

Abstract
The crawling movement of nematode sperm requires coordination of leading edge protrusion with cell body retraction, both of which are powered by modulation of a cytoskeleton based on major sperm protein (MSP) filaments. We used a cell-free in vitro motility system in which both protrusion and retraction can be reconstituted, to identify two proteins involved in cell body retraction. Pharmacological and depletion-add back assays showed that retraction was triggered by a putative protein phosphatase 2A (PP2A, a Ser/Thr phosphatase activated by tyrosine dephosphorylation). Immunofluorescence showed that PP2A was present in the cell body and was concentrated at the base of the lamellipod where the force for retraction is generated. PP2A targeted MSP fiber protein 3 (MFP3), a protein unique to nematode sperm that binds to the MSP filaments in the motility apparatus. Dephosphorylation of MFP3 caused its release from the cytoskeleton and generated filament disassembly. Our results suggest that interaction between PP2A and MFP3 leads to local disassembly of the MSP cytoskeleton at the base of the lamellipod in sperm that in turn pulls the trailing cell body forward.
INTRODUCTION
Amoeboid cell motility plays a central role in many processes, such as embryo development, wound healing, and immunological defense, and requires coordination of protrusion of the leading edge with retraction of the trailing cell body (reviewed by Rafelski and Theriot, 2004). In most eukaryotic cells, protrusion is powered by localized Arp2/3 complex-mediated actin assembly along their leading edge, whereas retraction is thought to be driven by actomyosin contractility (Svitkina et al., 1997; Pollard and Borisy, 2003). Although leading edge protrusion has been studied extensively in a variety of cells (Wang, 1985; Hug et al., 1995; Svitkina et al., 1997), reconstituted motility systems from cell extracts (reviewed in Cameron et al., 2000; Pantaloni et al., 2001), and even in defined protein mixtures (Loisel et al., 1999; Co et al., 2007; Akin and Mullins, 2008), much less is known about the mechanism of cell body retraction. The molecular complexity of actin-based motility and the lack of simpler reconstituted systems have hindered exploration of this important component of cell migration.
Nematode sperm provide an alternate opportunity for investigating the mechanism underlying cell motility. Although in these cells locomotion is based on a dynamic cytoskeleton based on major sperm protein (MSP) instead of actin, the general features of leading edge protrusion and cell body retraction are so remarkably similar to those observed in actin-based cells (Theriot, 1996; Roberts and Stewart, 2000) that the mechanisms for generating locomotion must be comparable in both systems. MSP-based motility has been reconstituted in vitro in sperm extracts in which vesicles derived from the plasma membrane at the leading edge induce ATP-dependent assembly of columnar meshworks of MSP filaments called fibers. Polymerization of MSP filaments at the surface of the vesicle elongates the fiber and pushes the vesicles forward in the same way as cytoskeletal assembly along the leading edge drives protrusion of the lamellipodia in intact sperm (Italiano et al., 1996). Fibers assembled in vitro can be manipulated to reconstitute retraction much more easily than in actin-based systems. Perfusion with Yersinia tyrosine phosphatase (YOP) in sperm extract triggers rapid shrinkage of the fiber, which generates sufficient force to pull an artificial cargo attached to retracting fiber (Miao et al., 2003). Ascaris sperm do not undergo the periodic lamellipodial extension–retraction cycles exhibited by some actin-based crawling cells (e.g., see Dubin-Thaler et al., 2008). Thus, fiber shrinkage reconstitutes the forces that pull the cell body forward when sperm crawl. Unlike microfilaments, MSP filaments have no structural polarity and no motor proteins have been identified in the MSP motility apparatus. Thus, the retraction of fibers in vitro and of the cell body of crawling sperm do not seem to require the chemomechanical activity of a protein such as myosin.
The reconstituted Ascaris in vitro motility system has proven to be valuable for identifying the molecular components of motility and for understanding how polymerization dynamics can generate protrusive forces (LeClaire et al., 2003; Buttery et al., 2003; Yi et al., 2007; Miao et al., 2008). For example, a phosphorylated integral membrane protein, MPOP, that recruits a soluble casein kinase 1 called MSP polymerization–activating kinase (MPAK) to the vesicle surface, generates the localized polymerization that results in movement. MPAK, in turn, phosphorylates a second cytosolic component, MSP fiber protein MFP) 2, which then binds to filaments and accelerates their assembly. These biochemical events are essential for leading edge protrusion in sperm (LeClaire et al., 2003; Buttery et al., 2003; Yi et al., 2007). In contrast, little is known about the biochemical basis of retraction. YOP only induces retraction in the presence of a cell-free extract of sperm (S100), suggesting that another component is required (Miao et al., 2003). Here, we report that a protein phosphatase (a putative PP2A homologue) in S100 is activated by YOP and then triggers retraction of MSP fibers. Furthermore, we show that MFP3, a structural component in the MSP motility apparatus, is released from the cytoskeleton when it is dephosphorylated by PP2A.
MATERIALS AND METHODS
Sperm Collection and Preparation of Sperm Extracts
Ascaris sperm were collected and activated as described previously (Sepsenwol et al., 1989; Italiano et al., 1996). For immunofluorescence studies, activated sperm were pipetted onto glass coverslips, allowed to settle for 5 min, and fixed in 2% glutaraldehyde, 0.5% Triton X-100 in HKB buffer (50 mM HEPES, 65 mM KCl, and 10 mM NaHCO3, pH 7.6) for 30 min. To prepare cell-free extracts (S100), pellets of sperm were lysed by three freeze-thaw cycles and then centrifuged at 100,000 × g for 45 min at 4°C (LeClaire et al., 2003). The resulting supernatant (S100) was aliquoted and stored at −70°C until use.
