

Abstract
Th17 cells play an important role in mediating autoimmune diseases, but the molecular mechanism underlying Th17 differentiation is incompletely understood. We show here that NF-κB–inducing kinase (NIK), which is known to regulate B-cell maturation and lymphoid organogenesis, is important for the induction of Th17 cells. NIK-deficient naive CD4 T cells are attenuated in the differentiation to Th17 cells, although they are competent in committing to the other effector lineages. Consistently, NIK knockout mice are resistant to experimental autoimmune encephalomyelitis, a disease model that involves the function of Th17 cells. This phenotype was also detected in Rag2 knockout mice reconstituted with NIK-deficient T cells, confirming a T-cell intrinsic defect. We further show that NIK mediates synergistic activation of STAT3 by T-cell receptor and IL-6 receptor signals. NIK deficiency attenuates activation of STAT3 and induction of STAT3 target genes involved in Th17-commitment program. These findings establish NIK as an important signaling factor that regulates Th17 differentiation and experimental autoimmune encephalitis induction.
Introduction
CD4 T cells play a central role in shaping the immune system for effective response to microbial infections. Upon activation by an antigen, naive CD4 T cells differentiate into subsets of effector T cells, T helper (Th)1, Th2, and Th17 cells, which are characterized by the production of specific cytokines and engagement in specialized immune functions.1 Th1 cells produce interferon-γ (IFN-γ) and mediate cellular immune responses against infection by intracellular pathogens, whereas Th2 cells produce IL-4, IL-5, and IL-13 and play an important role in antibody responses to extracellular pathogens.2,3 A signature cytokine produced by the Th17 cells is IL-17A, which mediates inflammatory responses by recruiting immune cells and inducing the production of proinflammatory cytokines.4–7 Strong evidence suggests that Th17 cells are involved in various autoimmune and inflammatory diseases,7,8 such as experimental autoimmune encephalitis (EAE)5,9 and rheumatoid arthritis.10–12 Moreover, activated CD4 T cells can also differentiate into inducible regulatory T cells (iTregs), which suppress the function of effector T cells, thereby keeping an immune response under control
As seen with Th1 and Th2 cells,2,3 the development of Th17 cells is regulated by the specific cytokine microenvironment.13,14 IL-6 and transforming growth factor-β (TGF-β) are critical cytokines that, together with the T-cell receptor (TCR) signal, initiate the differentiation of Th17 cells from naive CD4 T cells.15–17 Another cytokine, IL-21, is induced by IL-6 in the course of Th17 cell differentiation and may function to sustain the Th17 polarizing signal in an autocrine manner.18–20 IL-21 also induces the expression of IL-23 receptor (IL-23R), rendering the Th17 cells responsive to IL-23, a cytokine that is involved in the maintenance of the Th17 population.5,21 In addition, recent studies suggest that Th17 cell differentiation also involves TL1A and its receptor DR3, members of the TNF and TNF receptor (TNFR) superfamilies, respectively.22,23 Notably, several other TNF/TNFR family members are well-known costimulatory molecules involved in the activation and differentiation of T cells, although their role in Th17 production is not well characterized.24
The intracellular signaling mechanism mediating Th17 differentiation is still incompletely understood. Nevertheless, the primary signaling event induced by IL-6 is activation of STAT3, a critical transcription factor involved in the initiation of the Th17 commitment program.25 IL-6 stimulates the tyrosine phosphorylation of STAT3, a common mechanism that triggers the dimerization, nuclear translocation, and DNA binding activity of STAT proteins.26 Activated STAT3 regulates Th17 cell differentiation by participating in the transcriptional activation of several Th17-regulatory genes, including those encoding IL-21, IL-23R, and the orphan nuclear hormone receptor RORγt.20,27,28 Induction of RORγt represents a central step in the Th17-commitment program, because RORγt functions as a Th17 lineage-specific transcription factor that regulates the expression of the IL-17A gene locus.19,20,27 The transcriptional induction of RORγt, as well as the other Th17 regulatory and effector genes, requires not only IL-6 and TGF-β but also the TCR signal. However, the signaling molecules connecting the TCR signal to the Th17-commitment pathway are poorly defined.
MAP kinase (MAPK) signaling pathways participate in diverse biological processes, including the differentiation of Th1 and Th2 cells.29 Activation of MAPKs involves a signaling cascade initiated from activation of a family of upstream kinases, the MAPK kinase kinases (MAP3Ks).30 In addition to mediating the typical MAP3K signaling cascades, MAP3Ks also possess other signaling functions. One MAP3K, the NF-κB–inducing kinase (NIK), has a central role in mediating a noncanonical NF-κB signaling pathway.31 This pathway involves processing of an NF-κB precursor protein, p100, to a mature NF-κB subunit, p52, and the nuclear translocation of p52/RelB NF-κB complex. The noncanonical NF-κB pathway is stimulated by a subset of TNFR family members and plays a critical role in B-cell maturation and lymphoid organogenesis.31 In addition to regulating noncanonical NF-κB, NIK appears to possess other signaling functions. At least in cell line models, NIK phosphorylates MEK1, thereby causing activation of the downstream MAPK, ERK.32 Moreover, overexpressed NIK induces the phosphorylation of STAT3 in a prostate cancer cell line.33 Notably, the catalytic activity of NIK is rapidly stimulated upon ligation of TCR and the costimulatory molecule CD28,34 suggesting a role for NIK in regulating the signaling events involved in T-cell activation or differentiation.
