, can enhance non-amyloidogenic APP processing through upregulation of the P2Y2R in neurons.
INTRODUCTION
Inflammation is a central component of several chronic neurological diseases including Alzheimer’s disease (Skaper, 2007). Various inflammatory mediators, such as cytokines, chemokines, proteases and protease inhibitors, have been detected in the brains of patients with Alzheimer’s disease (Parihar and Hemnani, 2004). Interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) are rapidly upregulated in a variety of tissues in response to pro-inflammatory stimuli and amplify downstream signaling cascades that mediate inflammation (Stoll et al., 2002). In Alzheimer's disease, IL-1β regulates the increased expression and processing of amyloid precursor protein (APP) (Griffin et al. 1994), the phosphorylation of tau (Sheng et al., 2000), the induction of secretory phospholipase in astrocytes (Moses et al., 2006) and the increased production of acetylcholinesterase (AChE) in neurons (Li et al., 2000; Sue and Griffin, 2006). These inflammatory processes result in neuronal stress and injury, which in turn promotes microglial cell activation and IL-1β overexpression in a self-propagating cycle (reviewed by Standridge, 2006). Recent studies, however, indicate that certain aspects of the inflammatory response, specifically constitutive IL-1β production, may have therapeutic potential for alleviating amyloid pathologies (Wyss-Coray et al., 2001; Shaftel et al., 2007).
Nucleotides co-released with neurotransmitters can activate a widely distributed family of nucleotide receptors in the central nervous system (CNS). P2 nucleotide receptors (i.e., ionotropic P2X receptors and G protein-coupled P2Y receptors) have been implicated in the regulation of numerous functions in the mammalian nervous system, including neurotransmission, glial cell migration, cell proliferation, ion transport, and neuronal cell apoptosis and survival (Burnstock, 2000; Ciccarelli et al., 2001; Weisman et al., 2005). The P2Y2 nucleotide receptor (P2Y2R) is distinguished among other human P2 receptors by its ability to be activated equipotently by ATP and UTP. The P2Y2R is upregulated in response to stress or injury in various cell types, including activated thymocytes, salivary gland epithelial cells, and vascular smooth muscle and endothelial cells (Koshiba et al., 1997; Turner et al., 1997; Seye et al., 1997 & 2002).
Previously, we demonstrated that the heterologously expressed G protein-coupled P2Y2R mediates the α-secretase-dependent release of sAPPα from human 1321N1 astrocytoma cells that are devoid of endogenous P2Y receptors (Camden et al., 2005). Cleavage of APP by α-secretase results in the shedding of nearly the entire ectodomain of APP to yield a large soluble protein called sAPPα, and essentially precludes the generation of neurotoxic Aβ peptide from the same APP molecule (Kojro and Fahrenholz, 2005). Several zinc metalloproteinases, including a disintegrin and metalloproteinase (ADAM) 9 and 10, tumor necrosis factor-alpha convertase (TACE/ADAM17), and the aspartyl protease β-secretase beta-site APP cleaving enzyme (BACE2) can cleave APP at the α-secretase site (Allinson et al., 2003). While the proteolytic cleavage of APP is constitutive, it can also be increased through activation of protein kinase C (PKC) or other signaling pathways by substances such as G protein-coupled receptor (GPCR) agonists (Nitsch et al., 1997; Kirazov et al., 1997). It has been postulated that the proteolytic products of APP can modulate neuronal excitability, synaptic plasticity, neurite outgrowth, synaptogenesis, and cell survival (Mattson, 1997).
The aim of this study was to determine whether cytokines such as IL-1β regulate functional expression of the P2Y2R in rat primary cortical neurons and whether P2Y2R activation can modulate APP processing in these cells.
MATERIALS AND METHODS
Materials
Fetal bovine serum (FBS) was obtained from Hyclone (Logan, UT). Dulbecco’s modified Eagle’s medium (DMEM), minimum essential medium (MEM), Neurobasal medium, penicillin (100 units/ml), streptomycin (100 units/ml) and B27-AO Neurobasal medium (B27 medium without cortex antioxidants) were obtained from Gibco-BRL (Carlsbad, CA). Rabbit anti-rat extracellular signal-regulated kinase 1 and 2 (ERK1/2), horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG, and HRP-conjugated goat anti-mouse IgG antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-rat sAPPα antibody was obtained from Signet Laboratories (Dedham, MA). All other antibodies were obtained from Cell Signaling Technology (Beverly, MA). The Dual Color Protein Standards and nitrocellulose membranes (0.45 mm) were obtained from Bio-Rad (Hercules, CA). LumiGLO chemiluminescent substrates were obtained from New England Biolabs (Beverly, MA). The RNeasy Plus Mini Kit was obtained from Qiagen (Chatsworth, CA). The First Strand cDNA Synthesis Kit was obtained from Roche (Indianapolis, IN). Real-time polymerase chain reaction (PCR) was performed on an Applied Biosystems 7500 Real-Time PCR machine with TaqMan Gene Expression assay probes (P2Y2R NM_017255.1 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), NM_017008.3) and TaqMan Universal PCR Master Mix (2X) (Foster City, CA). The GenCarrier™2 Transfection Reagent Kit was obtained from Epoch Biolabs (Sugar Land, TX). Nucleotides and all other biochemical reagents were obtained from Sigma Chemical Co. (St. Louis, MO).
Primary cell culture of cortical neurons
Experimental procedures for cell cultures of 95% pure rat primary cortical neurons (rPCNs) were carried out as previously described (Kong et al., 2005). Briefly, cerebral cortices from 18-day-old embryos of Sprague-Dawley rats were removed and the meninges were discarded. The brain tissue was mechanically dissociated in HGDMEM comprised of DMEM, 10% (v/v) FBS, 2 mM glutamine, 100 IU/ml penicillin, 100 mg/ml streptomycin, and 7.5 mg/ml fungizone. The tissue clumps were dispersed with a 10 ml pipette and suspended in 6 ml of 0.25% (w/v) trypsin at 37 °C for 10 min. Then, 2 ml of heat-inactivated horse serum were added to neutralize trypsin activity. The cell suspension was centrifuged at 900 g for 5 min and the cell pellet was suspended in HGDMEM. The resulting cell suspension was filtered through a sterilized 75 mm cell strainer (Becton Dickinson, Franklin Lakes, NJ) and the cells were seeded on plastic culture plates precoated with poly-D-lysine (0.1 µg/ml). After 16–18 h and every 3 days thereafter, the medium was replaced with B27-AO Neurobasal medium (2 mM glutamine, 100 IU/ml penicillin, 100 mg/ml streptomycin, 7.5 mg/ml fungizone, 10 ml of B27-AO Neurobasal medium, and Neurobasal medium to 500 ml) and 15 µg/ml 5-fluoro-2’-deoxyuridine was added to retard glial cell proliferation. The neurons were used for experiments after 7 days in culture (DIV7). Unless otherwise stated, DIV7 cells were treated for 24 h at 37 °C in serum-free DMEM with or without IL-1β at the indicated concentration. The next day, cells were either harvested or the medium was replaced with fresh serum-free DMEM containing UTP, PMA, or other compounds, as indicated in the figure legends.
Real-Time and Reverse Transcription-PCR analysis of P2Y2R mRNA expression
Total RNA was isolated from rPCNs using the RNeasy Plus Mini Kit (Qiagen). cDNA was synthesized from 500 ng purified RNA using the First Strand cDNA Synthesis Kit for RT-PCR (AMV; Roche). Specific oligonucleotide primers were designed to selectively amplify cDNA for P2Y2R and GAPDH, as previously described (Wang et al., 2005). The amplification was performed using 35 cycles of denatuation for P2Y2R and 30 cycles for GAPDH at 95 °C for 30 sec, with annealing at 60 °C for 30 sec and extension at 72 °C for 1 min. The resulting PCR products were resolved on a 1% (w/v) agarose gel containing 10 mg/ml ethidium bromide and photographed under UV illumination. Ten percent of the synthesized cDNA was used as a template in a 50 µl real-time PCR. For TaqMan quantitative real-time PCR analysis, the probes were labeled on the 5’ end with 6-carboxy-fluorescein phosphoramidite (FAM) (for P2Y2R) or VIC (for GAPDH), and at the 3’ end with minor groove binder (MGB) dye as the quencher. The GAPDH gene was used as a stable endogenous control. The samples were run in quadruplicate for the P2Y2R target and the endogenous GAPDH control. After computing the relative amounts of target gene and endogenous control for each sample, the final amount of target gene in the sample was calculated as a ratio of the amounts of P2Y2R to GAPDH, using Applied Biosystems software. Relative mRNA levels of the control were normalized to 1.
Single cell calcium assay
The intracellular free calcium concentration, [Ca2+]i, was quantified in single cells using the Ca2+-sensitive fluorescent dye fura-2, and the InCyt Dual-Wavelength Fluorescence Imaging System (Intracellular Imaging, Cincinnati, OH). Briefly, rat PCNs cultured on poly-D-lysine-coated coverslips were incubated with 2.5 mM fura-2-acetoxymethylester at 37 °C for 30 min in physiological salt solution (PSS) containing (mM) NaCl 138, KCl 5, CaCl2 2, MgCl2 1, HEPES 10, glucose 10, pH 7.4, and washed with PSS. The coverslips with fura-2-loaded cells were positioned on the stage of an inverted epifluorescence microscope (Nikon; model TMD) and stimulated with agonist at 37 °C, as described in the figure legends. The percentage of cells that responded to agonist was determined, as previously described (Kong et al., 2005). Viable cells were identified by responsiveness to carbachol and non-responding cells were eliminated.
Overexpression of P2Y2 receptors in rPCNs
Rat primary cortical neurons at DIV7 expressing undetectable levels of endogenous P2Y2R were transiently transfected with 1 µg of human P2Y2 receptor cDNA using the GenCarrier™2 Transfection Reagent Kit (Epoch Biolabs), according to the manufacturer’s instructions. The transiently-transfected cells were cultured for an additional 24–48 h in Neurobasal medium plus B27-AO Neurobasal medium and used to detect sAPPα release.
Western blot analysis
rPCNs were plated on 6-well plates and grown until DIV7. Cells were serum-starved and incubated in 1 ml of serum-free DMEM with or without various compounds for specified time periods, as indicated in the figure legends. At the end of the incubation period, the medium was collected and centrifuged at 12,000 × g for 5 min to remove cellular debris. The supernatant, representing conditioned medium, was stored at −80 °C until further use. To measure the level of secreted APPα, 200 µl of supernatant were diluted 4:1 with 50 µl of 5X Laemmli sample buffer (187.5 mM Tris-HCl, pH 6.8, 6% (w/v) SDS, 1.8% (v/v) β-mercaptoethanol and 0.003% (w/v) bromophenol blue). The cells were washed twice with ice-cold PBS, and lysed in 2X Laemmli sample buffer (200 µl/well).
Cell lysate and conditioned medium were heated for 5 min at 65 °C, subjected to 7.5% (w/v) SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes for protein immunoblotting. After overnight blocking at 4 °C with 5% (w/v) fat-free milk in TBS-T (10 mM Tris-HCl, pH 7.4, 120 mM NaCl, and 0.1% (v/v) Tween-20), membranes were incubated with either 1:1000 dilution of anti-ERK1/2, anti-phospho-ERK1/2, or anti-rat actin antibodies for 2 h at room temperature, or 1:1000 dilution of anti-sAPPα antibodies overnight at 4 °C followed by incubation with HRP-conjugated anti-rabbit or anti-mouse IgG antibodies (1:1000 dilution in TBS-T containing 5% (w/v) fat-free milk) for 1 h at room temperature. Protein immunoreactivity was visualized on autoradiographic film using the LumiGlo Chemiluminescence System (New England BioLabs), according to the manufacturer’s instructions. The protein bands detected on X-ray film were quantified using a computer-driven scanner and Quantity One software (Bio-Rad, Hercules, CA). The activation levels of kinases or sAPPα release were expressed as a percentage of controls (i.e., total kinase or actin, respectively).
Statistical analysis
Results are expressed as the means ± S.E.M. of data obtained from at least 3 experiments. Statistical analysis of data was performed using Graph Pad Prism version 5.0. Statistical significance was determined by one-way ANOVA followed by Newman-Keuls multiple comparison tests or 2-way ANOVA followed by Bonferroni post-tests. Differences were considered to be statistically significant when p < 0.05 (*) and not significant when p > 0.05 (n.s.).