Fiber Retraction Assays
Fibers were grown by adding 1 mM ATP to one fifth diluted S100 and pipetting 8–10 μl of the extract into chambers formed by mounting a 22- × 22-mm glass coverslip onto a glass slide with two parallel strips of double-stick tape. After 15–20 min, the fibers were washed by perfusion of 25 μl of KPM buffer (LeClaire et al., 2003) through the chamber followed by perfusion of 30-μl of test solutions. Phase contrast images were obtained at 30-s intervals with an Orca 12-bit charge-coupled device camera (Hamamatsu, Bridgewater, NJ). Images were captured and processed with MetaMorph software (Molecular Devices, Sunnyvale, CA).
Protein phosphatases tested for effects on fiber retraction were PP2A (Millipore, Billerica, MA) and YOP (New England Biolabs, Ipswich, MA). Phosphatase inhibitors used included calyculin A (Cell Signaling Technology, Danvers, MA), okadaic acid (EMD Biosciences, San Diego, CA), PP1 inhibitor 2 (New England Biolabs), and sodium orthovanadate (Sigma-Aldrich, St. Louis, MO).
Antibodies
The following antibodies were used: anti-phosphoserine (Millipore Bioscience Research Reagents, Temecula, CA), anti-phospho-threonine (Cell Signaling Technology), anti-PP2A c subunit (clone 1D6; Millipore), peroxidase-conjugated goat anti-mouse or rabbit antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA), AlexaFluor 568- or AlexaFluor 488-conjugated goat anti-mouse or goat anti-rabbit (Invitrogen, Carlsbad, CA) and 10-nm gold-conjugated anti-rabbit immunoglobulin G (IgG) antibody (Sigma-Aldrich). Protein A/G-conjugated agarose was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Polyclonal antibody to MFP3 was generated by injecting the New Zealand White rabbits with 1 mg of MFP3, obtained by electro-elution from SDS-polyacrylamide gel electrophoresis (PAGE) gels, in combination with 1 ml of complete Freund's adjuvant. A booster injection of antigen in incomplete Freund's adjuvant was administered 4 wk later. Antiserum was collected after another 4 wk and stored at −70°C. Antibodies were purified from immune serum by using an ImmunoPure Plus Immoblized Protein A/IgG purification kit (Pierce Chemical, Rockford, IL).
Immunofluorescence
Glutaraldehyde-fixed sperm or fibers grown in vitro were washed in phosphate-buffered saline (PBS) buffer, treated three times with 10 mM NaBH4 in PBS for 20 min to quench unreacted aldehydes, and blocked with 0.5% bovine serum albumin (BSA) in PBS for at least 4 h. The specimens were then incubated in primary antibodies, washed, treated with appropriate secondary antibodies, and washed again. All antibodies were used at 5 μg/ml for 3 h at 25°C or overnight at 4°C. For controls, primary antibodies were omitted. Immunostained cells or fibers were imaged with a QFM510 laser scanning confocal microscope (Carl Zeiss, Thornwood, NY) equipped with dual HeNe laser with appropriate filters or with an Axioskop2 microscope (Carl Zeiss) equipped with a 40× acroplan/phase objective with appropriate filters.
Immunodepletion and Add-Back Assay
To deplete PP2A from extracts of Ascaris sperm, anti-PP2A catalytic subunit (PP2Ac) antibody was coupled to immobilized Protein G Plus beads using the bifunctional cross-linking agent disuccinimidyl suberate (Pierce Chemical). S100 was diluted up to 40-fold into PBS buffer and incubated with the beads. Unbound, immunodepleted fractions were collected by centrifugation after 2 h and subjected to three additional rounds of immunodepletion. Mock depletion followed the same procedure by using beads coupled with normal mouse IgG. The depletion efficiency was monitored by SDS-PAGE and immunoblot analysis with anti-PP2A antibody. After dialysis into KPM buffer, the immunodepleted material was concentrated to its original volume and stored at −70°C until use in retraction assays. A commercial PP2A was used to replenish depleted extracts to test for the recovery of retraction-triggering activity.
Immunoprecipitation and Immunoblot
S100 was incubated in KPM with 0.5% Triton X-100 for 30 min at 25°C and then mixed with equal volume of 2× immunoprecipitation (IP) buffer (2% Triton X-100, 300 mM NaCl, 20 mM Tris, pH 7.4, and 2 mM EGTA). The lysates were precleared on protein A/G-coupled agarose beads for 1 h and then incubated with 3 μg of anti-PP2A or anti-MFP3 antibody at 4°C overnight or for 3 h on a rotating wheel. Protein G beads (25 μl) were added and the mixture was incubated at 25°C for 3 h. The beads were harvested by centrifugation at 1000 × g for 1 min and then washed extensively with IP buffer. Attached proteins were then eluted with either 3.0 M potassium chloride or 100 mM glycine-HCl buffer, pH 2.7.
For immunoblot analysis, protein samples was resolved on a SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories, Hercules, CA). After being blocked with Tris-buffered saline/Tween 20 containing 0.5% BSA, the membranes were incubated with appropriate primary antibody overnight in blocking buffer, incubated with peroxidase-conjugated secondary antibody for 3 h, and developed by enhanced chemiluminescence (Pierce Chemical). In some cases, blot membranes were stripped and reprobed with another antibody.
Immuno-electron Microcopy (EM)
MSP filaments for immuno-EM were prepared on a 5- × 7-mm glass coverslips in S100 diluted 1:5 with KPM plus 1 mM ATP for 20 min, fixed with 2.5% glutaraldehyde for 20 min, and washed with KPM buffer. The fixed MSP filaments were then incubated in three changes of 10 mM NaBH4 (20 min each) and blocked with 0.5% BSA in PBS for at least 4 h. The samples were incubated at 4°C in PBS containing 5 μg/ml rabbit anti-MFP3 plus 0.5% BSA overnight, washed three times for 10 min each in 20 mM Tris buffer, pH 8.0, and probed with 10-nm colloidal gold-conjugated goat anti-rabbit Ig G antibodies in Tris buffer, pH 8.0 at 25°C for at least 4 h. As controls, primary antibodies were omitted. After three washes in Tris buffer, pH 8.0, the samples were fixed in 2.5% glutaraldehyde, dehydrated in ethanol, and critical point dried as described previously (Ris, 1985). Platinum replicas were prepared as described previously (Svitkina et al., 1995, LeClaire et al., 2003) and examined on a CM 120 electron microscope (Philips, Wilmington, DE) operated at 80 kV.
