

Abstract
A quinazoline that decreases polyglutamine aggregate burden in a cell-based assay was identified from a high-throughput screen of a chemical-compound library, provided by the NIH Molecular Libraries Small Molecule Repository (MLSMR). A structure and activity study yielded leads with submicromolar potency.
Huntington’s disease (HD), a member of the trinucleotide-repeat neurological disorders, is caused by a heritable, polyglutamine-expansion mutation in the NH3-terminus of the huntingtin protein (Htt).1–3 The intracellular accumulation of insoluble aggregates of mutant Htt is a hallmark of HD, but the precise mechanism of toxicity remains a matter of conjecture.4 In mouse models of HD, expression of the NH3-terminal portion of the mutant Htt protein recapitulates key features of the disease with mice displaying a phenotype of progressive neurological impairment. Neurons in these models5–6 show an intrinsic ability to clear polyQ aggregates, and accompanying this clearance, a reversal of the HD-like neurologic symptoms is observed. Macroautophagy, the lysosome-mediated degradation of cytosolic proteins, is implicated as the cellular pathway responsible for aggregate clearance.7–10 Using a unique two-tiered functional genetic screen, we previously found that macroautophagy-mediated clearance of accumulated mutant protein can be increased through IRS-2 stimulation in a cell-based model of HD.7 In order to search for small-molecule activators of Htt clearance, we used this model in a high-throughput fluorescent cell-based assay format to monitor the aggregation and clearance of a tet-regulatable11, conditionally expressed mutant protein consisting of the first 17 NH3-terminal amino acids of Htt followed by a polyQ stretch of 103 residues fused to a monomeric cyan fluorescent protein (mCFP).7 These stable transfectants do not manifest the acute polyQ-length-dependent cell death observed after transient transfection7 and thus provided an excellent platform for the identification of modulators of protein degradation which act over a period of days.
The degree of aggregation in this assay is measured by the presence and magnitude of fluorescent intracellular inclusions. We screened a 10K chemical compound library, provided by the NIH-sponsored MLSCN, for diminution of aggregation of mutant protein after incubation for 72h. We identified six hits: five tetracycline derivatives, likely suppressing tet-regulatable gene expression, and quinazoline 1a (Figure 1), with an IC50 of approximately 4 μM. The structure of 1a was confirmed by synthesis and dose-dependent activity was demonstrated with an IC50 of 2.2 μM (Table 1). A structure-activity relationship (SAR) study was conducted to identify more potent derivatives. Three regions of the quinazoline were selected for modification: A, the distal piperazine N-alkyl group; B, the piperazine ring itself; and C, the 6-bromo group on the aromatic core. We envisioned that analogs would be easily synthesized from common 4-chloro-6-halo-quinazoline intermediates (Scheme 1).12 As this work proceeded, Sarkar et al., seeking enhancers of autophagy, reported several bromo-substituted quinazolines13 and we synthesized three of their compounds, 12–14, for comparison (Table 3).
Figure 1.
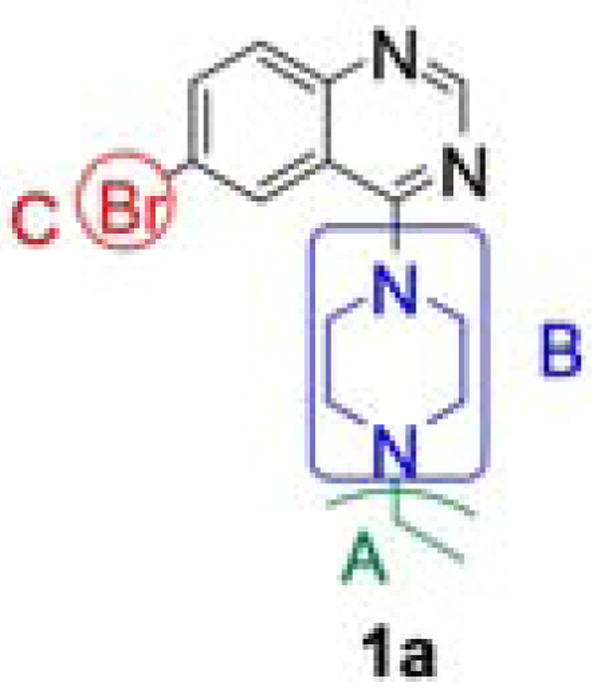
Structure analysis of our lead compound
Table 1.
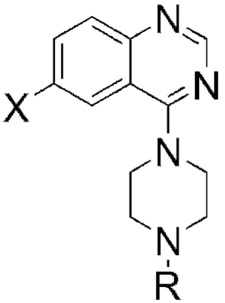
Modifications of Regions A and C: IC50 values of our piperazine analogs 1–4.
 | |||
|---|---|---|---|
| Compds | X | R | IC50, μM |
| 1a | Br | Ethyl | 2.21 |
| 1b | Cl | Ethyl | 3.63 |
| 1c | F | Ethyl | 5.26 |
| 1d | H | Ethyl | > 10 |
| 2a | Br | Methyl | 1.31 |
| 2b | Cl | Methyl | > 10 |
| 2c | F | Methyl | > 10 |
| 2d | H | Methyl | > 10 |
| 3a | Br | H | 2.74 |
| 4a | Br | 4-(1,3-benzodioxol-5- ylmethyl) | Inactive |
| 4b | Cl | 4-(1,3-benzodioxol-5- ylmethyl) | 1.07 |
| 4c | F | 4-(1,3-benzodioxol-5- ylmethyl) | > 10 |
| 4d | H | 4-(1,3-benzodioxol-5- ylmethyl) | > 10 |
Scheme 1.