RESULTS
Upregulation of functional P2Y2Rs in rat primary cortical neurons (rPCNs) by IL-1β
Several subtypes of P2 nucleotide receptor mRNA are expressed in rPCNs, including P2Y1, P2Y4, P2Y6, P2X3, P2X5, P2X6 and P2X7 (Kong et al., 2005; Weisman et al., 2005). However, P2Y2R mRNA and receptor activity are barely detectable in these cells (Weisman et al., 2005, Kong et al., 2005). Since previous studies demonstrated that IL-1β causes an increase in P2Y2R mRNA expression in mouse primary striatal astrocytes (Stella et al., 1997), we used real-time PCR analysis to examine whether this cytokine could also increase P2Y2R mRNA expression in rPCNs. After overnight incubation with IL-1β (0–100 ng/ml), P2Y2R mRNA expression was upregulated in a dose-dependent manner (Figures 1A and B). IL-1β did not significantly increase the expression of P2Y4R or P2Y6R mRNA (data not shown). Furthermore, P2Y2R mRNA upregulation was abrogated in a concentration-dependent manner by a 30 min pre-incubation with Bay 11–7082 (Figure 1B), an IκB-α phosphorylation inhibitor (Pierce et al., 1997), which suggests that IκB/NF-κB signaling pathway regulates transcriptional activation of the P2Y2R. Stimulation of IL-1β-treated rPCNs with the P2Y2R agonist UTP caused an increase in [Ca2+]i (Figure 1C) and activation of the mitogen-activated protein kinases (MAPKs) ERK1/2 (Figures 2A and B), indicating that functional expression of the P2Y2R is also upregulated by IL-1β in rPCNs. Co-incubation of rPCNs with TNF-α and IL-1β did not further enhance P2Y2R-mediated calcium mobilization (data not shown) or ERK1/2 phosphorylation as compared to IL-1β alone (Figures 2C and D).
Figure 1. IL-1β upregulates P2Y2 receptor mRNA and function in rPCNs.
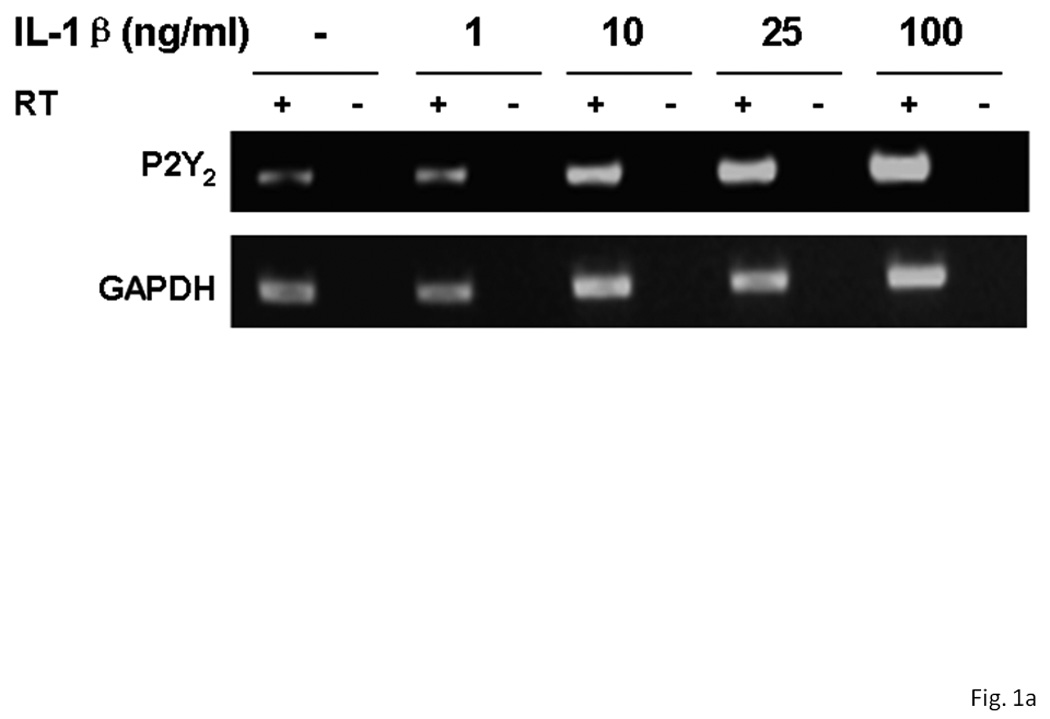
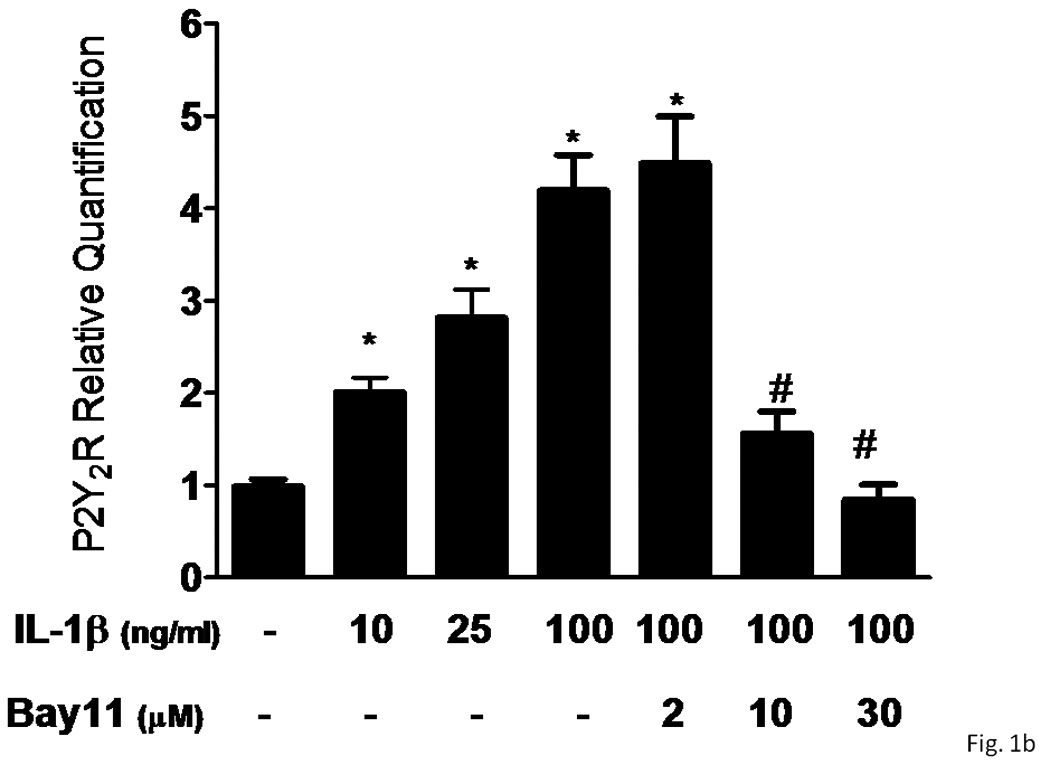
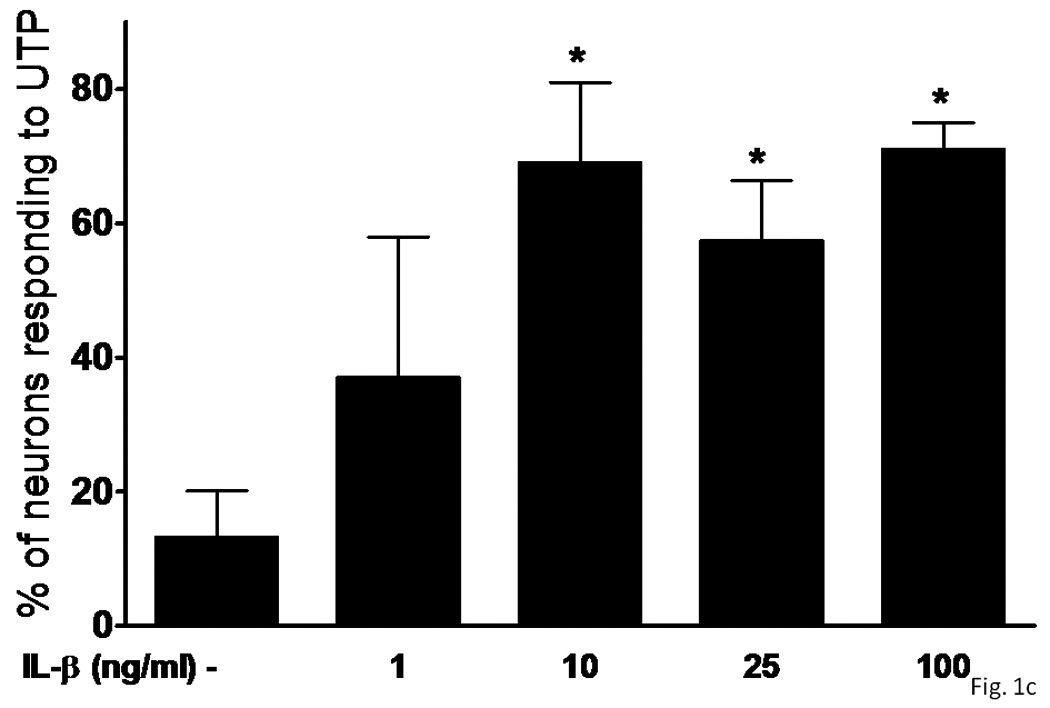
(A) Rat primary cortical neurons (rPCNs) at DIV7 were treated with the indicated concentration of IL-1β for 24 h at 37 °C in serum-free DMEM and P2Y2R mRNA levels were detected by real-time PCR using GAPDH as a control. RT + or – indicates usage of reverse transcriptase in the PCR. (B) The effect on IL-1β-induced P2Y2R mRNA expression of a 30 min pretreatment of rPCNs with the indicated concentration of the IκB-α phosphorylation inhibitor Bay11 is shown. Real-time PCR data was expressed as a ratio of P2Y2R to GAPDH mRNA levels (n=6). Bay11 preincubation alone did not significantly change P2Y2R mRNA expression levels as compared to the untreated control (data not shown). * represents significant differences (p < 0.05), as compared to control conditions, and # represents significant differences (p < 0.05), as compared to rPCNs treated with 100 ng/ml IL-1β. (C) IL-1β-treated cells on coverslips were loaded with fura-2 and coverslips were positioned on the stage of an inverted epifluorescence microscope for measurement of [Ca2+]i, as described in the “Materials and Methods”. Then, rPCNs were incubated in PSS with UTP (100 µM), and the time-dependent increase in [Ca2+]i was determined. Results are expressed as the percentage of rPCNs responding to UTP. Data represent the means ± S.E.M. of results from at least three experiments where “n.s.” represents no significant differences (p > 0.05) and * represents significant differences (p < 0.05), as compared to rPCNs treated without IL-1β.
Figure 2. UTP stimulates phosphorylation of ERK1/2 in IL-1β-treated rPCNs.