Peptide Sequencing and cDNA Cloning
Bands containing MSP fiber proteins (MFP3) were excised from SDS-PAGE gels and digested with trypsin (Hoffman-La Roche, Nutley, NJ). Separation of the resulting peptide digests and N-terminal sequence analysis of selected peptides were performed as described previously for other sperm motility proteins (Buttery et al., 2003).
MFP3 cDNA was obtained by reverse transcription-polymerase chain reaction (RT-PCR) by using mRNA isolated from Ascaris testes and primers designed from the peptide sequences obtained as described method. The cDNA was then inserted into a pCR 2.1 TOPO vector (Invitrogen) for sequencing of both strands. The full-length cDNA sequence (NCBI accession AY326288) was translated by ExPASy translation program.
Identification of Phosphorylation Sites in MFP3 by Fourier Transform Ion Cyclotron Resonance Tandem Mass Spectrometry (FTICR MSMS)
All solvents were purchased from Mallinckrodt Baker (Philipsburg, NJ) in the highest purity. Trypsin was purchased from Sigma-Aldrich. A C18 (5 cm × 75 μm; 15-μm i.d. spray tip) nanoscale capillary column was purchased from New Objective (Woburn, MA).
For in-gel tryptic digestion, protein gel spots were excised and diced into ∼1- × 1-mm pieces and digested with 100 pmol of trypsin following a standard in-gel digestion protocol (Shevchenko et al., 2006). After digestion, peptides were extracted with 300 μl of acetonitrile:H2O:formic acid (75:20:5), speed vacuumed to dryness, and stored at −80°C for future analysis. Before MS analysis, the samples were dissolved in 10 μl of H2O:methanol:formic acid (97.5:2:0.5).
Mass analysis was performed with a modified hybrid linear quadrupole ion trap/FT-ICR mass spectrometer (LTQ-FT; Thermo Fisher Scientific, Bremen, Germany) equipped with an actively shielded 14.5 T superconducting magnet (Magnex, Oxford, United Kingdom). The in-gel digested samples were analyzed by liquid chromatography (LC)-MS/MS with the 14.5 T LTQ FTICR MS operated in top-5 data-dependent mode (high-resolution FTICR MS, low-resolution LTQ MS/MS). For each precursor ion measurement, 1 million ions were accumulated in the LTQ before transfer to the ICR cell for measurement. The five most abundant ions were collisionally dissociated in the LTQ (3 microscans; 10,000 target ions, 2.0-Da isolation width, 35.0 normalized collision energy, 0.250 activation Q, 30-ms activation period, and dynamic exclusion list size of 60 for 1 min). Automatic gain control provided <0.500 ppb FT-ICR rms mass error with external calibration. Data were collected with Xcalibur software (Thermo Fisher Scientific).
Raw data were extracted with a custom peak picking algorithm at a 10% signal-to-noise ratio threshold. Peak-list files were searched with MASCOT (Matrix Science, Cambridge, United Kingdom) against a database that contained the MPF3 sequence (1 ppm mass tolerance for parent ions; 1-Da tolerance for MS/MS; trypsin cleavage specificity, one missed cleavage; and oxidation and carbamidomethyl as allowed modifications for methionine and cysteine, respectively). Phosphorylation was limited to serine, threonine, and tyrosine.
RESULTS
Role of a Putative PP2A, a Ser/Thr Phosphatase, in the Biochemical Pathway That Generates Fiber Retraction
Previous studies established that although MSP fibers shortened dramatically when treated with YOP in the presence of sperm extracts (S100), YOP had little effect when added in buffer alone (Miao et al., 2003). These data suggest that YOP may induce retraction by acting on a component in S100. To search for this component, we screened a range of agents for effects on YOP-S100–induced fiber retraction. We found that retraction was blocked by inhibitors of PP2A but not by inhibitors of other Ser/Thr phosphatases. Thus, calyculin A, a selective PP1/PP2A phosphatase inhibitor, and okadaic acid (OA), a PP2A specific inhibitor, blocked the effect of YOP-S100. In contrast, a PP1-specific inhibitor, inhibitor 2 (I2), had no effect on retraction (Figure 1, A and B). In addition, treatment of fibers with KPM buffer containing commercial PP2A induced retraction at rates comparable with that observed after treatment with YOP-S100. Moreover, OA blocked the effect of added PP2A on fibers (Figure 1, C and D). Fibers treated with KPM buffer alone shortened only very slightly (data not shown). Together, these data suggest that YOP activates PP2A, which, in turn, triggers retraction. To test this hypothesis, we performed Western blot analysis of S100 with a specific antibody against PP2Ac. The antibody labeled a single band with a molecular weight of ∼36 kDa (Figure 2A), which is the typical molecular weight of well-conserved PP2Ac subunit shown on a SDS-PAGE gel. Immunofluorescence microscopy showed that the same antibody detected PP2A localized to the cell body and concentrated at the junction of lamellipod-cell body (Figure 2B), where the force for cell body retraction is produced (Italiano et al., 1999).
Figure 1.

Retraction of MSP fibers is initiated by PP2A phosphatase. (A) Time-lapse sequences of fiber retraction triggered by YOP-treated S100 (top) and the inhibitory effect of 10 nM OA (bottom). Bar, 5 μm. (B) Retraction rate of fibers perfused with YOP-S100 in the presence of 1 mM OV, 100 nM calyculin A (CA), and 100 ng/μl I2 or 10 nM OA. (C) Time-lapse sequences of PP2A-triggered retraction (top) and its inhibition by OA. Bar, 5 μm. (D) Retraction rate of fibers induced by PP2A (0.1 U/perfusion) with or without 10 nM OA. Entries are the means of 10–15 fibers measured ± SD.