In the present study, we investigated the function of NIK in T-cell differentiation using NIK knockout mice. We show that NIK has a critical role in Th17 cell differentiation and EAE induction, although this MAP3K is dispensable for the differentiation of Th1, Th2, and iTreg cells. We provide evidence that NIK is required for synergistic activation of STAT3 by the TCR and IL-6 signals. Thus, our data establish NIK as a critical regulator of Th17 differentiation and shed light on a new function of NIK in mediating TCR signaling.
Methods
Mice
NIK knockout mice, from a 129Sv/Ev background,35 were provided by Amgen (Thousand Oaks, CA) and maintained in the specific pathogen-free facility of M. D. Anderson Cancer Center. NIK+/– heterozygous mice were bred to generate age- and sex-matched NIK+/+ and NIK−/− mice that were used in the experiments. Rag2 knockout (Rag2−/−) mice, from a 129Sv/Ev background, were obtained from Taconic (Germantown, NY). All animal experiments were in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Texas M. D. Anderson Cancer Center.
Antibodies, reagents, and plasmids
Functional grade anti–mouse (m) CD3ϵ (145-2C11) and anti-mCD28 (37.51) antibodies as well as the blocking antibodies for mIFN-γ (XMG1.2) and mIL-4 (11B11) were from eBioscience (San Diego, CA). Fluorescence-labeled antibodies for mCD4-Pacific blue (L3T4), mCD25-PE-Cy7 (PC61.5), mCD62L-APC (MEL-14), mCD44-APC-Cy7 (IM7), mIL-17A-PE (eBio17B7), mIFN-γ-FITC (XMG1.2), mIL-4-PE (11B11), and mFoxp3-FITC (FJK-16s) were also purchased from eBioscience. PE-conjugated anti–phospho-STAT3 (pY705) was from BD Biosciences (San Jose, CA). The recombinant mIL-6 and hTGF-β were purchased from PeproTech (Rocky Hill, NJ), and recombinant LIGHT (lymphotoxin-like, exhibits inducible expression, and competes with HSV glycoprotein D for HVEM, a receptor expressed by T lymphocytes) was from R&D Systems (Minneapolis, MN). Phorbol 12-myristate 13-acetate (PMA) and ionomycin were from Sigma (St Louis, MO), and monensin was from eBioScience. The retroviral vector pCLXSN(GFP) was a modified version of pCLXSN,36 in which the neomycin gene was replaced with a GFP gene. pCLXSN(GFP)-NIK was created by inserting the human NIK cDNA into the pCLXSN(GFP) vector.
CD4 T-cell differentiation and proliferation assays
CD4 T cells were isolated from splenocytes using a CD4 T-cell Isolation Kit (Miltenyi Biotec, Auburn, CA) and then subjected to flow cytometric cell sorting (FACSAria, BD Biosciences) to purify naive CD4 T cells (CD4+CD25−CD44loCD62Lhi). Purified naive CD4 T cells were stimulated with the indicated amounts of plate-bound anti-CD3 and anti-CD28 under Th0 (10 μg/mL anti–IL-4, 10 μg/mL anti–IFN-γ), Th1 (10 ng/mL IFN-γ, 10 ng/mL IL-12, 10 μg/mL anti–IL-4), Th2 (10 ng/mL IL-4, 10 μg/mL anti–IFN-γ), Th17 (20 ng/mL IL-6, 5 ng/mL TGF-β, 10 μg/mL anti–IL-4, 10 μg/mL anti–IFN-γ), or iTreg (10 units/mL IL-2, 5 ng/mL TGF-β) conditions. When indicated, recombinant LIGHT (500 ng/mL) was added to the culture as a costimulatory molecule. After the indicated times, the cells were subjected to intracellular cytokine staining (ICS) and real-time RT-PCR analyses.
For proliferation assays, naive CD4 T cells were labeled with carboxyl fluorescent succinimidyl ester (CFSE, 1.25 μg/mL in PBS) for 5 minutes at room temperature. After 2 washes with RPMI medium or PBS (supplemented with 5% FCS), the cells were stimulated with anti-CD3/CD28 under Th0 or Th17 conditions, as described above. After 72 hours, CFSE intensity was determined by flow cytometry.
Retroviral infection of CD4 T cells
Naive CD4 T cells were activated with plate-bound anti-CD3 (4 μg/mL) plus anti-CD28 (4 μg/mL) in 24-well plates for 36 hours and then infected with pCLXSN(GFP) or pCLXSN(GFP)-NIK retroviruses. After 36 hours of infection, infected cells were enriched by cell sorting based on GFP expression and subjected to Th17 differentiation assays.
ICS
T cells isolated from spleen and central nervous system (CNS; brain and spinal cord) of immunized mice or from in vitro cultures were stimulated for 4 hours with PMA (50 ng/mL) and Ionomycin (500 ng/mL) in the presence of monensin (10 μg/mL). After the stimulation period, cells were fixed in 2% paraformaldehyde and permeablized in 0.5% saponin before staining for relevant cytokines. The stained cells were analyzed by flow cytometry.