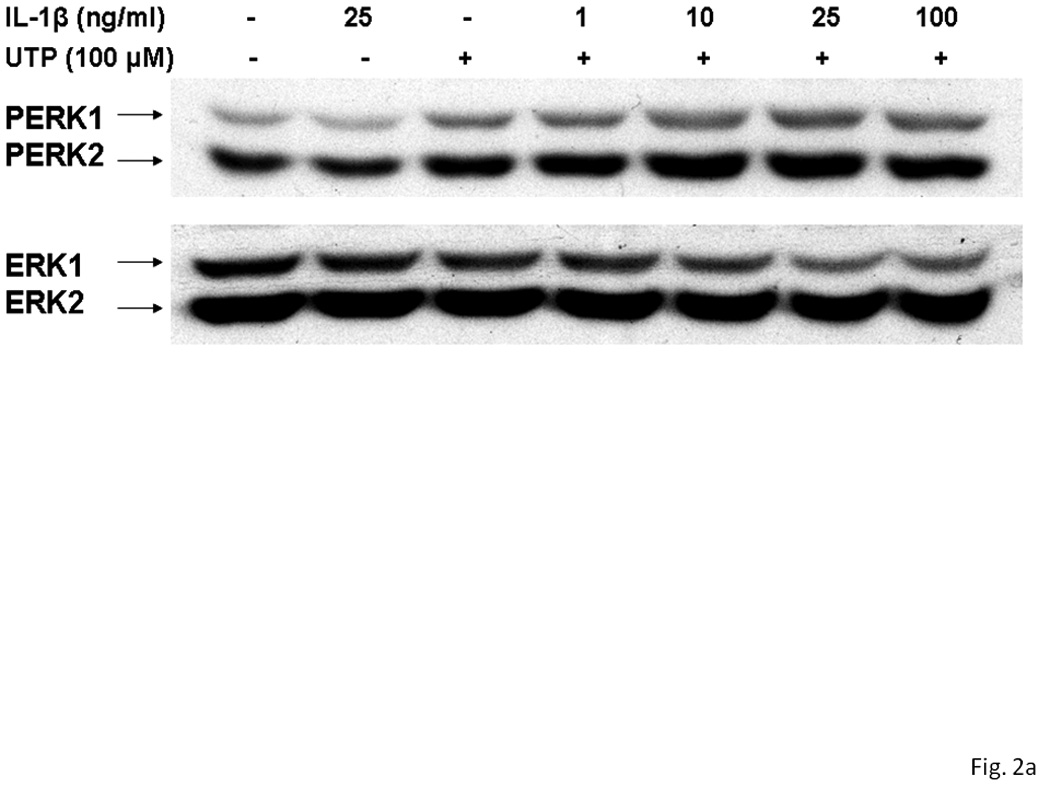
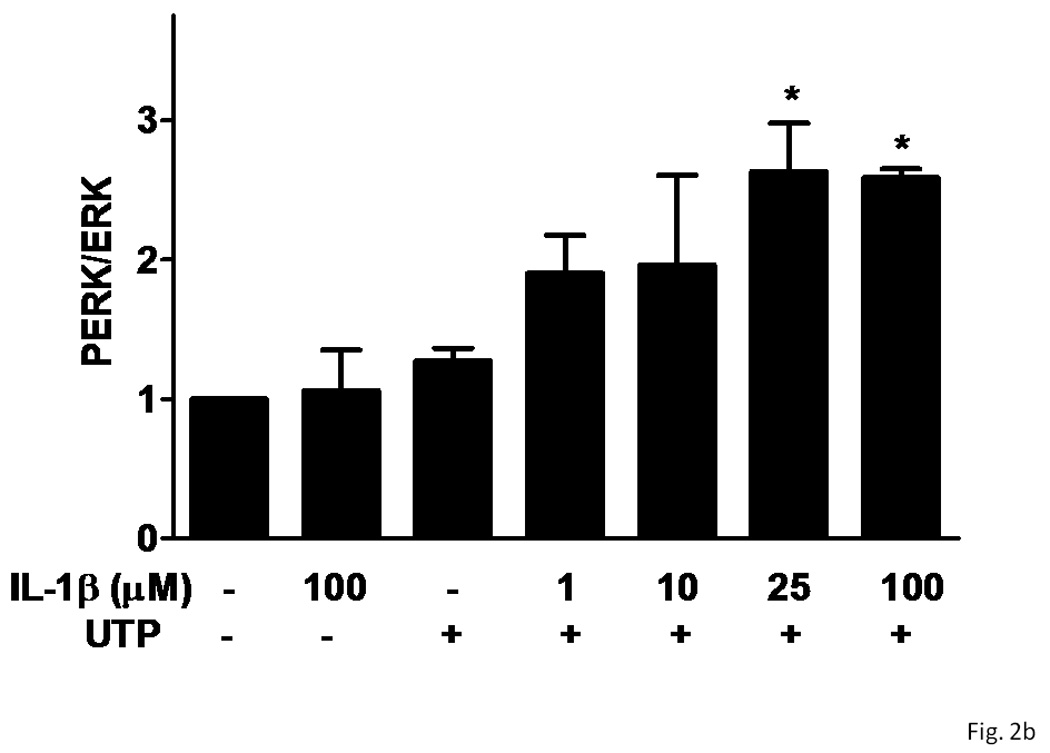
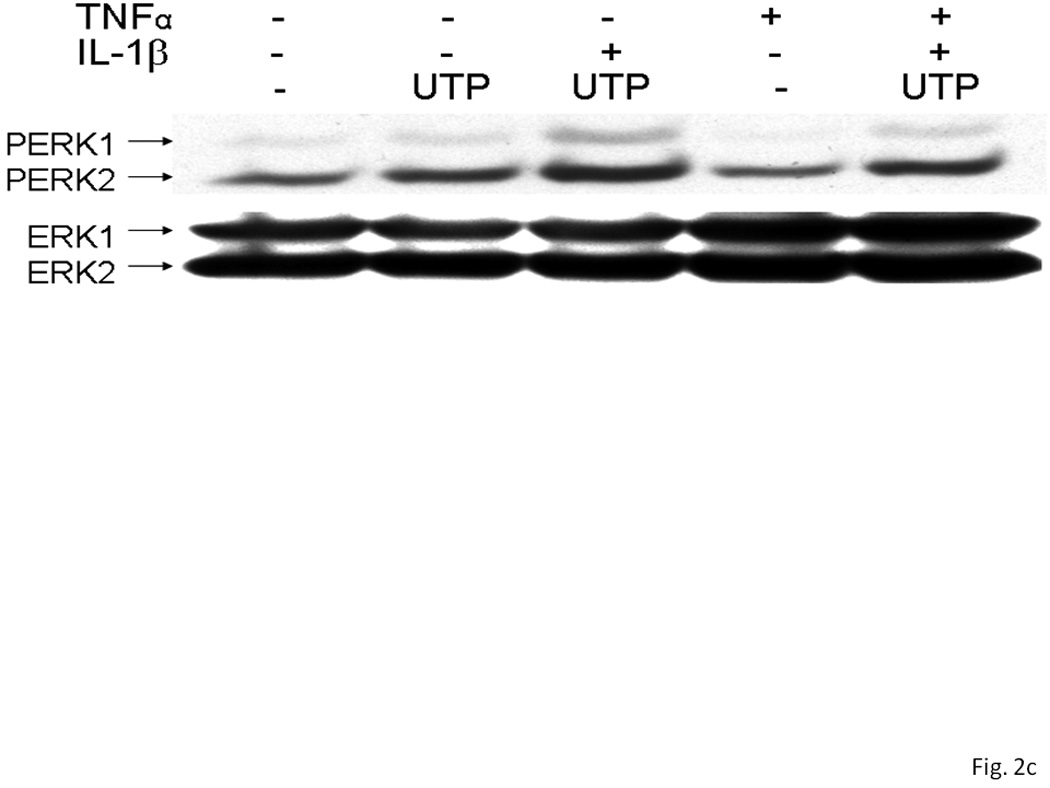
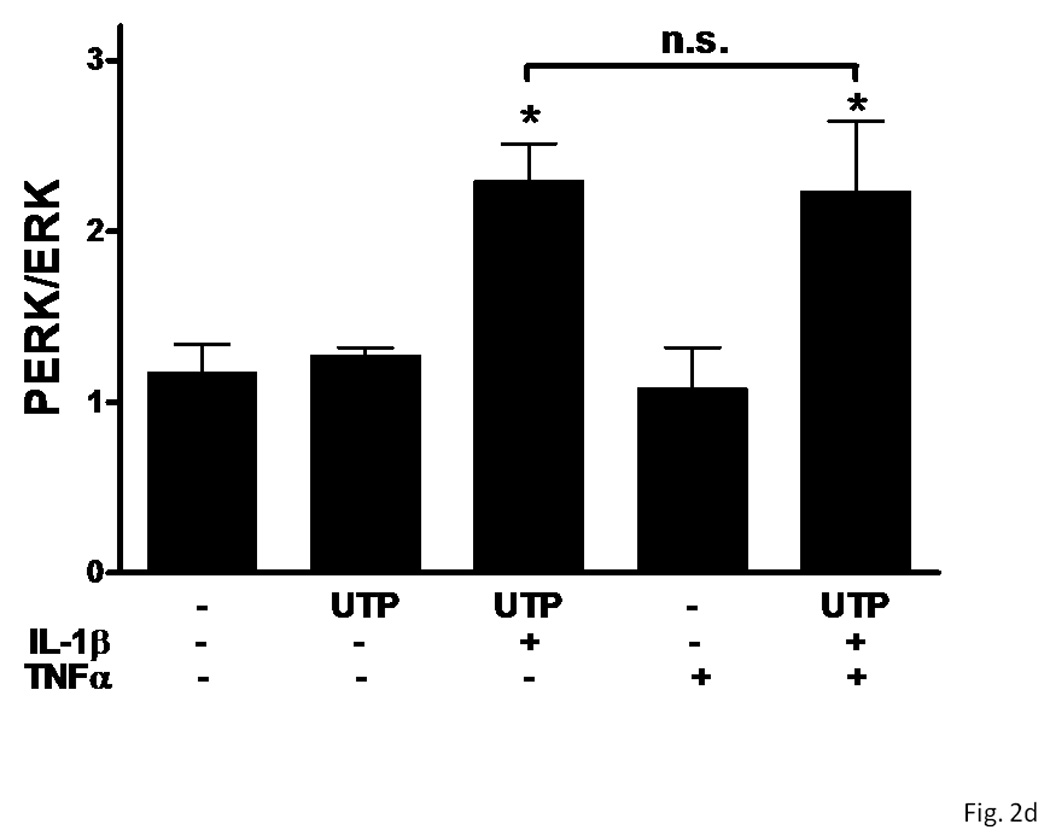
rPCNs were pretreated with the indicated concentration of IL-1β and/or TNF-α in serum-free DMEM for 24 h at 37 °C. Then, the medium was removed and cells were incubated for 10 min in serum-free DMEM with or without 100 µM UTP at 37 °C. (A, C) Cell lysates were subjected to Western blot analysis for phosphorylated ERK1/2 and total ERK1/2. (B, D) UTP-induced ERK1/2 phosphorylation was expressed as the ratio of phosphorylated ERK1/2 to total ERK1/2 after subtracting the basal level of ERK1/2 phosphorylation in the absence of UTP. The increase of ERK1/2 phosphorylation induced by IL-1β and/or TNF-α in rPCNs was expressed as a ratio of the UTP-stimulated rPCNs in the absence of cytokine pretreatment. Data represent the means ± S.E.M. of results from three experiments where “n.s.” represents no significant differences and * represents significant differences (p < 0.05), as compared to rPCNs treated without cytokines.
P2Y2R-mediated release of sAPPα from rPCNs treated with IL-1β
Overnight treatment of rPCNs with IL-1β (1–100 ng/ml) caused a dose-dependent increase in sAPPα release when Il-1β treated neurons were stimulated with UTP (100 µM) (Figure 3). UTP also increased the release of sAPPα from rPCNs transfected with P2Y2R cDNA, as compared to untransfected rPCNs (Figure 4), confirming the hypothesis that UTP enhances sAPPα release from rPCNs through activation of the P2Y2R subtype.
Figure 3. UTP induces sAPPα release in rPCNs pre-treated with IL-1β.
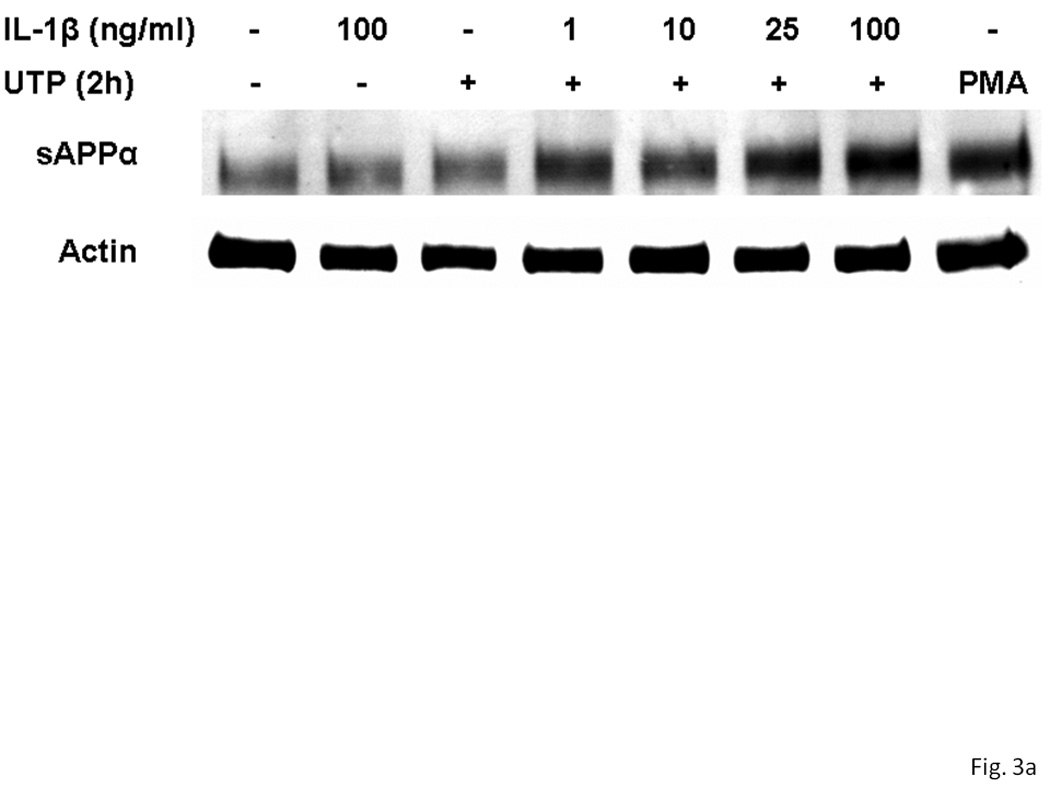
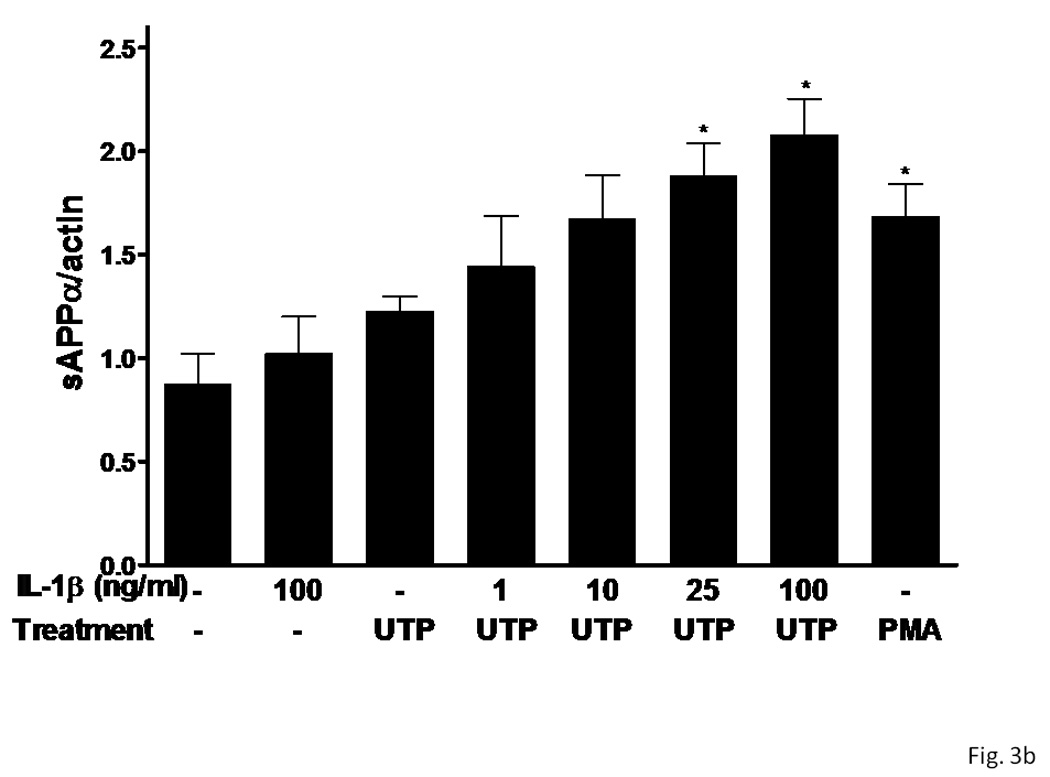
rPCNs were pretreated with the indicated concentration of IL-1β in serum-free DMEM for 24 h at 37 °C. Then, the medium was removed and cells were incubated for 2 h at 37 °C in serum-free DMEM with or without 100 µM UTP or 1 µM PMA, a positive control. (A) The medium was collected and assayed by Western blot analysis for sAPPα release, whereas the actin level in cell lysate was determined as a control. (B) UTP- and PMA-induced sAPPα release in rPCNs with or without IL-1β pretreatment is expressed relative to the actin level in cell lysate, after subtracting basal sAPPα release in the absence of IL-1β and UTP treatment. Data represent the means ± S.E.M. of results from at least three experiments, where * represents significant differences (p < 0.05), as compared to UTP-treated rPCNs in the absence of IL-1β pretreatment.