Figure 2.

PP2A is present in Ascaris sperm. (A) Anti-PP2Ac antibody identified a 36-kDa of single band on Western blots (WB) of S100; CBB, an identical lane stained with Coomassie Brilliant Blue. (B) Differential interference contrast (left), anti-PP2Ac antibody immunofluorescence staining (center), and merged images (right) of an Ascaris sperm. The dotted line marks the boundary between the lamellipod and the cell body. PP2A labeling was confined to the cell body and concentrated at its junction with the base of lamellipod. Bar, 10 μm.
To determine whether the native PP2A present within sperm triggers retraction, we screened several batches of S100 and found that ∼10% of them induce retraction of fibers without addition of the tyrosine phosphatase YOP. The retraction-triggering activity of these S100, termed retraction-competent S100 (RC-S100) was inhibited by OA but not I2 or orthovanadate (OV), consistent with intrinsic PP2A in these S100 preparations inducing MSP fiber retraction (Figure 3, A and B). To test this hypothesis, we isolated PP2A from RC-S100 by immunoprecipitation using a PP2Ac antibody bound to Sepharose beads. The immunoprecipitated material was enriched in PP2A and induced retraction when dialyzed into KPM buffer and perfused onto fibers. Moreover, OA inhibited the retraction induced by this material (Figure 3C). We also compared the retraction capability of the extracts subjected to PP2A depletion with those of mock depletion. The rate of fiber retraction triggered by RC-S100 in which the PP2A concentration was reduced (Figure 3E) was ∼23% of that observed in batches of RC-S100 subjected to mock immunoprecipitation. Addition of commercial PP2A to the immunodepleted RC-S100 restored the rate of retraction to that observed in mock immunodepletion (Figure 3D).
Figure 3.

Intrinsic PP2A triggers retraction and is regulated by tyrosine dephosphorylation. (A) Time-lapse sequences showing retraction induced by perfusion of fibers with RC-S100 (top). OA blocked this retraction (bottom). Bar, 5 μm. (B) Comparison of rates of retraction induced by RC-S100 alone or with added OA (10 nM), OV (1 mM), or I2 (100 ng/μl). Entries represent the mean retraction rate for 10–15 fibers ± SD. (C) Rate of retraction induced by sperm PP2A, isolated by immunoprecipitation, in the presence and absence of OA. Material obtained by mock IP with mouse IgG served as a control. Entries are the means of seven to 10 fibers ± SD. (D) Effect of PP2A depletion on the rate of retraction induced by RC-S100. PP2A-depletion reduced the rate of retraction to ∼23% of that obtained using mock-depleted extracts. Add-back of a commercially obtained PP2A restored the retraction-triggering activity of depleted extracts. (E) SDS-PAGE and Western blot analysis of immunodepleted S100. Top, portion of a Western blot probed with anti-PP2Ac. Bottom, portion of an equivalent Coomassie Blue-stained gel containing MSP to show the relative amounts of sample loaded in each lane. The untreated control (left) and PP2A-depleted (center) lanes were loaded with equal amounts of total protein; the right lane was loaded with twofold more sample. Approximately 80% of PP2A was removed by immunodepletion. (F) SDS-PAGE gels of PP2A immunoprecipitated from RC-S100 (left), S100 (center), and YOP-treated S100 (right) and immunoblotted with either anti-PP2Ac (top) or anti-pY antibody (bottom). The PP2A from S100 was labeled by anti-pY but that from RC-S100 was not. YOP treatment abolished anti-pY labeling of the PP2A from S100.
Many forms of PP2A are activated by tyrosine dephosphorylation (Chen et al., 1992, 1994). To determine whether this occurs in sperm, we compared PP2A immunoprecipitated from S100 with that from RC-S100 on Western blots probed with anti-phosphotyrosine (anti-pY) (Figure 3F). We were unable to detect anti-pY labeling of the PP2A immunoprecipitated from RC-S100. In contrast, this antibody labeled PP2Ac isolated from S100, indicating that at least part of PP2Ac was tyrosine phosphorylated. Pretreatment of materials with YOP phosphatase eliminated the anti-pY signal. These data indicate that the retraction competence of S100 is directly correlated with tyrosine dephosphorylation of PP2A. This explains, at least in part, the requirement for a tyrosine phosphatase to render most batches of S100 retraction-competent and, conversely, the insensitivity of RC-S100 to a tyrosine phosphatase inhibitor (OV, Figure 3B).
MFP3 Is a Primary Target for PP2A
To identify substrates of PP2A involved in MSP fiber retraction, we used Western blots to compare the serine and threonine protein phosphorylation patterns in fibers isolated after treatment with PP2A to those in fibers treated with buffer alone (Figure 4A). Although PP2A treatment did not alter the labeling obtained with anti-phosphoserine antibody (anti-pS), Western blots of buffer-treated fibers contained a prominent labeled band at molecular weight ∼50 kDa when probed with anti-pT. No anti-pT labeling of this band could be detected after PP2A treatment of fibers. To identify this protein, the corresponding band in SDS-PAGE gel was excised and digested into peptides with trypsin. We used the sequences of two peptides (shown in Figure 4B, underlined), obtained from peptide sequencing with a Procise cLC protein sequencing system, to design primers for RT-PCR. The cDNA fragment generated in this way was then extended by standard 3′ and 5′ rapid amplification of cDNA ends to acquire the corresponding full-length cDNA (AY326288), which encoded a polypeptide of 472 amino acids (Figure 4B). The protein, which we designated MFP3, is unique to nematode sperm and exhibits homology to the uncharacterized Caenorhabditis elegans sperm-enriched proteins ssq1-4. As shown in Figure 4C, MFP3 contains 19 tandem repeats of a 15-residue motif in the middle of its sequence flanked by its ∼90-amino acid N domain and C domain. These 19 tandem repeats differ only marginally in sequence (Figure 4D) and show five different variants. The two flanking domains shared high identity (61%) in their amino acid sequences (Figure 4E).