Real-time quantitative RT-PCR
Total RNA was isolated from T cells using TRI reagent (Molecular Research Center, Cincinnati, OH) and subjected to cDNA synthesis using MMLV reverse transcriptase (Invitrogen, Carlsbad, CA) and oligo (dT) primers. Real-time quantitative PCR was performed using iCycler Sequence Detection System (Bio-Rad, Hercules, CA) and iQ SYBR Green Supermix (Bio-Rad). The expression of individual genes was calculated by a standard curve method and normalized to the expression of GAPDH. The gene-specific primer sets (all for murine genes) were: IL-17A, 5′-CTCAGACTACCTCAACCGTTC-3′, and 5′-TGAGCTTCCCAGATCACAGAG-3′; IL-17F, 5′-CCCATGGGATTACAACATCACTC-3′, and 5′-CACTGGGCCTCAGCGATC-3′; RORγt, 5′-CAAGTCATCTGGGAT-CCACTAC-3′, and 5′-TGCAGGAGTAGGCCACATTACA-3′; IL-21, 5′-ATCCTGAACTTCTATCAGCTCCAC-3′, and 5′-GCATTTAGCTATGTGCTTCTGTTTC-3′; IL-22, 5′-TCCGAGGAGTCAGTGCTAAA-3′, and 5′-AGAACGTCTTCCAGGGTGAA-3′; IL-23R, 5′-GCCAAGAAGAC CATTCCCGA-3′, and 5′-TCAGTGCTACAATCTTCTTCAGAGGACA-3′; and GAPDH, 5′-CTC ATG ACC ACA GTC CAT GCC ATC-3′, and 5′-CTG CTT CAC CAC CTT CTT GAT GTC-3′.
Adoptive transfer of T and B cells
B220+ B cells and CD90.2+ T cells were isolated from the splenocytes of NIK+/+ and NIK−/− mice using magnetic beads (Miltenyi Biotec). The purity of the isolated cells was more than 95%, as determined by flow cytometry. NIK+/+ B cells (5 × 106) were mixed with either NIK+/+ or NIK−/− T cells (10 × 106) and then injected into the tail vein of nonirradiated Rag2−/− mice. Recipient mice were subjected to EAE studies 16 hours after the adoptive transfer.
Induction and evaluation of EAE
The encephalitogenic peptide (residues 35-55, Met-Glu-Val-Gly-Trp-Tyr-Arg-Ser-Pro-Phe-Ser-Arg-Val-Val-His-Leu-Tyr-Arg-Asn-Gly-Lys) of myelin oligodendrocyte glycoprotein (MOG) was purchased from Genemed Synthesis Inc. (San Francisco, CA, 95% purity). To induce acute EAE, mice were injected s.c. (in the back region) with 300 μg of the MOG35-55 peptide in CFA containing 5 mg/mL heat-killed Mycobacterium tuberculosis (H37Ra strain; BD Diagnostics, Franklin Lakes, NJ). On the day of immunization and 48 hours later, the mice were also injected i.v. with Pertussis toxin (200 ng/mouse; List Biological Laboratories, Campbell, CA) in PBS. This immunization procedure was repeated once on day 8 after the initial immunization. Mice were examined daily for EAE disease symptoms, which were scored using a standard method: 0, no clinical signs; 1, limp tail; 2, paraparesis (weakness, incomplete paralysis of 1 or 2 hind limbs); 3, paraplegia (complete paralysis of 2 hind limbs); 4, paraplegia with fore limb weakness or paralysis; and 5, moribund state or death.
Analysis of in vivo T-cell differentiation and CNS infiltration
At the indicated times after MOG35-55 immunization, the mice were killed for splenocyte preparation and CNS infiltration analysis. CD90.2+ T cells were isolated from the splenocytes using magnetic beads (Miltenyi Biotec) and subjected to ICS as described above. For the preparation of CNS lymphocytes, brains and spinal cords were excised and dissociated for1 hour at 37°C by digestion with collagenase IV (0.5 mg/mL; Invitrogen) and DNase I (10 μg /mL; Roche, Indianapolis, IN) in RPMI medium. Dispersed cells were passed through a 40-μm nylon mesh and collected by centrifugation. The cells were then resuspended in RPMI medium, layered onto a Percoll density gradient (Biochrom, Berlin, Germany), and centrifuged for 30 minutes (625 g, 22°C). CNS lymphocytes were isolated by collection of the interphase fraction between 30% and 70% Percoll. After intensive washing in Hanks balanced-salt solution, cells were analyzed by flow cytometry.
Statistical analysis
Two-tailed unpaired t test statistical analysis was performed using the Prism software. P values less than .05 and less than .01 mean significant and very significant, respectively.
Results
NIK deficiency in mice attenuates in vivo generation of Th17 cells and induction of EAE
To assess the in vivo role of NIK in regulating T-cell differentiation and immune responses, we analyzed the effect of NIK deficiency on the induction of a T-cell-mediated autoimmune disease, EAE.5,9 Because the NIK knockout mice are from a genetic background (129Sv) known to be less susceptible to EAE, we used a protocol that involved 2 rounds of immunization with MOG35-55 and Pertussis toxin. After 2 weeks of the initial immunization, the NIK+/+ mice started to display EAE disease symptoms (Figure 1A, and Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). By day 25, all of these control mice developed EAE (Figure S1). The average disease score initially peaked on day 17 and lasted until at least day 35 (Figure 1A). In contrast to the NIK+/+ mice, none of the NIK−/− mice had EAE symptoms at least until day 35 (Figures 1A, S1).
Figure 1.