Figure 4. P2Y2 receptor expression in rPCNs enhances α-secretase-mediated APP processing induced by UTP.
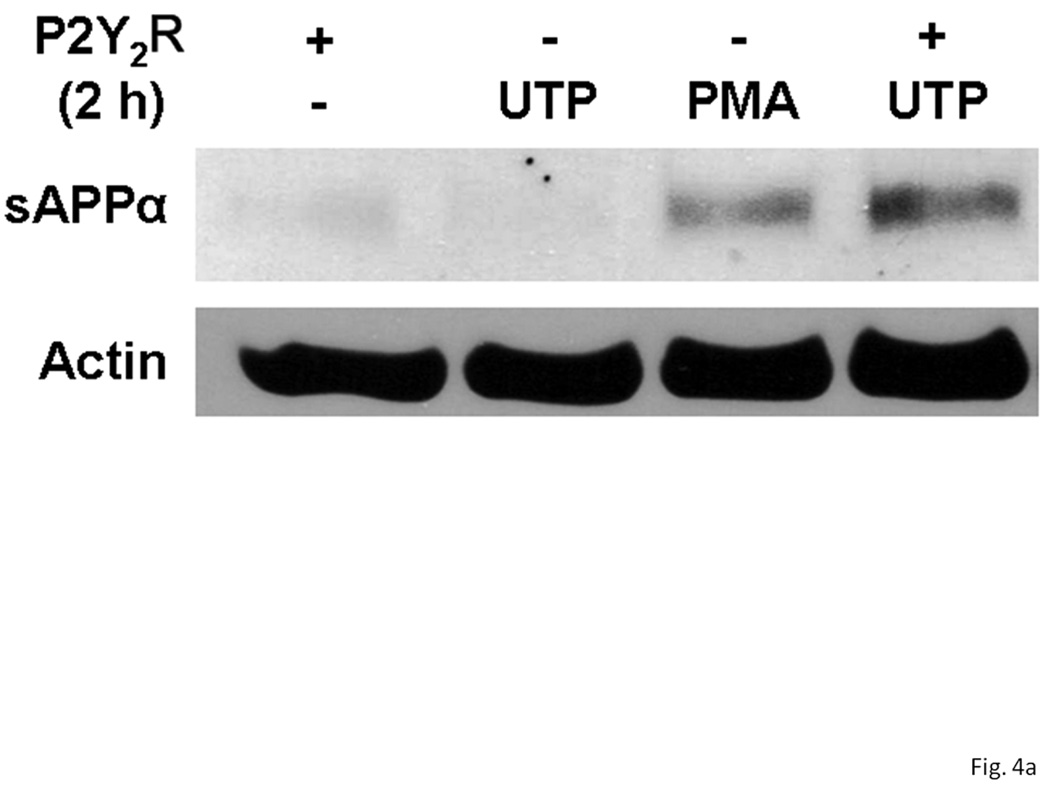
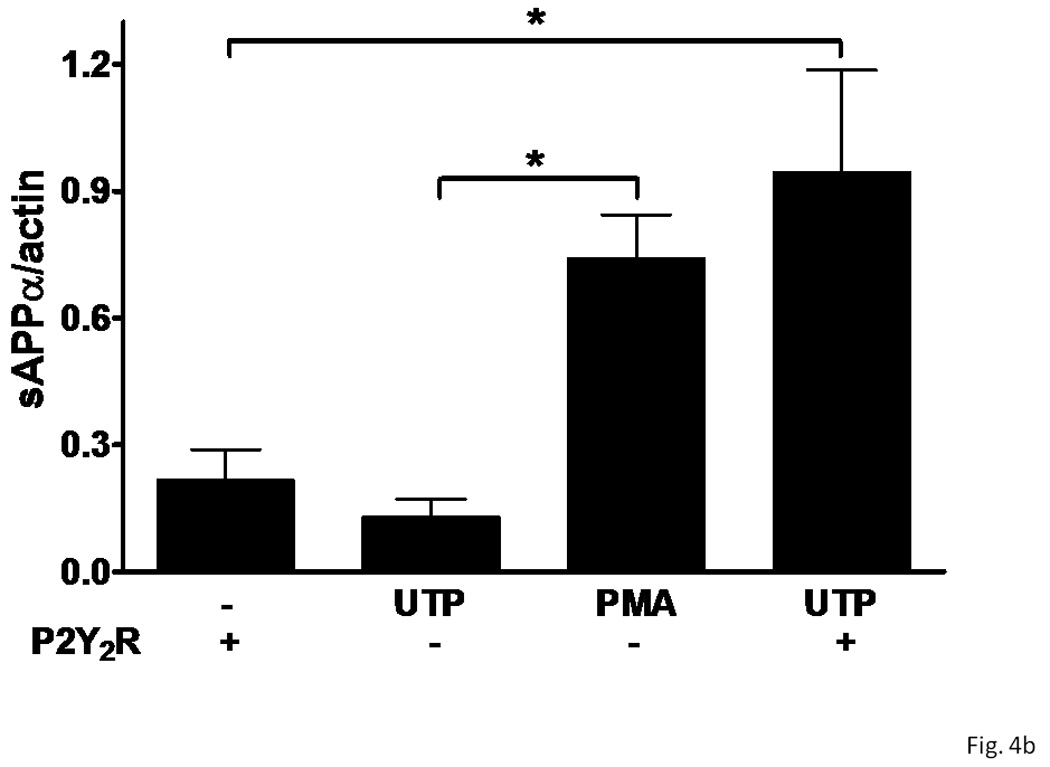
On DIV5, rPCNs were transfected with human P2Y2R cDNA (− indicates untransfected controls). On DIV7, the neurons were incubated for 2 h in serum-free DMEM with or without 100 µM UTP or 1 µM PMA at 37 °C. (A) sAPPα release into the medium was determined by Western blot analysis. (B) sAPPα release is expressed relative to the actin level in cell lysate. Data represent the means ± S.E.M. of results from three independent experiments, where * represents significant differences (p < 0.05) between the indicated sample groups.
P2Y2R-mediated release of sAPPα is dependent on ADAM10/17
Currently, several zinc metalloproteinases have been identified as potential α-secretases, including TACE/ADAM17 that catalyzes the shedding of the ectodomain of APP and other transmembrane proteins (Black et al., 1997; Moss et al., 1997), and ADAM10 and MDC9 that also catalyze sAPPα production (Lammich et al., 1999; Koike et al., 1999). In IL-1β-treated rPCNs, UTP-induced release of sAPPα was inhibited by the selective metalloproteinase inhibitor TAPI-2 (Figure 5), suggesting that P2Y2R-mediated APP processing in rPCNs is likely mediated by ADAM10/17.
Figure 5. A metalloprotease inhibitor prevents UTP-induced sAPPα release from rPCNs.
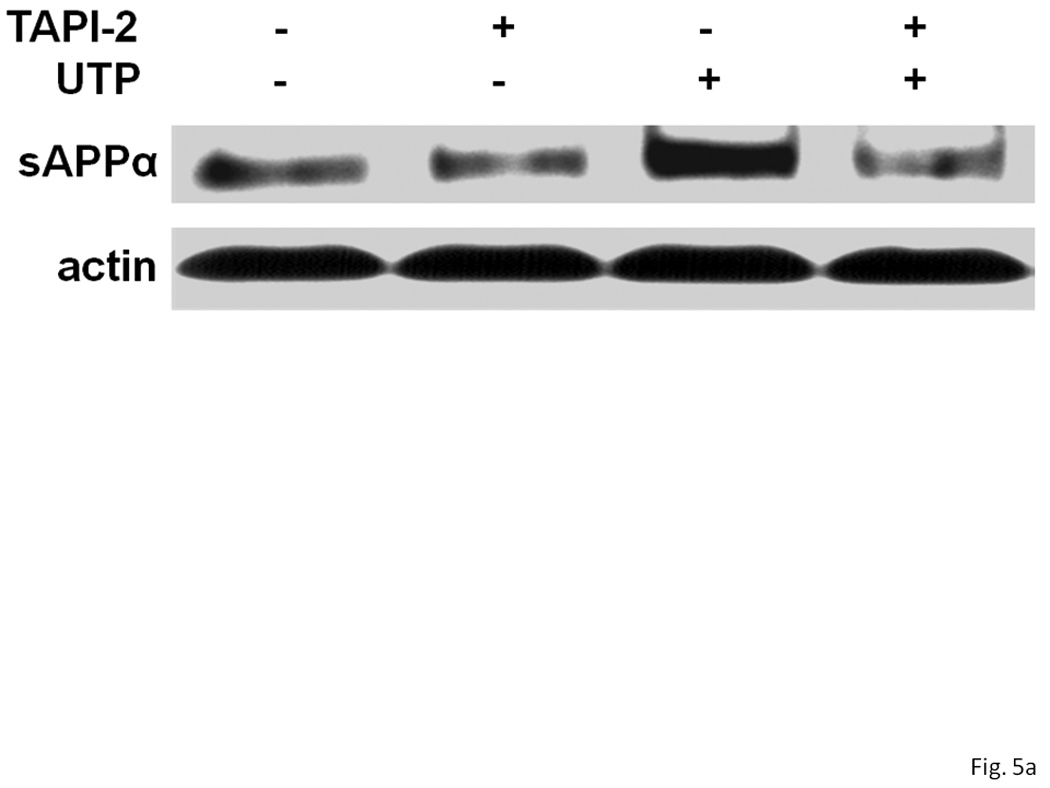
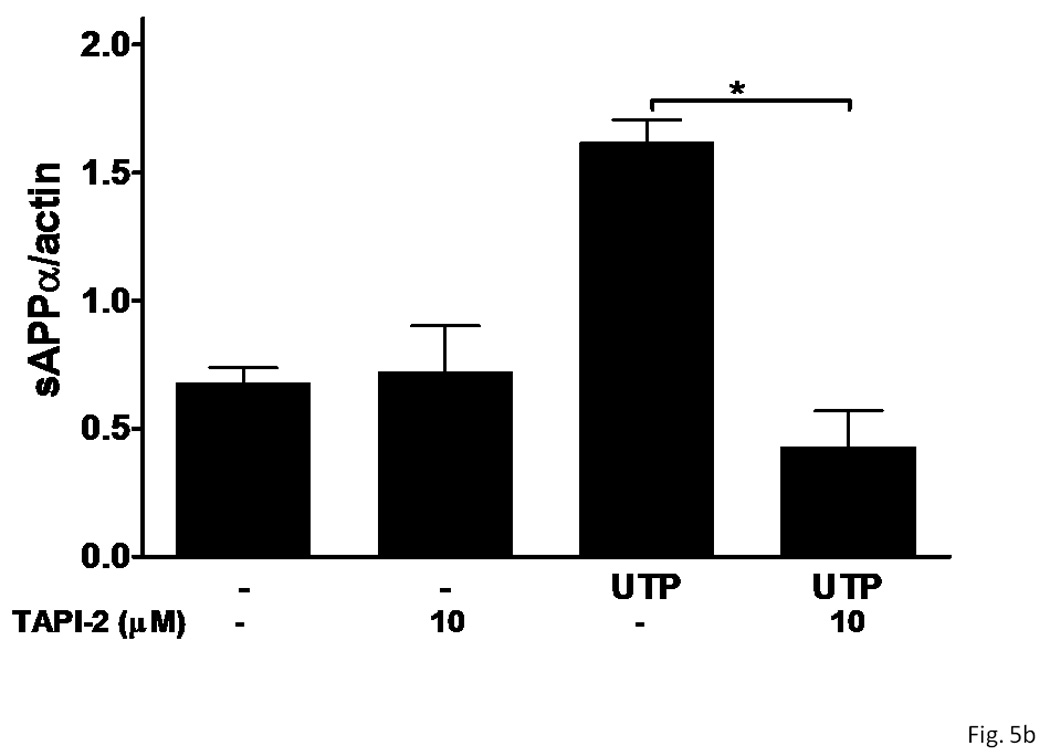
rPCNs were pretreated with 25 ng/ml IL-1β in serum-free DMEM for 24 h at 37 °C. The medium was replaced with fresh serum-free DMEM and the cells were incubated at 37 °C for 30 min with or without the metalloprotease inhibitor TAPI-2 (10 µM) followed by a 2 h incubation with or without UTP (100 µM). (A) The release of sAPPα into the medium was analyzed by Western blot analysis. (B) sAPPα release is expressed relative to the actin level in cell lysate. Data represent the means ± S.E.M. of results from six experiments, where * represents significant differences (p < 0.05) between the indicated sample groups analyzed by 2-way ANOVA followed by Bonferroni post-tests.