Figure 4.

Identification of MFP3 as the primary substrate for PP2A in fibers. (A) Western blot assays of MSP fibers obtained after treatment with KPM buffer (Ctrl) or with PP2A. Blots of duplicate gels were probed with anti-pS or anti-pT antibody. The arrow indicates the band that was excised from duplicate Coomassie-stained gels for peptide sequencing. (B) The deduced protein sequence of MFP3 from full-length cDNA, translated by proteomic tools at ExPasy (http://ca.expasy.org). Underlining shows the peptide sequences used for construction of primers for RT-PCR. (C) Diagram showing the domain organization of MFP3. Segments A–E are the tandem repeats, numeral indicate the numbers of each repeat. (D) Peptide compositions of tandem repeats in MFP3 sequence. Phosphorylation sites identified by FT-ICR MS/MS are highlighted in red. (E) The alignment of N-domain and C-domain of MFP3, which exhibited 61% amino acid identity.
To identify the phosphorylation sites in MFP3, Coomassie Blue-stained MFP3 two-dimensional gel spots (∼50 kDa) were in-gel trypsin-digested and subjected to nano-LC-MS/MS. The results showed phosphothreonine in three unique peptides, detected by the neutral loss of 80 Da (HPO3) upon collision-induced dissociation (Gibson and Cohen, 1990). Comparison of the mass of b- and y-ions in MS/MS spectrum to their calculated values confirmed the assignment of phosphorylation sites (Perkins et al., 1999). Parent ion mass was determined by FTICR mass spectrometry (Marshall et al., 1998). The mass errors for all assigned peptides are sub-ppm (Schaub et al., 2008). Lower resolution LTQ MS/MS spectra were used to sequence the peptides, whose assigned masses of 1496.695, 1528.667, and 1528.801 Da identified the tryptic peptides pTAIGAPVGMSGLDTK, pTAIGAPMGMSGLDTK, and 376pTAIGAPVGAPAVGAAVK392. Because it is unique, the peptide, 376pTAIGAPVGAPAVGAAVK392, can be specifically located in MFP3. The other two peptides that contain phosphothreonines are present in multiple copies, and we cannot discriminate which copies are phosphorylated. All of the peptides share the LDTK*TAIGA motif, in which *T is the phosphorylated residue.
We generated a specific polyclonal antibody against MFP3 protein, which recognized a single band in the extracts of Ascaris sperm (Figure 5A). Indirect immunofluorescence labeling of sperm with this antibody showed that MFP3 was located in the fiber complexes in the lamellipod (Figure 5B), which coincided with the pattern of anti-pT labeling (Figure 5C). We also observed anti-MFP3 labeling throughout the fibers assembled in vitro (Figure 5D). Anti-MFP3 immunogold labeling of individual filaments obtained via polymerization in Triton X-100 treated S100 indicated that MFP3 binds to filaments, predominantly to their sides (Figure 5E).
Figure 5.

MFP3 is a component of MSP motility apparatus. (A) Western blot and Coomassie-stained lanes of S100. The polyclonal antibody raised against MFP3 specifically recognized a single band. (B–D) Paired phase contrast and indirect immunofluorescence micrographs of a sperm probed with anti-MFP3 (B), a sperm labeled with anti-pT (C), and a fiber grown in vitro with anti-MFP3 (D). Bar, 10 μm. (E–G) Immunogold labeling of MSP filaments labeled with anti-MFP3 antibody and 10-nm colloidal gold particles coated with secondary antibodies. Gold particles are shown as black dots (E) and are highlighted in red pseudocolor in F. (G) A control MSP filament with the primary antibody omitted. Bar, 150 nm.
Dephosphorylation of MFP3 Is Associated with Fiber Retraction
We examined the phosphorylation status of MFP3 during fiber retraction by obtaining samples of fibers at successive intervals after addition of PP2A to trigger retraction. Western blot analysis showed that the amount of sedimentable, fiber-associated MFP3 decreased as retraction progressed (Figure 6A). The MFP3 retained in these fibers still labeled with anti-pT, indicating that at least some of the MFP3 in the shortening fibers was still phosphorylated. By contrast, the amount of MFP3 released from retracting fibers to the soluble supernatant increased with time (Figure 6C), and we were unable to detect phosphorylation of this released fraction of MFP3 with anti-pT on Western blots. When fibers were treated with PP2A in the presence of OA, they did not retract (Figure 1C). There was no significant decrease in either sedimentable MFP3 or labeling with anti-pT in the PP2A-OA-treated fibers (Figure 6B). These results suggest that MFP3 dephosphorylation is associated with release of the protein from fiber during retraction.
Figure 6.