NIK knockout mice are resistant to EAE induction and defective in Th17 differentiation. (A) Age- and sex-matched NIK−/− and NIK+/+ mice (7 +/+ and 5 −/−) were immunized with MOG35-55 peptide on day 0 and day 8 and monitored daily for EAE disease symptoms. Data are representative of 3 experiments. (B,C) MOG35-55-immunized mice were killed on day 14. Splenic T cells were stimulated for 4 hours with PMA plus ionomycin and subjected to ICS and flow cytometry to determine the frequency of Th1 and Th17 cells among CD4+CD44+ cells based on their production of IFN-γ and IL-17A, respectively. Data are presented as a representative flow cytometry graph (B) and mean value of multiple mice (each circle or square in this and all the subsequent figures represents an individual mouse). (D,E) MOG35-55-immunized mice (7 NIK+/+ and 5 NIK−/−) were killed on day 35. The frequency of Th1 and Th17 cells in the spleen was analyzed as described in panels B and C. (F) Splenic T cells from day 35 MOG35-55-immunized mice were stimulated for 4 hours with PMA plus ionomycin. RNA was prepared from the cells and subjected to real-time PCR assays to detect the relative expression of the indicated genes (fold relative to one of the NIK+/+ samples). Data are representative of 2 experiments and are presented as mean value of multiple (7 NIK+/+ and 5 NIK−/−) mice.
To examine MOG35-55-induced T-cell differentiation in vivo, we analyzed the different subsets of CD4 effector T cells during the early and late phases of the EAE disease. We performed ICS to detect the in vivo frequency of Th17 and Th1 cells in the spleen of NIK+/+ and NIK−/− mice. During the early phase of EAE (day 14), a significant level of IL-17A-producing Th17 cells (around 1%) and IFNγ-producing Th1 cells (around 1%) was detected in the spleen of NIK+/+ mice (Figure 1B,C). The production of Th1 cells was not inhibited, but rather enhanced, in the NIK−/− mice (Figure 1B,C). In contrast, these mutant animals displayed a severe reduction in the population of Th17 cells (Figure 1B,C). Compared with the early phase of EAE, the late phase (day 35) produced a higher level of Th17 cells in the NIK+/+ spleen (Figure 1D,E). Importantly, the frequency of Th17 cells in the NIK−/− spleen was still low at this late time point (Figure 1D,E).
In addition to IL-17A, Th17 cells produce several other cytokines, including the IL-17A homologue, IL-17F, and IL-22. To further confirm the critical role of NIK in Th17 cell production in vivo, we analyzed the expression of these Th17-expressing genes by real-time PCR using splenic T cells derived from MOG-immunized (day 35) NIK+/+ and NIK−/− mice. Consistent with the ICS results, the expression of IL-17A, but not of IFN-γ, was severely attenuated in the NIK−/− T cells (Figure 1F). Moreover, the NIK−/− T cells also failed to express IL-17F and IL-22 (Figure 1F). Together, these results suggest that NIK has a critical role in mediating MOG35-55-induced Th17 cell production and EAE pathogenesis.
NIK expression in T cells is required for EAE induction
NIK has an important signaling role in lymphoid stromal cells, which is required for the development of lymph nodes and Peyer patches.31 However, previous studies suggest that these lymphoid organs are not required for immune responses involved in the induction of EAE or antigen-induced arthritis.37,38 Nevertheless, it is important to determine whether the EAE-resistant phenotype of the NIK−/− mice was due to their lymphoid organ abnormalities or the T-cell intrinsic defect in Th17 differentiation. To address this question, we performed lymphocyte transfer studies using Rag2−/− mice as recipients. Because the Rag2−/− mice lack lymphocytes, they are completely resistant to EAE induction. We transferred NIK+/+ or NIK−/− T cells, together with NIK+/+ B cells, into the Rag2−/− recipients. As expected, transfer of NIK+/+ T cells into Rag2−/− mice rendered these mice susceptible to EAE induction (Figure 2A). Similar to the wild-type mice, the Rag2−/− mice that had been transferred with NIK+/+ T cells began to develop EAE symptoms around 2 weeks of immunization and all of them became sick on day 22 (Figure 2A). In contrast, none of the NIK−/− T-cell recipients developed EAE (Figure 2A). Furthermore, the disease score of the NIK+/+ T-cell recipients was steadily increasing until day 25, but no disease symptom was observed in the NIK−/− T-cell recipients throughout of the 26-day experimental period (Figure 2B). Thus, NIK expression in T cells is critical for the induction of EAE.
Figure 2.

NIK-deficient T cells are defective in mediating EAE induction when adoptively transferred to Rag2−/− mice. (A,B) T cells isolated from NIK+/+ and NIK−/− mice were mixed with B cells derived from NIK+/+ mice and adoptively transferred into Rag2−/− mice. One day after the cell transfer, recipient mice were immunized with MOG35-55 peptide as described in Figure 1A. EAE incidence (A) and disease scores (B) were monitored daily. Mice transferred with NIK+/+ and NIK−/− donor T cells are indicated as filled circles and squares, respectively. (C) Rag2−/− mice were transferred with the indicated donor T cells together with wild-type B cells. MOG35-55-immunized mice were killed on day 14 or day 24. Splenic T cells were stimulated for 4 hours with PMA plus ionomycin and subjected to ICS and flow cytometry to determine the frequency of Th17 cells among CD4+CD44+ cells based on IL-17A production. Data are mean value of the indicated number of mice (each circle or square represents 1 individual mouse). (D) Rag2−/− mice were transferred with the indicated donor T cells together with wild-type B cells. After 14 or 24 days of MOG immunization, flow cytometry was performed to determine the frequency of CD4+CD45+ cells infiltrating to the brain and spinal cord. Data are mean value of the indicated number of mice. (E) MOG-immunized Rag2−/− recipients were killed on day 24 for isolating RNA from total CNS tissue. Real-time PCR was performed to determine the relative expression of the indicated genes as described in Figure 1F.