P2Y2R-mediated sAPPα release is partially dependent on PKC
P2Y2R activation causes the phospholipase C-dependent stimulation of PKC, and the activation of several other GPCRs has been reported to induce sAPPα release through PKC-dependent and -independent pathways (Nitsch et al., 1994 & 1995; Checler, 1995; LeBlanc et al., 1998). Therefore, we determined whether P2Y2R-mediated sAPPα release in rPCNs is dependent on activation of PKC. The general PKC inhibitor, GF109203, partially inhibited UTP-induced sAPPα release in IL-1β-treated neurons (Figure 6). Similarly, GF109203 significantly inhibited PMA-induced sAPPα release (Figure 6). These results indicate that P2Y2R-mediated sAPPα release is partially dependent on PKC.
Figure 6. UTP-induced sAPPα release is partially dependent on PKC activation in IL-1β-treated rPCNs.
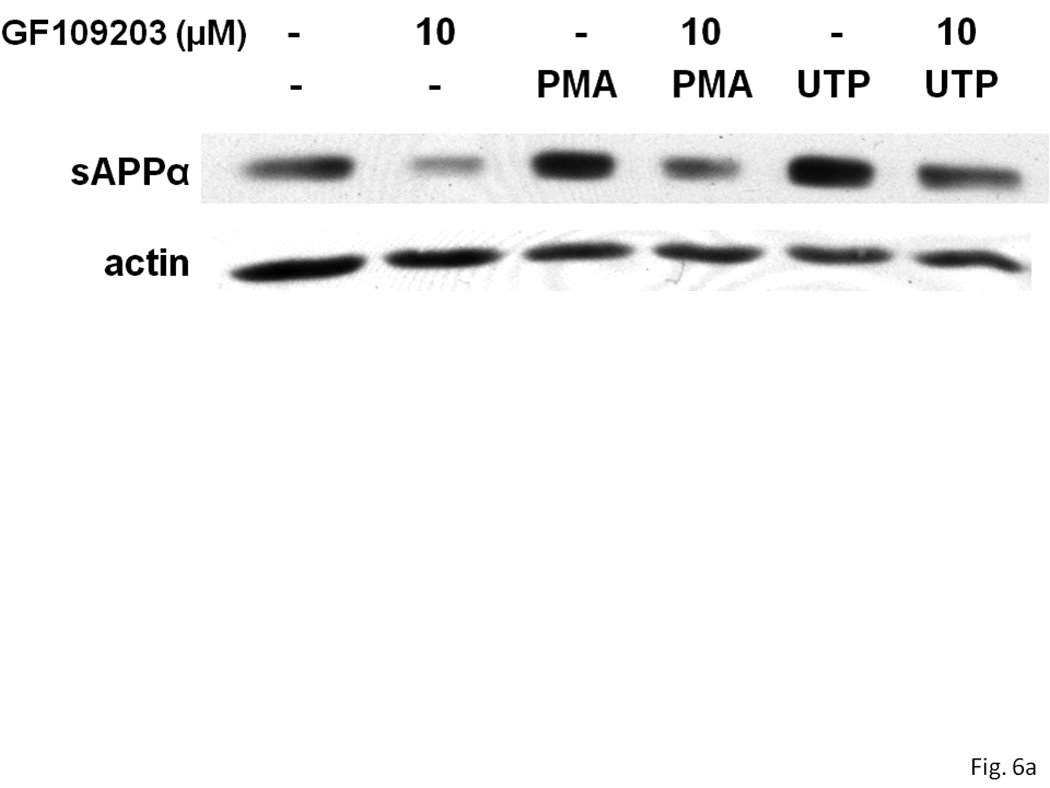
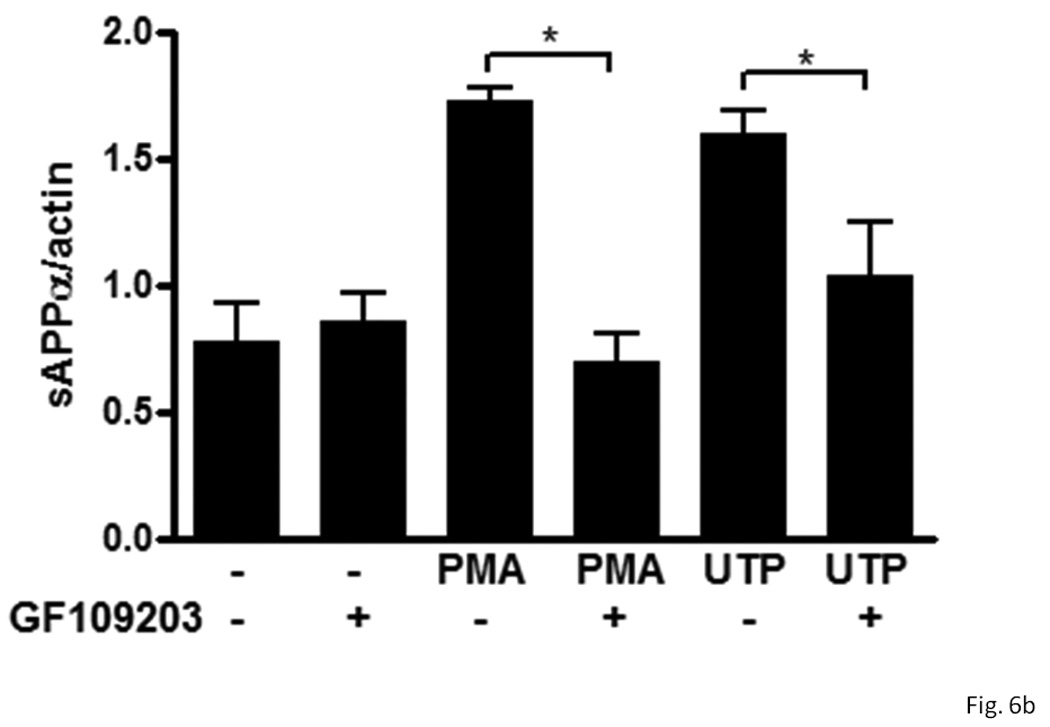
rPCNs were pretreated with 25 ng/ml IL-1β in serum-free DMEM for 24 h at 37 °C. The medium was replaced with fresh serum-free DMEM and the cells were incubated at 37 °C for 30 min with or without GF109203 (10 µM) followed by a 2 h incubation with or without UTP (100 µM) or PMA (1 µM). (A) The release of sAPPα into the medium was analyzed by Western blot analysis. (B) sAPPα release is expressed relative to the actin level in cell lysate. Data represent the means ± S.E.M. of results from three experiments, where * represents significant differences (p < 0.05) between the indicated sample groups analyzed by 2-way ANOVA followed by Bonferroni post-tests.
P2Y2R-mediated sAPPα release is dependent on PI3K
The release of sAPPα induced by insulin requires activation of the PI3K/Akt pathway (Solano et al., 2000). Since P2Y2Rs activate PI3K in HeLa cells, endothelial cells and rat primary astrocytes (Muscella et al., 2003; Kaczmarek et al., 2005; Wang et al., 2005), we determined whether PI3K/Akt regulates P2Y2R-mediated sAPPα release in IL-1β-treated rPCNs. As shown in Figure 7, incubation of IL-1β-treated rPCNs with the selective PI3K inhibitor LY294002 completely inhibited UTP-induced sAPPα release, but had no effect on constitutive release of sAPPα in IL-1β-treated rPCNs. These results suggest that UTP-induced sAPPα release in rPCNs is mediated through the PI3K/Akt signaling pathway.
Figure 7. Inhibition of PI3K prevents UTP-induced sAPPα release in IL-1β-treated rPCNs.
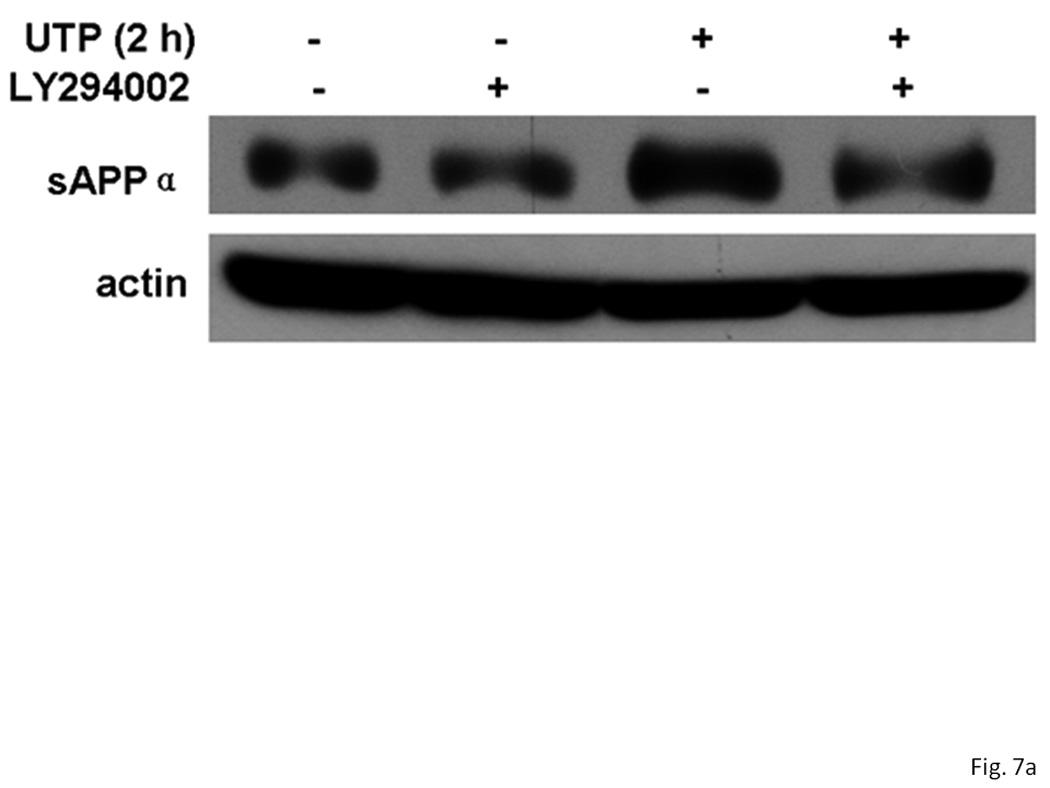
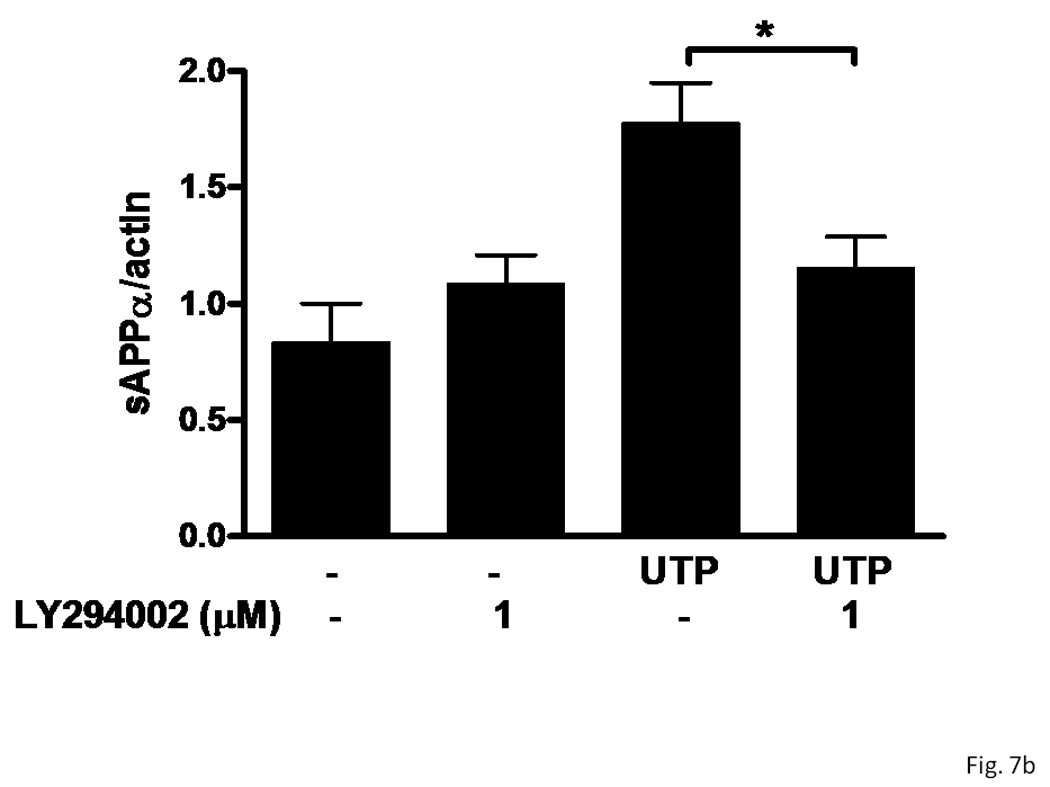
rPCNs were pretreated with 25 ng/ml IL-1β in serum-free DMEM for 24 h at 37 °C. The medium was replaced with fresh serum-free DMEM and the cells were incubated at 37 °C for 30 min with or without LY294002 (1 µM) followed by a 2 h incubation with or without UTP (100 µM). (A) The release of sAPPα into the medium was analyzed by Western blot analysis. (B) sAPPα release is expressed relative to the actin level in cell lysate. Data represent the means ± S.E.M. of results from three experiments, where * represents significant differences (p < 0.05) between the indicated sample groups analyzed by 2-way ANOVA followed by Bonferroni post-tests.