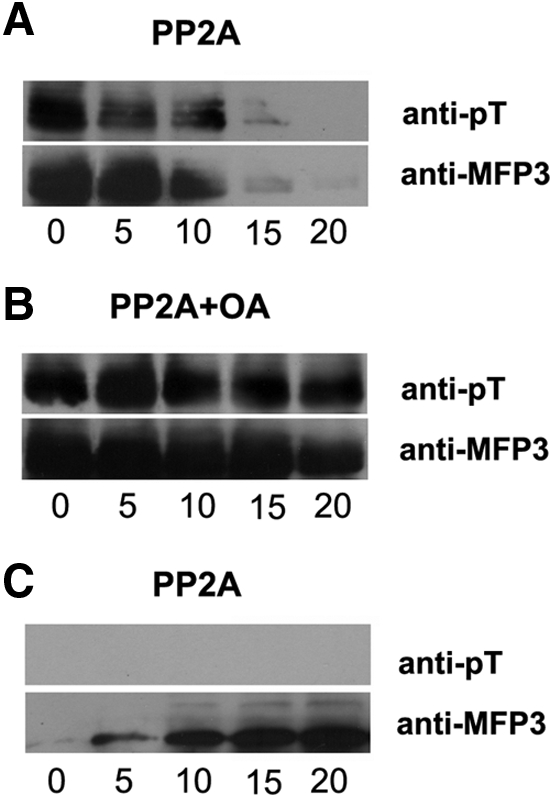
MFP3 dephosphorylation is associated with fiber retraction. Western blot analysis of the MFP3 content of MSP fibers pelleted at successive intervals after addition of PP2A (A) or PP2A with OA (B). Numerals indicate time (minutes) after addition of the test solution. Top panels were probed with anti-pT and the bottom panels with anti-MFP3. (A) The amount of sedimentable, fiber-associated MFP3 decreased as retraction progressed. The MFP3 retained in these fibers still labeled with anti-pT, indicating that at least some of the MFP3 in the shortening fibers was phosphorylated. (B) When fibers were treated with PP2A in the presence of OA, there was no significant decrease in the amount in either sedimentable MFP3 or labeling with anti-pT. (C) Western blot analysis of the supernatants obtained from the samples of pelleted fiber shown in A. The amount of MFP3 released from retracting fibers to the supernatant increased with time, but no MFP3 phosphorylation could be detected in these fractions with anti-pT.
DISCUSSION
Assembly of the MSP motility apparatus in cell-free extracts of Ascaris sperm has provided a reliable and versatile method for identifying the accessory components of locomotion. For example, biochemical dissection of fiber elongation led to the discovery of MPOP, MPAK, and MFP2 as proteins involved in leading edge protrusion in sperm (LeClaire et al., 2003; Buttery et al., 2003; Yi et al., 2007). Here, we exploited methods devised to study fiber retraction to identify two proteins that are involved in cell body retraction. PP2A, a ubiquitously expressed evolutionarily conserved Ser/Thr phosphatase, triggered retraction of MSP fibers by dephosphorylation of MFP3, a structural component in the MSP motility apparatus (Figure 7).
Figure 7.

Schematic illustration of a side view of a crawling sperm showing the components involved in phosphorylation/dephosphorylation of MFP3 and the relationship of this cycle to the MSP filaments dynamics that power sperm motility. See text for details.
Previous work showed that YOP tyrosine phosphatase only triggered retraction when added to fibers in the presence of S100, suggesting that it operated via a pathway involving at least one additional soluble downstream component (Miao et al., 2003). Several lines of evidence indicate that intrinsic PP2A is that key element of the retraction pathway. First, both commercial and sperm-derived PP2A in absence of any other soluble components in S100-triggered retraction. Second, retraction induced by YOP plus S100 and the intrinsic retraction activity of RC-S100 were blocked by OA, a PP2A-specific inhibitor, but not by inhibitors of other Ser/Thr phosphatases. Third, depletion of PP2A from RC-S100 decreased its retraction activity, and this activity was restored by adding back a commercial PP2A. Furthermore, our demonstration that the retraction competence of S100 depends on tyrosine dephosphorylation of its PP2A establishes a linkage between the activity of YOP in our assays and the regulation of PP2A. However, the native tyrosine phosphatase in sperm remains to be identified.
Although we cannot rule out interactions between PP2A and other sperm proteins, MFP3 is clearly the primary target of the phosphatase in fibers. This was the only fiber protein on which we were able to detect either Ser or Thr dephosphorylation by Western blot upon retraction. Consistent with the Western blot patterns, which showed MFP3 labeled by anti-pT and not anti-pS, MS/MS analysis detected only phosphothreonine residues in MFP3. Like other structural motility proteins in MSP system, MFP3 has substantial sequence similarity to a protein family of unknown function in C. elegans and shows no homology to known proteins in other types of cell. Consequently, the sequence by itself provides little insight into the function of the protein. However, our experimental data indicate that MFP3 seems to be multiply phosphorylated, binds primarily to the sides of MSP filaments, and is released upon dephosphorylation. The release of MFP3 is associated with filament disassembly during fiber retraction, suggesting MFP3, in its phosphorylated form, may be involved in stabilizing MSP filaments. This function would be consistent with the behavior of the fiber complexes in sperm, which undergo little, if any remodeling as they flux rearward through the lamellipod, but then disassembles rapidly at the lamellipod–cell body junction. PP2A is concentrated at this site and thus has access to the MSP filaments only at the location where cytoskeletal disassembly and generation of the force for retraction take place.
The key role of protein dephosphorylation in retraction is also consistent with previous studies that showed that, in the MSP system, protrusion and retraction are reciprocal events. The assembly of the MSP cytoskeleton that drives protrusion of the leading edge requires phosphorylation of two proteins, MPOP (LeClaire et al., 2003) and MFP2 (Yi et al., 2007). The kinase MPAK that targets MFP2 is recruited to the leading edge membrane by binding to phosphorylated MPOP. The interaction between PP2A and MFP3 links cytoskeletal disassembly and retraction to protein dephosphorylation at the opposite end of the lamellipod. In many other cellular functions, PP2A holoenzymes are directed to specific subcellular locations by binding to a regulatory subunit (Virshup, 2000). It is likely that a similar mechanism restricts the distribution of PP2A in sperm, thereby defining the site of cytoskeletal disassembly in a manner analogous to the way that recruitment of MPAK to the leading edge specifies the site of cytoskeletal assembly.