To examine whether NIK has a T-cell intrinsic role in the differentiation of Th17 cells in vivo, we analyzed the frequency of Th17 cells in the spleen of immunized mice at 2 different time points. The NIK−/− T-cell recipients produced a significantly lower level of Th17 cells compared with the NIK+/+ T-cell recipients (Figure 2C). This defect was particularly striking on day 24, thus suggesting the requirement of NIK in Th17 cell production in vivo.
We next analyzed the infiltration of T cells into the CNS during the initial and a later stage of EAE. Consistent with their development of EAE, the NIK+/+ T-cell recipients displayed infiltration of CD4+ T cells into the CNS on both day 14 and day 24 (Figure 2D). On the other hand, a significantly lower level of CNS-infiltrating T cells were detected in the NIK−/− T-cell recipients, particularly during the early stage (day 14; Figure 2D). To assess the existence of Th17 and Th1 effector T cells within the CNS of the immunized Rag2−/− recipient mice, we performed real-time PCR assays to detect the expression of IL-17A and IFN-γ genes in CNS tissue. As expected, a high level of IL-17A and IFN-γ expression was detected within the CNS of NIK+/+ T-cell recipients (Figure 2E). In sharp contrast, the CNS of the NIK−/− T-cell recipients had a considerably lower level of IL-17A expression (Figure 2E). Interestingly, these mutant recipients also expressed a substantially lower level of the Th1 cytokine, IFN-γ, in the CNS (Figure 2E). These results suggest that in addition to regulating Th17 cell differentiation, NIK may play a role in the CNS infiltration or activation of Th1 cells. Because both Th17 and Th1 cells contribute to the pathogenesis of EAE,39,40 these findings also explain the drastic EAE-resistance phenotype of the NIK−/− T-cell recipient mice.
NIK is required for Th17 cell differentiation in vitro
To further examine the T-cell intrinsic role of NIK in regulating Th17 cell differentiation, we used an in vitro T-cell differentiation model. The NIK+/+ and NIK−/− naive CD4 T cells were stimulated by anti-CD3/anti-CD28 under Th1, Th2, Th17, and iTreg polarizing conditions. Loss of NIK did not affect the differentiation of naive CD4 T cells to Th1 or Th2 cells (Figure 3A). Previous studies suggest that NIK mutant (aly) and NIK−/− mice have reduced numbers of nTregs, probably due to the NF-κB signaling defect in thymic stromal cells.41,42 Interestingly, the expression of NIK in naive T cells appeared to be less important for the differentiation of iTregs, as the NIK−/− T cells only displayed a moderate reduction in the iTreg induction (Figure 3B). Overall, these results suggest that NIK is largely dispensable for naive T-cell differentiation to Th1, Th2, and iTreg lineages. In contrast, a parallel experiment revealed that the production of Th17 cells was severely attenuated in the NIK−/− T cells (Figure 3B bottom panels). These findings suggest that NIK has a specific role in regulating naive CD4 T-cell differentiation to the Th17 lineage.
Figure 3.

NIK is required for differentiation of Th17 cells but is dispensable for the differentiation of other CD4 effector cells. (A) Naive CD4 T cells isolated from NIK+/+ and NIK−/− mice were stimulated for 72 hours with plate-bound anti-CD3 and anti-CD28 (1 μg/mL) under Th0, Th1, or Th2 conditions followed by flow cytometry to measure the frequency of IFN-γ producing Th1 cells and IL-4 producing Th2 cells. (B) Naive CD4 T cells were stimulated for 72 hours with plate-bound anti-CD3 and anti-CD28 (1 μg/mL) under Th0, Th17, or iTreg conditions and then subjected to flow cytometry to determine the frequency of Foxp3 producing iTregs and IL-17A producing Th17 cells. Data in both panels A and B are representative of 3 independent experiments.