P2Y2R-mediated release of sAPPα is partially dependent on PKC
Previous studies in our laboratory have shown that inhibition of MAPK (i.e., ERK1/2) partially inhibits UTP-stimulated sAPPα release in human 1321N1 astrocytoma cells expressing human P2Y2R cDNA (Camden et al., 2005). Therefore, we investigated the role of the MAPK pathway in UTP-stimulated sAPPα release in IL-1β-treated rPCNs. U0126 (1 µM), an inhibitor of the MAPK/ERK kinase (MEK), partially inhibited UTP-induced sAPPα release, but not constitutive release of sAPPα in IL-1β-treated rPCNs (Figure 8). These results suggest that P2Y2R-mediated sAPPα release in neurons is partially dependent on ERK1/2 activation.
Figure 8. UTP-stimulated sAPPα release in IL-1β-treated rPCNs is partially dependent on ERK1/2 activation.
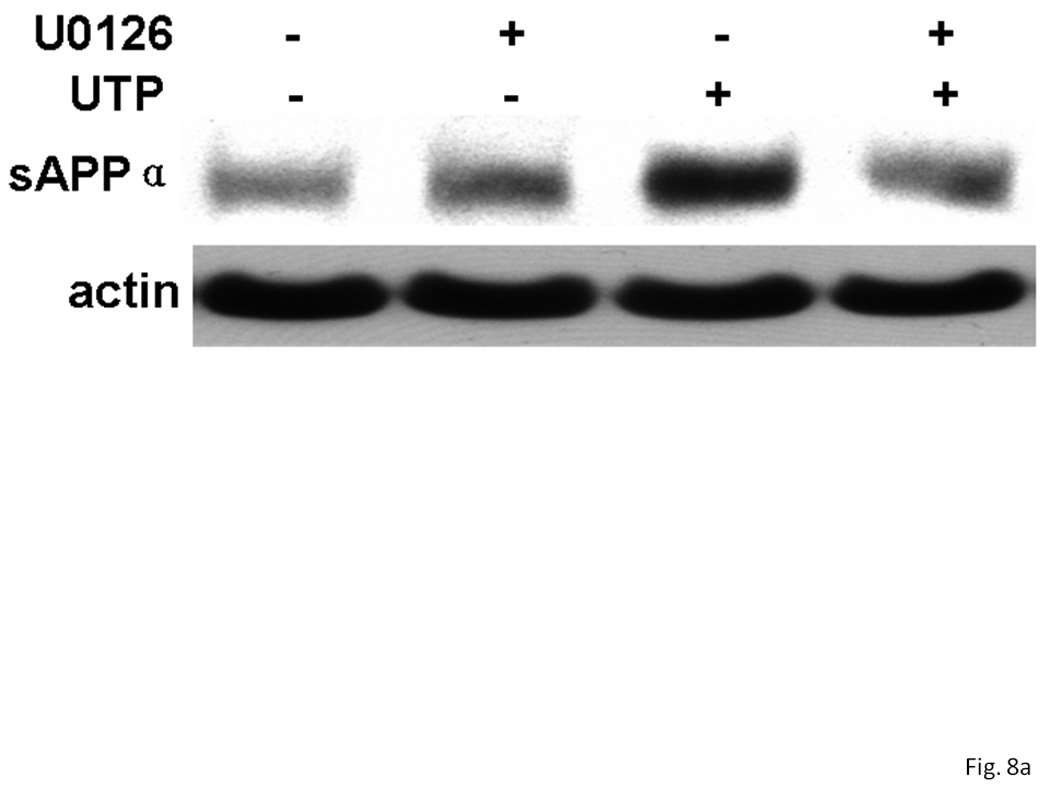
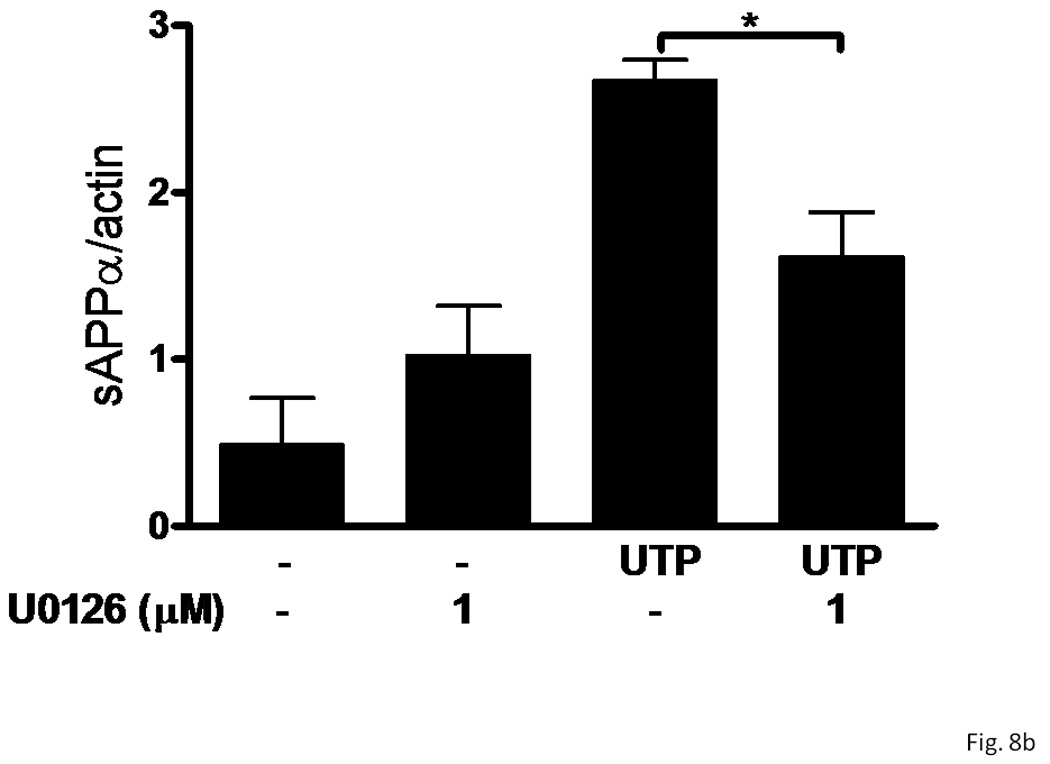
rPCNs were pretreated with 25 ng/ml IL-1β in serum-free DMEM for 24 h at 37 °C. The medium was replaced with fresh serum-free DMEM and the cells were incubated at 37 °C for 30 min with or without U0126 (1 µM) followed by a 2 h incubation with or without UTP (100 µM). (A) The release of sAPPα into the medium was analyzed by Western blot analysis. (B) sAPPα release is expressed relative to the actin level in cell lysate. Data represent the means ± S.E.M. of results from three experiments, where * represents significant differences (p < 0.05) between the indicated sample groups analyzed by 2-way ANOVA followed by Bonferroni post-tests.
DISCUSSION
In the present study, we demonstrated that the pro-inflammatory cytokine IL-1β upregulated expression of the P2Y2R via the IκB-α/NF-κB signaling pathway in rat primary cortical neurons. Furthermore, we showed that activation of the P2Y2R in IL-1β-treated neurons enhanced the release of sAPPα, a non-amyloidogenic cleavage product of APP (Li et al., 1997; Luo et al., 2001; Stein et al., 2004), by stimulating α-secretase activity mediated by the metalloproteinases ADAM10/17.
Previous studies have demonstrated that P2Y2R mRNA expression and function are upregulated in stressed or injured tissue in salivary gland epithelium (Turner et al., 1998, Ahn et al., 2000, Schrader et al., 2005), in intimal lesions of rat aorta (Seye et al., 1997) and in endothelial and smooth muscle cells of collared rabbit carotid arteries (Seye et al., 2002). P2Y2R mRNA expression is also upregulated in smooth muscle cells by IL-1β (Hou et al., 2000), a cytokine that mediates CNS inflammation in a mouse model of Alzheimer’s disease (Brugg et al., 1995; Hauss-Wegrzyniak et al., 1998). Furthermore, activation of the P2Y2R expressed in human 132N1 astrocytoma cells induces sAPPα release (Camden et al., 2005). The present study demonstrates for the first time that IL-1β-mediated P2Y2R upregulation and subsequent P2Y2R-mediated sAPPα release occurs in primary neurons. These results are consistent with reports that chronic inflammatory responses can enhance microglial cell-mediated amyloid-beta clearance and reduce plaque burden in murine models of AD (Wyss-Coray et al., 2001; Shaftel et al., 2007).
APP is a transmembrane glycoprotein that can be detected in most tissues but is abundantly expressed in the brain (Anderson et al., 1989). Post-translational processing of APP occurs by the action of at least three proteases called α-, β- and γ-secretases (Esler and Wolfe, 2001). Cleavage of APP by β- and γ-secretases results in the release of several protein fragments, including amyloid β (Aβ), the main component of senile plaques found in the brains of patients with Alzheimer’s disease (Martins et al., 1991). Cleavage of APP by α- and γ-secretases causes the release of sAPPα, thereby preventing secretion of amyloidogenic Aβ (Furukawa et al. 1996; Mattson, 1997; Kojro and Fahrenholz, 2005). Although sAPPα is constitutively secreted, release of this protein is modulated by neural activity and can be increased by agonists of various receptors (Robert et al., 2001; Canet-Aviles et al., 2002; Camden et al., 2005). The studies presented here show that both IL-1β-induced upregulation of P2Y2Rs in primary neurons (Figure 3) and transfection of neurons with P2Y2R cDNA (Figure 4) significantly increase UTP-stimulated sAPPα release, suggesting an important physiological role for the P2Y2R in Alzheimer’s disease pathology. IL-1β alone also significantly enhances sAPPα release and reduces sAPPβ and Aβ40/42 release in human neuroblastoma SK-N-SH cells (Tachida et al., 2008), although IL-1β alone did not enhance sAPPα release in rPCNs (Figure 3).