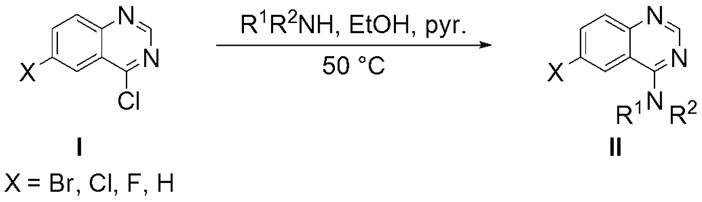
General procedure for the synthesis of our quinazoline compounds
Table 3.
Quinazoline derivatives 12–14 of Sarkar et al.: IC50 values in our assay.
 | ||
|---|---|---|
| Compds | R | IC50, μM |
| 12 | Propyl | 8.13 |
| 13 | Allyl | 5.36 |
| 14 | Benzyl | 6.13 |
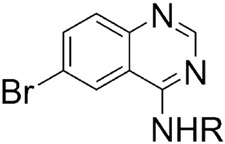
Substitution at the N-ethyl group of quinazoline 1a with either smaller or bulkier groups did not alter activity (Table 1). In contrast, substitution at the 6-position with fluorine or hydrogen was not well tolerated compared to substitution with bromine or chlorine. This pattern for substitution at the 6-position was also observed in the series based on replacement of the piperazine ring with 4′-substituted phenylethylene amine. That is, the activity of the 6-fluoro- and dehalogenated derivatives was generally lower (Table 2). In general, the secondary amines of Table 2 were superior to the corresponding piperazines of Table 1. Quinazoline itself is inactive in our assay and any substitutions of the quinazoline core with other cores resulted in loss of activity. Substitution of the amino group with an alkoxy group also resulted in loss of activity. The most potent inhibitor of accumulation of aggregated mutant Htt fragment was compound 5b with an IC50 of 0.71 μM. Alkylation of the 4′-hydroxyl group of compound 5b was not tolerated, c.f. compounds 7–10.
Table 2.
Modifications of Regions B and C: IC50 values of our secondary amine analogs 5–11.
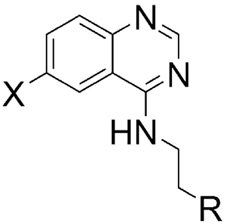 | |||
|---|---|---|---|
| Compds | X | R | IC50, μM |
| 5a | Br | Phenol | 1.11 |
| 5b | Cl | Phenol | 0.71 |
| 5c | F | Phenol | 6.38 |
| 5d | H | Phenol | > 15.8 |
| 6a | Br | 4-Chlorophenyl | Inactive |
| 6b | Cl | 4-Chlorophenyl | 3.44 |
| 6c | F | 4-Chlorophenyl | Inactive |
| 6d | H | 4-Chlorophenyl | 10.5 |
| 7 | Cl | 4-Phenoxyphenyl | Inactive |
| 8 | NH2 | 4-Phenoxyphenyl | Inactive |
| 9 | Cl | 4-(Cyclopentyloxy)phenyl | Inactive |
| 10 | Cl | 4-(Benzyloxy)phenyl | Inactive |
| 11 | Cl | Ethyl (4-phenoxy)acetate | 4.48 |
Sarkar compounds 12–14 were reported to have EC50 values of approximately 50 μM in a yeast assay. While no rank order was reported for this assay, the potency of the compounds in PC12 cells was 12 > 13 > 14 and in COS-7 cells was 14 > 13 > 12.13 In contrast, we found low micromolar potency in our system with a rank order of 12 > 14 > 13 (Table 3). It remains to be formally demonstrated that our assay and that of Sarkar et al. are directed to the same target.
The decrease in the number of aggregates per cell, observed with our compound may be due to the clearance of inclusions since the number of inclusions after the administration of compounds such as 5b is less than the number before treatment. Nonetheless, other factors may be at play, such as enhanced cell division leading to cytoplasmic dilution, and thus an apparent decrease in the number of inclusions, or enhanced cell death leading to a preferential loss of aggregate-containing cells. To eliminate these possibilities, we examined cell numbers and cell death after administration of compound 5b. As shown in Figure 2, despite the clearance of inclusions, neither signs of cytotoxicity nor differences in the total number of cells were observed.
Figure 2.