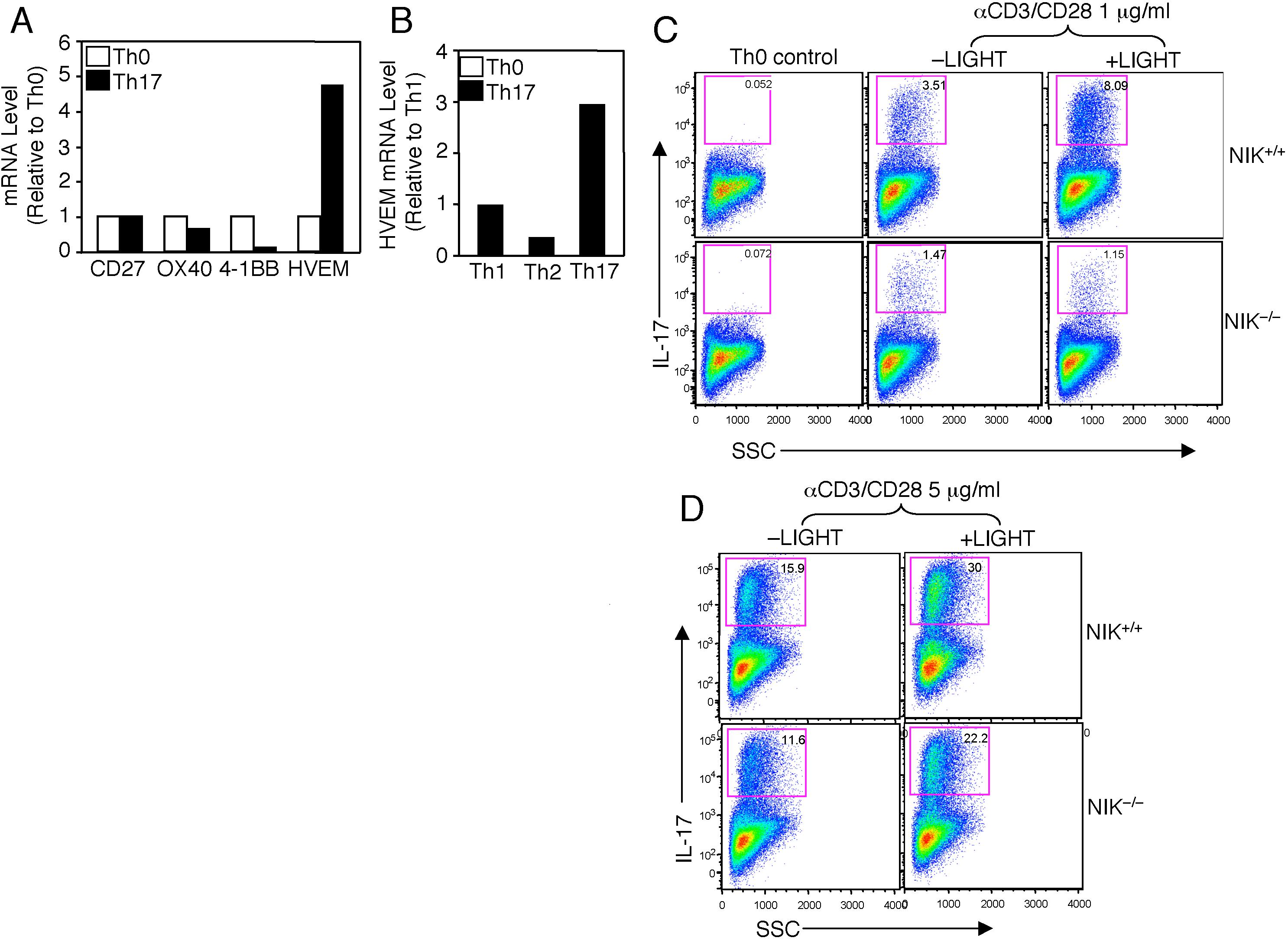
Because NIK can be activated by both the TCR34 and certain TNFRs involved in T-cell costimulation,31,43 we examined whether specific TNFR members are involved in Th17 cell differentiation and whether NIK is required for such costimulation. Stimulation of the wild-type T cells under Th17 conditions either did not affect or even down-regulated the expression of several TNFRs (Figure S2A). Interestingly, however, one TNFR member, herpes virus entry mediator (HVEM), was strongly induced when naive T cells were stimulated under Th17 conditions compared with the Th0 or Th1 conditions (Figure S2A,B). Furthermore, the HVEM ligand, LIGHT, greatly promoted Th17 cell differentiation, and this costimulatory effect was largely blocked in the NIK−/− T cells (Figure S2C). Interestingly, however, NIK became less important for Th17 induction when the T cells were stimulated with a high dose of TCR stimuli either in the presence or absence of LIGHT (Figure S2D). Thus, it appears that NIK regulates the strength of the TCR and costimulatory signals involved in Th17 differentiation.
NIK is dispensable for T-cell proliferation but is required for the induction of genes involved in Th17 commitment and effector functions
To further characterize the molecular mechanism by which NIK regulates Th17 cell differentiation, we examined the effect of NIK deficiency on the induction of T-cell proliferation and expression of genes involved in Th17 lineage commitment and effector functions. When stimulated under Th0 conditions, NIK+/+ and NIK−/− naive CD4 T cells displayed comparable proliferation efficiency (Figure 4A). Similarly, NIK+/+ and NIK−/− T cells did not show significant difference in proliferation upon stimulation under Th17 conditions (Figure 4A). Thus, the NIK deficiency has no significant effect on the proliferation potential of naive CD4 T cells, a finding that is consistent with the dispensable role of NIK in the induction of Th1 and Th2 cells (Figure 3A).
Figure 4.

NIK is dispensable for naive CD4 T-cell proliferation but is required for the induction of Th17-specific genes. (A) Naive CD4 T cells from NIK+/+ and NIK−/− mice were labeled with CFSE and stimulated for 72 hours with plate-bound anti-CD3 and anti-CD28 under Th0 or Th17 conditions. Cell proliferation was measured by flow cytometry and determined based on the dilution of CFSE during cell division. The intensity of CFSE is reduced to one-half after each cell division (indicated by numbers). Data are representative of 3 independent experiments. (B) Naive NIK+/+ and NIK−/− CD4 T cells were stimulated with plate-bound anti-CD3 and anti-CD28 (1 μg/mL) under Th0 or Th17 conditions for 18 hours. Real-time quantitative RT-PCR assays were performed to determine the relative expression of the indicated genes (fold to the NIK+/+ Th0 sample). Data are representative of 3 independent experiments. (C) Naive NIK+/+ and NIK−/− CD4 T cells were either directly subjected to Th17 differentiation assays as described in panel B (left) or preinfected with pCLXSN(GFP) or pCLXSN(GFP)-NIK retroviral vectors (right). The infected cells were enriched by cell sorting based on GFP expression and then subjected to differentiation assays. Relative expression of IL-17A was determined by real-time PCR and presented as fold relative to NIK+/+ Th0 sample (left panel) or to the Th0 GFP sample (right panel).