Responses to extracellular UTP in mammalian tissues can potentially be mediated by 3 P2Y receptor subtypes, P2Y2, P2Y4, and/or P2Y6 (Brunschweiger and Müller, 2006). P2Y4 receptors are endogenously expressed in rat neurons and have an agonist potency order of ITP = ATP = UTP = ATPγS = 2-MeSATP = Ap4A (Bogdanov et al., 1998). Therefore, it is reasonable to suggest that P2Y4Rs could contribute to the enhanced release of sAPPα evoked by UTP. In primary neurons without IL-1β treatment, P2Y4R mRNA is expressed at higher levels than P2Y2R mRNA (data not shown). However, there have been no studies to suggest that the P2Y4R can activate metalloproteinases, whereas P2Y2R activation has been shown to stimulate ADAM10/17 activity (Camden et al., 2005). The small percentage of rPCNs responding to UTP with an increase in [Ca2+]i in the absence of IL-1β stimulation (Figure 1C) may be due to P2Y4R activation. Nonetheless, in the absence of IL-1β pre-treatment, UTP did not significantly increase sAPPα release from rPCNs (Figure 3), suggesting that the P2Y4R does not contribute to APP processing. Moreover, since IL-1β does not increase P2Y4 or P2Y6 receptor mRNA expression in rPCNs (data not shown), it is most likely that the P2Y2R mediates UTP-induced sAPPα release in rPCNs treated with IL-1β.
There are three major isoforms of amyloid precursor protein (APP770, APP751 and APP695; Kitaguchi et al., 1988) resulting from alternative splicing of APP mRNA. APP695 is the predominant isoform in the brain (Tanaka et al., 1989), and has received the most attention in research on Alzheimer’s disease. The ratio of APP770:APP751:APP695 mRNA is 1:10:20 in the cerebral cortex of the brain (Tanaka et al., 1989). Compared with neurons that express high levels of APP695, astrocytes synthesize relatively higher levels of APP770 and APP751, as expected for these Kunitz protease inhibitor (KPI) containing isoforms. In our study, soluble APPα released into the UTP-containing medium of P2Y2R-expressing rPCNs was detected as a doublet of 100–120 kDa (data not shown), as has been described for proteolytic degradation of APP 695 (Johnson et al., 1988). These results support the notion that soluble APPα released from rPCNs is not due to glial cell contamination in the neuronal preparation (LeBlanc et al., 1991 & 1996 & 1997; Lahiri et al., 1994; Parvathy et al., 1998; Solano et al., 2000; Ma et al., 2005).
The data presented here also suggest that UTP induces APP processing in IL-1β-treated rPCNs through activation of the metalloproteases ADAM10/17 (Figure 5). Another enzyme that is inhibited by TAPI-2, TNF-convertase, is not involved in the regulated release of sAPPα from neuronal cells (Parkin et al., 2002).
Stimulation of GPCRs has been shown to regulate APP processing by protein kinase C (PKC)-dependent signaling pathways (Nitsch et al., 1992). However, APP processing can also be regulated by increases in [Ca2+]i, and activation of phospholipase A2 (PLA2), protein kinase A and tyrosine kinases (reviewed in Mills and Reiner, 1999). In human 1321N1 astrocytoma cells expressing human P2Y2R cDNA, UTP-induced sAPPα release was independent of P2Y2R-mediated activation of PKC (Camden et al., 2005). Our results with pharmacological inhibitors of PI3K, PKC and ERK1/2 indicated that P2Y2R-mediated sAPPα release from IL-1β-treated neurons requires PI3K activity (Figure 7) and is partially dependent on PKC and ERK1/2 activation (Figure 6 and Figure 8). The MAPK/ERK1/2 signaling pathway has been implicated in the regulation of both PKC-dependent and PKC-independent APP catabolism in PC12 cells, human embryonic kidney cells, and cortical neurons (Mills et al., 1997; Desdouits-Magnen et al., 1998). PI3K has also been reported to regulate α-secretase-dependent APP processing (Solano et al., 2000; Fu et al., 2002), and IL-1β increased the release of sAPPα via ERK1/2-dependent activation of α-secretase cleavage in neuroglioma U251 cells independent of PI3K (Ma et al., 2005). In SH-SY5Y cells, sAPPα release is induced by muscarinic receptor activation and regulated by arachidonic acid generation via PLA2 activation (Cho et al., 2006). In HEK239 cells, a direct role for Gαq/11 protein in the regulation of muscarinic M3 receptor-mediated sAPPα release was shown (Kim and Kim, 2005). Thus, there is significant divergence in agonist-induced signaling pathways that regulate sAPPα release in different cell types.
The results of this study strongly suggest that IL-1β via activation of the IκB/NF-κB pathway induces functional upregulation of the P2Y2R in primary cortical neurons and subsequent activation of the P2Y2R signaling cascade by UTP results in a significant increase in ADAM10/17-mediated sAPPα release which is dependent on PI3K and partially dependent on PKC and ERK1/2 activities. Together, these lines of evidence suggest that under inflammatory conditions in the brain where IL-1β levels are increased (Rothwell and Luheshi, 2000; Allan and Rothwell, 2001; Caquevel et al., 2004; Colangelo et al., 2003), functional P2Y2R expression in neuronal cells may serve a neuroprotective role by promoting non-amyloidogenic APP processing. Since P2Y2R expression in glial cells has been postulated to regulate chronic inflammatory responses seen in Alzheimer’s disease (Weisman et al., 2005), future studies are required to evaluate how glial cell-mediated chronic inflammation can be used to promote neuroprotective sAPPα release from neurons in response to P2Y2R activation during the progression of Alzheimer’s disease.
ACKNOWLEDGEMENT
This work was supported by NIH grant PO1 AG18357.
REFERENCES
- Ahn JS, Camden JM, Schrader AM, Redman RS, Turner JT. Reversible regulation of P2Y2 nucleotide receptor expression in the duct-ligated rat submandibular gland. Am J Physiol Cell Physiol. 2000;279:C286–C294. doi: 10.1152/ajpcell.2000.279.2.C286. [DOI] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–744. doi: 10.1038/35094583. Review. [DOI] [PubMed] [Google Scholar]
- Allinson TM, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res. 2003;74:342–352. doi: 10.1002/jnr.10737. [DOI] [PubMed] [Google Scholar]
- Anderson JP, Refolo LM, Wallace W, et al. Differential brain expression of the Alzheimer's amyloid precursor protein. EMBO J. 1989;8:3627–3632. doi: 10.1002/j.1460-2075.1989.tb08536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, et al. A metalloproteinase disintegrin that releases tumor-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- Bogdanov YD, Wildman SS, Clements MP, King BF, Burnstock G. Molecular cloning and characterization of rat P2Y4 nucleotide receptor. Br J Pharmacol. 1998;124:428–430. doi: 10.1038/sj.bjp.0701880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunschweiger A, Müller CE. P2 receptors activated by uracil nucleotides--an update. Curr Med Chem. 2006;13:289–312. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- Brugg B, Dubreuil YL, Huber G, Wollman EE, Delhaye-Bouchaud N, Mariani J. Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proc Natl Acad Sci U S A. 1995;92:3032–3035. doi: 10.1073/pnas.92.7.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. P2X receptors in sensory neurons. Br. J. Anaesth. 2000;84:476–488. doi: 10.1093/oxfordjournals.bja.a013473. [DOI] [PubMed] [Google Scholar]
- Cacquevel M, Lebeurrier N, Cheenne S, Vivien D. Cytokines in neuroinflammation and Alzheimer's disease. Curr Drug Targets. 2004;5:529–534. doi: 10.2174/1389450043345308. Review. [DOI] [PubMed] [Google Scholar]
- Camden JM, Schrader AM, Camden RE, Gonzalez FA, Erb L, Seye CI, Weisman GA. P2Y2 nucleotide receptors enhance α-secretase-dependent amyloid precursor protein processing. J Biol Chem. 2005;280:18696–18702. doi: 10.1074/jbc.M500219200. [DOI] [PubMed] [Google Scholar]
- Canet-Aviles RM, Anderton M, Hooper NM, Turner AJ, Vaughan PF. Muscarine enhances soluble amyloid precursor protein secretion in human neuroblastoma SH-SY5Y by a pathway dependent on protein kinase C(α), src-tyrosine kinase and extracellular signal-regulated kinase but not phospholipase C. Brain Res Mol Brain Res. 2002;102:62–72. doi: 10.1016/s0169-328x(02)00184-5. [DOI] [PubMed] [Google Scholar]
- Checler F. Processing of the beta-amyloid precursor protein and its regulation in Alzheimer's disease. J Neurochem. 1995;65:1431–1444. doi: 10.1046/j.1471-4159.1995.65041431.x. [DOI] [PubMed] [Google Scholar]
- Cho HW, Kim JH, Choi S, Kim HJ. Phospholipase A2 is involved in muscarinic receptor-mediated sAPPalpha release independently of cyclooxygenase or lypoxygenase activity in SH-SY5Y cells. Neurosci Lett. 2006;397:214–218. doi: 10.1016/j.neulet.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Ciccarelli R, Ballerini P, Sabatino G, Rathbone MP, D'Onofrio M, Caciagli F, Di Iorio P. Involvement of astrocytes in purine-mediated reparative processes in the brain. Int J Dev Neurosci. 2001;19:395–414. doi: 10.1016/s0736-5748(00)00084-8. [DOI] [PubMed] [Google Scholar]
- Colangelo V, Schurr J, Ball MJ, Pelaez RP, Bazan NG, Lukiw WJ. Gene expression profiling of 12633 genes in Alzheimer hippocampal CA1: transcription and neurotrophic factor down-regulation and up-regulation of apoptotic and pro-inflammatory signaling. J Neurosci Res. 2003;70:462–473. doi: 10.1002/jnr.10351. [DOI] [PubMed] [Google Scholar]
- Desdouits-Magnen J, Desdouits F, Takeda S, Syu LJ, Saltiel AR, Buxbaum JD, Czernik AJ, Nairn AC, Greengard P. Regulation of secretion of Alzheimer amyloid precursor protein by the mitogen-activated protein kinase cascade. J Neurochem. 1998;70:524–530. doi: 10.1046/j.1471-4159.1998.70020524.x. [DOI] [PubMed] [Google Scholar]
- Esler WP, Wolfe MS. A portrait of Alzheimer secretases--new features and familiar faces. Science. 2001;293:1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- Fu W, Lu C, Mattson MP. Telomerase mediates the cell survival-promoting actions of brain-derived neurotrophic factor and secreted amyloid precursor protein in developing hippocampal neurons. J Neurosci. 2002;22:10710–10719. doi: 10.1523/JNEUROSCI.22-24-10710.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP. Increased activity-regulating and neuroprotective efficac y o f alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem. 1996;67:1882–1896. doi: 10.1046/j.1471-4159.1996.67051882.x. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Gentleman SM, Graham DI, Mrak RE, Roberts GW. Microglial interleukin-1 alpha expression in human head injury: correlations with neuronal and neuritic beta-amyloid precursor protein expression. Neurosci Lett. 1994;176:133–136. doi: 10.1016/0304-3940(94)90066-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauss-Wegrzyniak B, Dobrzanski P, Stoehr JD, Wenk GL. Chronic neuroinflammation is a contributor to the pathophysiology of Alzheimer's disease. Brain Res. 1998;780:294–303. doi: 10.1016/s0006-8993(97)01215-8. [DOI] [PubMed] [Google Scholar]
- Hou M, Moller S, Edvinsson L, Erlinge D. Cytokines induced upregulation of vascular P2Y2 receptors and increased mitogenic responses to UTP and ATP. Arterioscler Thromb Vasc Biol. 2000;20:2064–2069. doi: 10.1161/01.atv.20.9.2064. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Pasinetti GM, May PC, Ponte PA, Cordell B, Finch CE. Selective reduction of mRNA for the beta-amyloid precursor protein that lacks a Kunitz-type protease inhibitor motif in cortex from Alzheimer brains. Exp Neurol. 1988;102:264–268. doi: 10.1016/0014-4886(88)90104-5. [DOI] [PubMed] [Google Scholar]
- Kaczmarek E, Erb L, Koziak K, Jarzyna R, Wink MR, Guckelberger O, Blusztajn JK, Trinkaus-Randall V, Weisman GA, Robson SC. Modulation of endothelial cell migration by extracellular nucleotides: involvement of focal adhesion kinase and phosphatidylinositol 3-kinase-mediated pathways. Thromb Haemost. 2005;93:735–742. doi: 10.1267/THRO05040735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kim HJ. Direct involvement of G protein alpha(q/11) subunit in regulation of muscarinic receptor-mediated sAPPalpha release. Arch Pharm Res. 2005;28:1275–1281. doi: 10.1007/BF02978212. [DOI] [PubMed] [Google Scholar]
- Kirazov L, Loffler T, Schliebs R, Bigl V. Glutamate-stimulated secretion of amyloid precursor protein from cortical rat brain slices. Neurochem Int. 1997;30:557–563. doi: 10.1016/s0197-0186(96)00119-2. [DOI] [PubMed] [Google Scholar]