A long-standing problem in the amoeboid motility of nematode sperm has been the source of energy that powers the process. Because MSP itself lacks any detectable ATPase activity, the energy to power locomotion must be supplied to other components of the system. For example, MSP polymerization and leading edge protrusion depend on phosphorylation of two proteins that define the site (LeClaire et al., 2003) and the rate of cytoskeletal assembly (Yi et al., 2007). On the basis of our present results, we hypothesize that phosphorylation of MFP3 may function to provide energy to power retraction. One plausible way in which this could be achieved could relate to the substantial overall positive charge of MSP (pI = 8.3–8.9; King et al., 1992), which would tend to result in MSP dimers repelling one another. Multiple phosphorylation of MFP3 would generate a molecule with a large negative charge that could lie in the grooves of the MSP helix (analogous to the way in which tropomyosin lies in the long-pitch grooves of the F-actin helix). In this orientation, MFP3 could span several MSP dimers so that its overall negative charge could effectively neutralize their positive charge and thereby stabilize the filaments. In this context, it is likely that the periodicity of the 15-residue tandem repeats in the central region of MFP3 matches a structural repeat in MSP filament helices (Stewart et al., 1993), although clearly it will be necessary to determine the structure of MFP3 to evaluate this hypothesis. We further propose that the energy stored by phosphorylating MFP3 would then be released by PP2A, and, by reducing the negative charge on the molecule, would render the filaments unstable because the repulsion between the positively charged MSP dimers would no longer be masked. Thus, through its modulation of filament stability the phosphorylation state of MFP3 could modulate the cytoskeletal dynamics of nematode sperm and provide the energy for retraction.
Although the precise mechanism is not fully understood, in many actin-based crawling cells cell body retraction has been proposed to be powered by the contractile activity of myosin (Svitkina et al., 1997; Iwadate and Yumura, 2008). In the MSP motility system, no motor proteins have been discovered and the apolar structure of MSP filament does not provide a suitable framework for motor function (Bullock et al., 1998; Roberts and Stewart, 2000). Instead, MSP-based retraction involves shrinkage of cytoskeletal gel volume due to disassembly and rearrangement of the constituent filaments (Miao et al., 2003). In actin-rich cells, retraction is also associated with cytoskeletal reorganization and disassembly (Heidemann and Buxbaum, 1998), and so the results obtained here in the Ascaris system make it pertinent to ask whether these processes also contribute to pulling the cell body forward in actin-rich cells. The discovery of two key components of the retraction machinery in nematode sperm provides a biochemical handle for exploring the biophysical basis of this process and assessing whether a similar mechanism may operate in other crawling cells.
ACKNOWLEDGMENTS
We thank Margaret Seavy, Lori McFadden, Kim Riddle, Tom Fellers, and Doris Terry for expert technical assistance; and our colleagues in Tallahassee and Cambridge, particularly Katsuya Shimabukuro and Andrew Malowney, for valuable discussions. This work was supported by National Institutes of Health grant R37 GM-29994, National Science Foundation Division of Materials Research through grant DMR-0654118, and the state of Florida.
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-03-0240) on May 20, 2009.
REFERENCES
- Akin O., Mullins R. D. Capping protein increases the rate of actin-based motility by promoting filament nucleation by the Arp2/3 complex. Cell. 2008;133:841–851. doi: 10.1016/j.cell.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock T., McCoy A. J., Kent H., Roberts T. M., Stewart M. Structural basis of amoeboid motility in nematode sperm. Nat. Struct. Biol. 1998;5:184–189. doi: 10.1038/nsb0398-184. [DOI] [PubMed] [Google Scholar]
- Buttery S. M., Ekman G. C., Seavy M., Stewart M., Roberts T. M. Dissection of the Ascaris sperm motility machinery identifies key proteins involved in major sperm protein-based amoeboid locomotion. Mol. Biol. Cell. 2003;14:5082–5088. doi: 10.1091/mbc.E03-04-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron L. A., Giardinia P. A., Soo F. S., Theriot J. A. Secrets of actin-based motility revealed by a bacterial pathogen. Nat. Rev. Mol. Cell. Biol. 2000;1:110–119. doi: 10.1038/35040061. [DOI] [PubMed] [Google Scholar]
- Chen J., Parsons S., Brautigan D. L. Tyrosine phosphorylation of protein phosphatase 2A in response to growth stimulation and v-src transformation of fibroblasts. J. Biol. Chem. 1994;269:7957–7962. [PubMed] [Google Scholar]
- Chen J., Martin B. L., Brautigan D. L. Regulation of protein serine-threonine phosphatase 2A by tyrosine phosphorylation. Science. 1992;257:1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- Co C., Wong D. T., Gierke S., Chang V., Taunton J. Mechanism of actin network attachment to moving membranes: barbed end capture by N-WASP WH2 domains. Cell. 2007;128:901–913. doi: 10.1016/j.cell.2006.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin-Thaler B. J., Hofman J. M., Cai Y., Xenias H., Spielman I., Shneidman A. V., David L. A., Döbereiner H. G., Wiggins C. H., Sheetz M. P. Quantification of cell edge velocities and traction forces reveals distinct motility modules during cell spreading. PLoS ONE. 2008;3:e3735. doi: 10.1371/journal.pone.0003735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson B. W., Cohen P. Liquid secondary ion mass spectrometry of phosphorylated and sulfated peptides and proteins. Methods Enzymol. 1990;193:480–501. doi: 10.1016/0076-6879(90)93434-m. [DOI] [PubMed] [Google Scholar]