We next examined the role of NIK in regulating the gene expression program involved in the induction of Th17 differentiation. As previously demonstrated,7 stimulation of wild-type naive CD4 T cells under Th17 conditions resulted in the induction of genes encoding IL-21, IL-23R, and the Th17 lineage specific transcription factor RORγt, as well as the Th17 effector cytokines IL-17A, IL-17F, and IL-22 (Figure 4B). Consistent with the ICS results (Figure 3B), the induction of IL-17A gene expression was largely blocked in the NIK-deficient T cells (Figure 4B). Similarly, the mutant T cells had a defect in the induction of IL-17F and IL-22 genes. Interestingly, the NIK deficiency also attenuated the induction of the major Th17-regulatory genes, IL-21, IL-23R, and RORγt (Figure 4B). Thus, NIK is not simply required for IL-17A gene induction but is rather involved in the signal transduction that mediates the Th17-specific gene expression program.
To further confirm that the loss of NIK was responsible for the defect of the NIK−/− T cells in Th17 differentiation, we reconstituted these mutant T cells in vitro with NIK by retroviral transduction. To assure efficient selection of the transduced cells, we used a retroviral vector expressing green fluorescence protein (GFP) and sorted the GFP-positive cells for the Th17 differentiation experiment. As expected, parallel experiment using uninfected cells revealed a severe defect of NIK−/− T cells in Th17 differentiation, as indicated by the defect in IL-17A induction (Figure 4C left panel). Furthermore, reconstitution of the NIK−/− T cells with NIK, but not GFP, rescued the induction of IL-17A (Figure 4C right panel). These results further emphasize the T-cell intrinsic role of NIK in the regulation of Th17 cell differentiation.
NIK mediates the synergistic induction of STAT3 tyrosine phosphorylation by TCR and IL-6R signals
In B cells, NIK is known to induce the processing of NF-κB2 precursor protein p100,44 a central step in noncanonical NF-κB activation.31 However, stimulation of T cells through the TCR signaling pathway does not lead to appreciable p100 processing.45 Because NIK activation by the TCR signal occurs rapidly,34 we reasoned that NIK might be involved in the regulation of the early signaling events involved in Th17 lineage commitment. In this regard, the transcription factor STAT3 is a key molecule that initiates the Th17 commitment program.7 STAT3 activation involves its phoshorylation at tyrosine 705, which triggers its dimerization and nuclear translocation.26 STAT3 phosphorylation can be stimulated by IL-6,7 although early studies also suggested the induction of this event by the TCR signal.46 To examine the role of NIK in regulating STAT3 phosphorylation, we used a flow cytometry approach that allows detection of STAT3 phosphorylation on a single-cell basis. Stimulation of naive CD4 T cells with IL-6 for 25 minutes led to the induction of STAT3 tyrosine phosphorylation (Figure 5, top left panel). This signaling event appeared to be independent of NIK, as it similarly occurred in NIK-deficient T cells. Furthermore, the IL-6 stimulated STAT3 phosphorylation was transient, which was almost reversed to the background level after 50 minutes (Figure 5 top right panel). The TCR signal also induced STAT3 tyrosine phosphorylation, albeit with a lower magnitude and more transient kinetics (Figure 5 middle panels). Remarkably, however, the TCR signal potently synergized with IL-6, leading to much stronger and prolonged induction of STAT3 tyrosine phosphorylation (Figure 5 bottom panels). Moreover, the synergistic induction of STAT3 phosphorylation by the TCR and IL-6 signals was substantially attenuated in the NIK−/− T cells. These results suggest that optimal activation of STAT3 in naive CD4 T cells requires both the IL-6R and TCR signals and that NIK plays a critical role in mediating this functional interplay.
Figure 5.

NIK regulates the synergistic induction of STAT3 tyrosine phosphorylation by the TCR and IL-6R signals. NIK+/+ and NIK−/− naive CD4 T cells were stimulated for the indicated times with IL-6 (20 ng/mL), anti-CD3/CD28 (2 μg/mL), or IL-6 plus anti-CD3/CD28. Site-specific tyrosine phosphorylation of STAT3 (Tyrosine 705) was analyzed by flow cytometry. Data are representative of 3 experiments.
Discussion
The results presented in this paper demonstrate a novel and unexpected function of NIK in the regulation of T-cell mediated immune functions. Using T-cell transfer and in vitro differentiation approaches, we have shown that NIK has a T-cell intrinsic function in the regulation of Th17 cell differentiation. Consistent with these findings, NIK deficiency renders mice resistant to the induction of EAE, an autoimmune disease that involves Th17 cells. Like the role of NIK in T-cell differentiation, the function of NIK in mediating EAE is T-cell intrinsic, as revealed by T-cell transfer into Rag2−/− mice. Our findings thus establish NIK as a critical signaling molecule that regulates Th17 differentiation and EAE induction.