- Kitaguchi N, Takahashi Y, Tokushima Y, Shiojiri S, Ito H. Novel precursor of Alzheimer's disease amyloid protein shows protease inhibitory activity. Nature. 1988;331:530–532. doi: 10.1038/331530a0. [DOI] [PubMed] [Google Scholar]
- Koike H, Tomioka S, Sorimachi H, et al. Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem J. 1999;343:371–375. [PMC free article] [PubMed] [Google Scholar]
- Kojro E, Fahrenholz F. The non-amyloidogenic pathway: structure and function of alpha-secretases. Subcell Biochem. 2005;38:105–127. doi: 10.1007/0-387-23226-5_5. [DOI] [PubMed] [Google Scholar]
- Kong Q, Wang M, Liao Z, Camden JM, Yu S, Simonyi A, Sun G, Gonzalez FA, Erb L, Seye CI, Weisman GA. P2X7 nucleotide receptors mediate caspase-8/9/3-dependent apoptosis in rat primary cortical neurons. Purinergic Signalling. 2005;1:337–347. doi: 10.1007/s11302-005-7145-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba M, Apasov S, Sverdlov V, Chen P, Erb L, Turner JT, Weisman GA, Sitkovsky MV. Transient up-regulation of P2Y2 nucleotide receptor mRNA expression is an immediate early gene response in activated thymocytes. Proc Natl Acad Sci U S A. 1997;94:831–836. doi: 10.1073/pnas.94.3.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri DK, Lewis S, Farlow MR. Tacrine alters the secretion of the beta-amyloid precursor protein in cell lines. J Neurosci Res. 1994;37:777–787. doi: 10.1002/jnr.490370612. [DOI] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc AC, Chen HY, Autilio-Gambetti L, Gambetti P. Differential APP gene expression in rat cerebral cortex, meninges, and primary astroglial, microglial and neuronal cultures. FEBS Lett. 1991;292:171–178. doi: 10.1016/0014-5793(91)80861-v. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC, Xue R, Gambetti P. Amyloid precursor protein metabolism in primary cell cultures of neurons, astrocytes, and microglia. J Neurochem. 1996;66:2300–2310. doi: 10.1046/j.1471-4159.1996.66062300.x. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC, Papadopoulos M, Belair C, Chu W, Crosato M, Powell J, Goodyer CG. Processing of amyloid precursor protein in human primary neuron and astrocyte cultures. J Neurochem. 1997;68:1183–1190. doi: 10.1046/j.1471-4159.1997.68031183.x. [DOI] [PubMed] [Google Scholar]
- LeBlanc AC, Koutroumanis M, Goodyer CG. Protein kinase C activation increases release of secreted amyloid precursor protein without decreasing Aβ production in human primary neuron cultures. J Neurosci. 1998;18:2907–2913. doi: 10.1523/JNEUROSCI.18-08-02907.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Roch JM, Sundsmo M, Otero D, Sisodia S, Thomas R, Saitoh T. Defective neurite extension is caused by a mutation in amyloid beta/A4 (A beta) protein precursor found in familial Alzheimer's disease. J Neurobiol. 1997;32:469–480. [PubMed] [Google Scholar]
- Li Y, Liu L, Kang J, Sheng JG, Barger SW, Mrak RE, Griffin WS. Neuronal-glial interactions mediated by interleukin-1 enhance neuronal acetylcholinesterase activity and mRNA expression. J Neurosci. 2000;20:149–155. doi: 10.1523/JNEUROSCI.20-01-00149.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JJ, Wallace MS, Hawver DB, Kusiak JW, Wallace WC. Characterization of the neurotrophic interaction between nerve growth factor and secreted alpha-amyloid precursor protein. J Neurosci Res. 2001;63:410–420. doi: 10.1002/1097-4547(20010301)63:5<410::AID-JNR1036>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Ma G, Chen S, Wang X, Ba M, Yang H, Lu G. Short-term interleukin-1(beta) increases the release of secreted APPα via MEK1/2-dependent and JNK-dependent alpha-secretase cleavage in neuroglioma U251 cells. J Neurosci Res. 2005;80:683–692. doi: 10.1002/jnr.20515. [DOI] [PubMed] [Google Scholar]
- Martins RN, Robinson PJ, Chleboun JO, Beyreuther K, Masters CL. The molecular pathology of amyloid deposition in Alzheimer's disease. Mol Neurobiol. 1991;5:389–398. doi: 10.1007/BF02935560. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77:1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- Mills J, Laurent Charest D, Lam F, Beyreuther K, Ida N, Pelech SL, Reiner PB. Regulation of amyloid precursor protein catabolism involves the mitogen-activated protein kinase signal transduction pathway. J Neurosci. 1997;17:9415–9422. doi: 10.1523/JNEUROSCI.17-24-09415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills J, Reiner PB. Regulation of amyloid precursor protein cleavage. J Neurochem. 1999;72:433–460. doi: 10.1046/j.1471-4159.1999.0720443.x. [DOI] [PubMed] [Google Scholar]
- Moses GS, Jensen MD, Lue LF, Walker DG, Sun AY, Simonyi A, Sun GY. Secretory PLA2-IIA: a new inflammatory factor for Alzheimer's disease. J Neuroinflam. 2006;7:3–28. doi: 10.1186/1742-2094-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss ML, Jin SL, Milla ME, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385:733–736. doi: 10.1038/385733a0. [DOI] [PubMed] [Google Scholar]
- Muscella A, Elia MG, Greco S, Storelli C, Marsigliante S. Activation of P2Y2 receptor induces c-FOS protein through a pathway involving mitogen-activated protein kinases and phosphoinositide 3-kinases in HeLa cells. J Cell Physiol. 2003;195:234–240. doi: 10.1002/jcp.10242. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Wurtman RJ, Growdon JH. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science. 1992;258:304–307. doi: 10.1126/science.1411529. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Slack BE, Farber SA, et al. Regulation of proteolytic processing of the amyloid beta-protein precursor of Alzheimer's disease in transfected cell lines and in brain slices. J Neural Transm Suppl. 1994;44:21–27. doi: 10.1007/978-3-7091-9350-1_2. [DOI] [PubMed] [Google Scholar]
- Nitsch RM, Wurtman RJ, Growdon JH. Regulation of proteolytic processing of the amyloid beta-protein precursor by first messengers. A novel potential approach for the treatment of Alzheimer's disease. Arzneim Forsch. 1995;45:435–438. [PubMed] [Google Scholar]
- Nitsch RM, Deng A, Wurtman RJ, Growdon JH. Metabotropic glutamate receptor subtype mGluR1alpha stimulates the secretion of the amyloid beta-protein precursor ectodomain. J Neurochem. 1997;69:704–712. doi: 10.1046/j.1471-4159.1997.69020704.x. [DOI] [PubMed] [Google Scholar]
- Parihar MS, Hemnani T. Alzheimer's disease pathogenesis and therapeutic interventions. J Clinical Neurosci. 2004;11:456–467. doi: 10.1016/j.jocn.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Parkin ET, Trew A, Christie G, Faller A, Mayer R, Turner AJ, Hooper NM. Structure-activity relationship of hydroxamate-based inhibitors on the secretases that cleave the amyloid precursor protein, angiotensin converting enzyme, CD23, and pro-tumor necrosis factor-alpha. Biochem (Mosc) 2002;41:4972–4981. doi: 10.1021/bi015936e. [DOI] [PubMed] [Google Scholar]
- Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM. Alzheimer's amyloid precursor protein alpha-secretase is inhibited by hydroxamic acid-based zinc metalloprotease inhibitors: similarities to the angiotensin converting enzyme secretase. Biochem. 1998;37:1680–1685. doi: 10.1021/bi972034y. [DOI] [PubMed] [Google Scholar]
- Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem. 1997;272:21096–21103. doi: 10.1074/jbc.272.34.21096. [DOI] [PubMed] [Google Scholar]
- Robert SJ, Zugaza JL, Fischmeister R, Gardier AM, Lezoualc’h F. The human serotonin 5-HT4 receptor regulates secretion of non-amyloidogenic precursor protein. J Biol Chem. 2001;276:44881–44888. doi: 10.1074/jbc.M109008200. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Luheshi GN. Interleukin 1 in the brain: biology, pathology and therapeutic target. Trends Neurosci. 2000;23:618–625. doi: 10.1016/s0166-2236(00)01661-1. [DOI] [PubMed] [Google Scholar]
- Schrader AM, Camden JM, Weisman GA. P2Y2 nucleotide receptor up-regulation in submandibular gland cells from the NOD.B10 mouse model of Sjogren's syndrome. Arch Oral Biol. 2005;50:533–540. doi: 10.1016/j.archoralbio.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Seye CI, Gadeau AP, Daret D, Dupuch F, Alzieu P, Capron L, Desgranges C. Overexpression of the P2Y2 purinoceptor in intimal lesion of the rat aorta. Arterioscler Thromb Vasc Biol. 1997;17:3602–3610. doi: 10.1161/01.atv.17.12.3602. [DOI] [PubMed] [Google Scholar]
- Seye CI, Kong Q, Erb L, Garrad RC, Krugh BW, Wang M, Turner JT, Sturek M, Gonzalez FA, Weisman GA. Functional P2Y2 nucleotide receptors mediate uridine 5'-triphosphate-induced intimal hyperplasia in collared rabbit carotid arteries. Circulation. 2002;106:2720–2726. doi: 10.1161/01.cir.0000038111.00518.35. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, O'Banion MK. Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci. 2007;27:9301–9309. doi: 10.1523/JNEUROSCI.1418-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Zhu SG, Jones RA, Griffin WS, Mrak RE. Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Exp Neurol. 2000;163:388–391. doi: 10.1006/exnr.2000.7393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD. The brain as a target for inflammatory processes and neuroprotective strategies. Ann N Y Acad Sci. 2007;1122:23–34. doi: 10.1196/annals.1403.002. [DOI] [PubMed] [Google Scholar]
- Solano DC, Sironi M, Bonfini C, Solerte SB, Govoni S, Racchi M. Insulin regulates soluble amyloid precursor protein release via phosphatidyl inositol 3 kinase-dependent pathway. FASEB J. 2000;14:1015–1022. doi: 10.1096/fasebj.14.7.1015. [DOI] [PubMed] [Google Scholar]
- Standridge JB. Vicious cycles within the neuropathophysiologic mechanisms of Alzheimer's disease. Curr Alzheimer Res. 2006;3:95–108. doi: 10.2174/156720506776383068. Review. [DOI] [PubMed] [Google Scholar]
- Stein TD, Anders NJ, DeCarli C, Chan SL, Mattson MP, Johnson JA. Neutralization of transthyretin reverses the neuroprotective effects of secreted amyloid precursor protein (APP) in APPSW mice resulting in tau phosphorylation and loss of hippocampal neurons: support for the amyloid hypothesis. J Neurosci. 2004;24:7707–7717. doi: 10.1523/JNEUROSCI.2211-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Estelles A, Siciliano J, Desagher S, Piomelli D, Glowinski J, Premont J. Interleukin-1 enhances the ATP-evoked release of arachidonic acid from mouse astrocytes. J Neurosci. 1997;17:2939–2946. doi: 10.1523/JNEUROSCI.17-09-02939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M. Detrimental and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system. Adv Exp Med Biol. 2002;513:87–113. doi: 10.1007/978-1-4615-0123-7_3. [DOI] [PubMed] [Google Scholar]
- Sue W, Griffin T. Inflammation and neurodegenerative diseases. Am J Clin Nutr. 2006;83:470S–474S. doi: 10.1093/ajcn/83.2.470S. Review. [DOI] [PubMed] [Google Scholar]
- Tachida Y, Nakagawa K, Saito T, et al. Interleukin-1 beta up-regulates TACE to enhance alpha-cleavage of APP in neurons: resulting decrease in Abeta production. J Neurochem. 2008;104:1387–1389. doi: 10.1111/j.1471-4159.2007.05127.x. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Shiojiri S, Takahashi Y, Kitaguchi N, Ito H, Kameyama M, Kimura J, Nakamura S, Ueda K. Tissue-specific expression of three types of beta-protein precursor mRNA: enhancement of protease inhibitor-harboring types in Alzheimer's disease brain. Biochem Biophys Res Commun. 1989;165:1406–1414. doi: 10.1016/0006-291x(89)92760-5. [DOI] [PubMed] [Google Scholar]
- Turner JT, Weisman GA, Camden JM. Upregulation of P2Y2 nucleotide receptors in rat salivary gland cells during short-term culture. Am J Physiol. 1997;273:C1100–C1107. doi: 10.1152/ajpcell.1997.273.3.C1100. [DOI] [PubMed] [Google Scholar]
- Turner JT, Park M, Camden JM, Weisman GA. Salivary gland nucleotide receptors: changes in expression and activity related to development and tissue damage. Ann N Y Acad Sci. 1998;842:70–75. doi: 10.1111/j.1749-6632.1998.tb09633.x. [DOI] [PubMed] [Google Scholar]
- Wang M, Kong Q, Gonzalez FA, Sun GY, Erb L, Seye CI, Weisman GA. P2Y2 nucleotide receptor interaction with αv integrin mediates astrocyte migration. J Neurochem. 2005;95:630–640. doi: 10.1111/j.1471-4159.2005.03408.x. [DOI] [PubMed] [Google Scholar]
- Weisman GA, Wang M, Kong Q, Chorna NE, Sun GY, Gonzalez FA, Seye CI, Erb L. Molecular determinants of P2Y2 nucleotide receptor function. Mol Neurobiol. 2005;31:169–183. doi: 10.1385/MN:31:1-3:169. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]