- Heidemann S., Buxbaum R. Cell crawling: first the motor, now the transmission. J. Cell Biol. 1998;141:1–4. doi: 10.1083/jcb.141.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug C., Jay P. Y., Reddy I., McNally J. G., Bridgman P. C., Elson E. L., Cooper J. A. Capping protein levels influence actin assembly and cell motility in Dictyostelium. Cell. 1995;81:591–600. doi: 10.1016/0092-8674(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Italiano J. E., Jr, Roberts T. M., Stewart M., Fontana C. A. Reconstitution in vitro of the motile apparatus from the amoeboid sperm of Ascaris shows that filament assembly and bundling move membranes. Cell. 1996;84:105–114. doi: 10.1016/s0092-8674(00)80997-6. [DOI] [PubMed] [Google Scholar]
- Italiano J. E., Jr, Stewart M., Roberts T. M. Localized depolymerization of the major sperm protein cytoskeleton correlates with forward movement of the cell body in the amoeboid movement of nematode sperm. J. Cell Biol. 1999;146:1087–1095. doi: 10.1083/jcb.146.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwadate Y., Yumura S. Actin-based propulsive forces and myosin-II–based contractile forces in migrating Dictyostelium cells. J. Cell Sci. 2008;21:1314–1324. doi: 10.1242/jcs.021576. [DOI] [PubMed] [Google Scholar]
- King K. L., Stewart M., Roberts T. M., Seavy M. Structure and macromolecular assembly of two isoforms of the major sperm protein (MSP) from the amoeboid sperm of the nematode, Ascaris suum. J. Cell Sci. 1992;101:847–857. doi: 10.1242/jcs.101.4.847. [DOI] [PubMed] [Google Scholar]
- Loisel T. P., Boujemaa R., Pantaloni D., Carlier M. F. Reconstitution of actin-based motility of Listeria and Shigella using pure proteins. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- LeClaire L. L. III, Stewart M., Roberts T. M. A 48 kDa integral membrane phosphoprotein orchestrates the cytoskeletal dynamics that generate amoeboid cell motility in Ascaris sperm. J. Cell Sci. 2003;116:2655–2663. doi: 10.1242/jcs.00469. [DOI] [PubMed] [Google Scholar]
- Marshall A. G., Hendrickson C. L., Jackson G. S. Fourier transform ion cyclotron resonance mass spectrometry: a primer. Mass Spectrom. Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Miao L., Vanderlinde O., Stewart M., Roberts T. M. Retraction in amoeboid cell motility powered by cytoskeletal dynamics. Science. 2003;302:1405–1407. doi: 10.1126/science.1089129. [DOI] [PubMed] [Google Scholar]
- Miao L., Vanderlinde O., Liu J., Grant R. P., Wouterse A., Shimabukuro K., Philipse A., Stewart M., Roberts T. M. The role of filament-packing dynamics in powering amoeboid cell motility. Proc. Natl. Acad. Sci. USA. 2008;105:5390–5395. doi: 10.1073/pnas.0708416105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantaloni D., Le Clainche C., Carlier M. F. Mechanism of actin-based motility. Science. 2001;292:1502–1506. doi: 10.1126/science.1059975. [DOI] [PubMed] [Google Scholar]
- Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Pollard T. D., Borisy G. G. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Rafelski S. M., Theriot J. A. Crawling toward a unified model of cell motility: spatial and temporal regulation of actin dynamics. Annu. Rev. Biochem. 2004;73:209–239. doi: 10.1146/annurev.biochem.73.011303.073844. [DOI] [PubMed] [Google Scholar]
- Ris H. The cytoplasmic filament system in critical point-dried whole mounts and plastic-embedded sections. J. Cell Biol. 1985;100:1474–1487. doi: 10.1083/jcb.100.5.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts T. M., Stewart M. Acting like actin. The dynamics of the nematode major sperm protein (MSP) cytoskeleton indicate a push-pull mechanism for amoeboid cell motility. J. Cell Biol. 2000;149:7–12. doi: 10.1083/jcb.149.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub T. M., Hendrickson C. L., Horning S., Quinn J. P., Senko M. W., Marshall A. G. High performance mass spectrometry: Fourier transform ion cyclotron resonance at 14.5 Tesla. Anal. Chem. 2008;80:3985–3990. doi: 10.1021/ac800386h. [DOI] [PubMed] [Google Scholar]
- Sepsenwol S., Ris H., Roberts T. M. A unique cytoskeleton associated with crawling in the amoeboid sperm of the nematode, Ascaris suum. J. Cell Biol. 1989;108:55–66. doi: 10.1083/jcb.108.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Stewart M., King K. L., Roberts T. M. Crystallization of the motile major sperm protein (MSP) of the nematode, Ascaris suum. J. Mol. Biol. 1993;232:298–300. doi: 10.1006/jmbi.1993.1383. [DOI] [PubMed] [Google Scholar]
- Svitkina T. M., Verkhovsky A. B., Borisy G. G. Improved procedures for electron microscopic visualization of the cytoskeleton of cultured cells. J. Struct. Biol. 1995;115:290–303. doi: 10.1006/jsbi.1995.1054. [DOI] [PubMed] [Google Scholar]
- Svitkina T. M., Verkhovsky A. B., McQuade K. M., Borisy G. G. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J. Cell Biol. 1997;139:397–415. doi: 10.1083/jcb.139.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot J. A. Worm sperm and advances in cell locomotion. Cell. 1996;84:1–4. doi: 10.1016/s0092-8674(00)80068-9. [DOI] [PubMed] [Google Scholar]
- Virshup D. M. Protein phosphatase 2A: a panoply of enzymes. Curr. Opin. Cell Biol. 2000;12:180–185. doi: 10.1016/s0955-0674(99)00074-5. [DOI] [PubMed] [Google Scholar]
- Wang Y. L. Exchange of actin subunits at the leading edge of living fibroblasts: possible role of treadmilling. J. Cell Biol. 1985;101:597–602. doi: 10.1083/jcb.101.2.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi K., Buttery S., Stewart M., Roberts T. M. A Ser/Thr kinase required for membrane-associated assembly of the major sperm protein motility apparatus in the amoeboid sperm of Ascaris. Mol. Biol. Cell. 2007;18:1816–1825. doi: 10.1091/mbc.E06-08-0741. [DOI] [PMC free article] [PubMed] [Google Scholar]