Compound 5b leads to a dose-dependent decrease in the number of aggregates per cell in the absence of increased proliferation or cell death. Cells were seeded in a 96-well format and treated with 2μg/mL doxycycline (Dox), 0.1% DMSO (No dox) as control, or compound 5b. A. Percent (%) cells with aggregates. i. The overall effect of 5b on % cells with aggregates with respect to Dox and No dox. ANOVA reveals a statistically significant effect of ‘Treatment’ on “% Cells with aggregates’ (F(2,37) =14.486, p<0.001. ‘*’ = p < 0.05 in Fisher Posthoc analysis when compared to No dox.) ii. 5b exerts a dose-dependent decrease of aggregates. B. % Dead cells. There is no effect on cell death of 5b as compared to Dox or No dox (i) or across dose (ii). (F(2,37) = 0.346; p = 0.7095) C. Number of cells per well. There is no effect on the total cell number of 5b as compared to Dox or No dox (i) or across dose (ii). (F(2,37) = 2.225; p = 0.1224).
In summary, we prepared and identified four potent inhibitors of huntingtin protein aggregation, 5b, 4b, 5a, and 2a. Our library of analogs provides a starting point for mechanistic studies to determine the target of action.
Acknowledgments
The authors would like to acknowledge Teresa Wojtasiewicz for technical support and would like to thank the NIH’s Molecular Libraries Initiative for funding our research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Zhang X, Smith DL, Meriin AB, Engemann S, Russel DE, Roark M, Washington SL, Maxwell MM, Marsh JL, Thompson LM, Wanker EE, Young AB, Housman DE, Bates GP, Sherman MY, Kazantsev AG. Proc Natl Acad Sci USA. 2005;102:892. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colby DW, Chu Y, Cassady JP, Duennwald M, Zazulak H, Webster JM, Messer A, Lindquist S, Ingram VM, Wittrup KD. Proc Natl Acad Sci USA. 2004;101:17616. doi: 10.1073/pnas.0408134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, Lehrach H, Wanker EE. Proc Natl Acad Sci USA. 2000;97:6739. doi: 10.1073/pnas.110138997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Nature. 2004;431:805. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto A, Lucas JJ, Hen R. Cell. 2000;101:57. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 6.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Cell. 1996;87:493. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto A, Cremona ML, Rothman JE. J Cell Biol. 2006;172:719. doi: 10.1083/jcb.200510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwata A, Riley BE, Johnston JA, Kopito RR. J Biol Chem. 2005;280:40282. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 9.Iwata A, Christianson JC, Bucci M, Ellerby LM, Nukina N, Forno LS, Kopito RR. Proc Natl Acad Sci USA. 2005;102:13135. doi: 10.1073/pnas.0505801102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EMC, Isaacs A, Brech A, Stenmark H, Simonsen A. J Cell Biol. 2007;179:485. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gossen M, Bujard H. Annu Rev Genet. 2002;36:153. doi: 10.1146/annurev.genet.36.041002.120114. [DOI] [PubMed] [Google Scholar]
- 12.1HNMR and 13C NMR spectra were recorded on a Varian Mercury 300 T NMR spectrometer. High-resolution mass spectrometry was done by Dr. Yasuhiro Itagaki at Columbia University. 4,6-Dichloroquinazoline (I, X = Cl) was stirred at room temperature in a 4:1 mixture of ethanol (4.0 mL) and pyridine (1.00mL). Tyramine (0.124 g, 0.904 mmol) was added to the white suspension which immediately upon addition of the amine turned dark bright orange and then gradually turned into a clear, bright orange solution. The solution was stirred overnight and was subsequently heated for 5h at 50 °C, after which it was allowed to cool to room temperature. The product was purified via column chromatography (1:1 hexanes : ethyl acetate) to give a white solid, 5b, (0.106 g, 0.354 mmol, 59 %): mp 178–180 °C; 1H NMR (300 MHz,(CD3)2SO) δ 9.19 (s, 1H), 8.49 (s, 1H), 8.39 (s, 1H), 7.78 (d, 1H, J = 9.0 Hz), 7.69 (d, 1H, J = 8.7 Hz), 7.05 (d, 2H, J = 8.1 Hz), 6.68 (d, 2H, J = 8.4 Hz), 3.67 (t, 2H, J = 7.5 Hz), 2.83 (t, 2H, J = 7.5 Hz); 13C NMR (75 MHz, (CD3)2SO) δ 158.4 (1C), 155.5 (1C), 155.4 (1C), 147.6 (1C), 132.7 (1C), 129.6 (2C), 129.4 (2C), 129.3 (1C), 121.9 (1C), 115.7 (1C), 115.0 (2C), 42.6 (1C), 33.6 (1C); ESI-MS (M++H): 300; FAB+ HRMS calcd for C16H15ON3 35Cl: 300.0904; Found: 300.0902; MS m/z 300 (M++H, 100 %), 235, 217, 192, 180, 176.
- 13.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, Webster JA, Lewis TA, O’Cane CJ, Schreiber SL, Rubinsztein DC. Nat Chem Biol. 2007;3:331. doi: 10.1038/nchembio883. [DOI] [PMC free article] [PubMed] [Google Scholar]