NIK is known as a central player in the noncanonical NF-κB signaling pathway, which involves the processing of NF-κB2 precursor protein p100 to p52 and nuclear translocation of the p52/RelB heterodimer.31 This NIK-specific pathway is stimulated primarily in B cells and lymphoid stromal cells by a subset of TNFR family members.31 On the other hand, activation of T cells by T-cell mitogens does not lead to appreciable processing of p100.45 We also failed to detect significant p100 processing in CD4 T cells stimulated with anti-CD3/CD28 under Th17 conditions (data not shown). Consistently, NIK deficiency does not lead to a global defect in naive CD4 T-cell activation or differentiation. Both the proliferation and Th1/Th2 differentiation are competent in the NIK−/− T cells. However, the fact that NIK is activated by the TCR signal34 suggests a signaling role for this kinase in T cells. Notably, NIK activation by the TCR signal occurs rapidly,34 which is in sharp contrast to the slow induction of the noncanonical NF-κB signaling by TNFRs.31 These previous findings, together with our observation that NIK is required for the induction of Th17-regulatory genes, suggest the involvement of NIK in early signaling events of the Th17 commitment program. Indeed, our current study demonstrates that NIK is required for the synergistic activation of STAT3 by the TCR and IL-6R signals. In naive T cells, the IL-6 stimulated STAT3 phosphorylation occurs weakly and transiently, but this signaling event is greatly potentiated and prolonged by TCR ligation. Moreover, the NIK deficiency severely attenuated the synergistic activation of STAT3 by the TCR and IL-6R signals. Given the critical role of STAT3 in the induction of Th17 commitment, the current finding suggests that regulation of STAT3 activation is an important mechanism by which NIK participates in the induction of Th17 differentiation. On the other hand, our study does not exclude the involvement of p100 processing or degradation in the Th17 induction. To address this possibility, mice expressing a processing-defective p100 would be important.
A recent study suggests that NIK is involved in LIGHT-stimulated STAT3 phosphorylation in a tumor cell line.34 Interestingly, our current study suggests that LIGHT has an important costimulatory role in the induction of Th17 cell differentiation. Although LIGHT stimulates both the lymphotoxin beta receptor and HVEM, only the latter is expressed in T cells.47 We have shown that the expression of HVEM is greatly promoted by the Th17-polarizing conditions and that, accordingly, LIGHT promotes the induction of Th17 cells in vitro. Our findings are consistent with the important function of the LIGHT/HVEM signaling pair in mediating inflammatory responses in vivo.48,49 A recent study demonstrates that HVEM expression in T cells is partially required for T-cell mediated induction of colitis, although HVEM has an opposite function when it is expressed in non-T cells.49 It is currently unclear whether T-cell-specific knockout of HVEM affects Th17 development in vivo. We have further shown that NIK is required for the induction of Th17 cell differentiation by the TCR and LIGHT signals. This function of NIK is particularly prominent when T cells are stimulated with low doses of anti-CD3/CD28 but becomes less striking in the presence of high doses of the TCR stimuli. It is important to note, though, that the role of NIK in Th17 cell differentiation is not limited to LIGHT costimualtion, as loss of NIK also affects the production of Th17 cells induced in the absence of LIGHT. It is thus unclear whether NIK mediates LIGHT-stimulated signaling in T cells or promotes the T-cell costimulation by acting on the TCR signal. On the other hand, our data clearly demonstrate a role for NIK in regulating the TCR signal, particularly in the activation of STAT3.
The effect of NIK deficiency on EAE induction is striking. Both regular NIK−/− mice and NIK−/− T cell-transferred Rag2−/− mice are completely resistant to EAE induction by MOG immunization. This phenotype cannot be solely attributed to the deficiency in IL-17 production, because IL-17A knockout mice show reduced severity, but not blocked incidence, of EAE disease9 and IL-17F-deficient mice only have a minor phenotype in EAE induction.44 However, because Th17 cells produce additional cytokines and possibly other factors, it is likely that the combinational function of the Th17-derived inflammatory factors is more significant than any of the single cytokine in mediating autoimmunity. Indeed, mice lacking the transcription factors RORγt and RORα, in which Th17 cell development is impaired, are completely resistant to EAE induction.50 Importantly, our data suggest that NIK is critical for Th17 cell development, rather than simply controls the expression of IL-17A and IL-17F. This finding at least partially explains the critical role of NIK in mediating EAE induction. However, it is also possible that NIK regulates additional functions of T cells involved in the pathogenesis of EAE. In this regard, we have shown that NIK deficiency in T cells reduces the expression of the Th1 cytokine, IFN-γ, during the effector phase of EAE. Because NIK is dispensable for Th1 cell production both in vivo (in spleen) and in vitro, this finding indicates that NIK may regulate the CNS migration or activation of Th1 cells. Notwithstanding, our current study clearly demonstrates a role for NIK in regulating the signaling program mediating Th17 cell differentiation, which may contribute to the resistance of NIK−/− mice to EAE induction.
Acknowledgments
We thank Amgen for providing the NIK knockout mice.
This study was supported by grants from the National Institutes of Health (NIH, Bethesda, MD; AI064639, GM084459, and AI057555).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: W.J. and X.-F.Z. performed research, analyzed data, and prepared figures; J.Y. contributed to experimental work; X.C. provided technical assistance; and S.-C.S. supervised the overall research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Shao-Cong Sun, Department of Immunology, The University of Texas M. D. Anderson Cancer Center, 7455 Fannin St, Box 902, Houston, TX 77030; e-mail: ssun@mdanderson.org.
References
- 1.Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 3.Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 2000;14:1693–1711. [PubMed] [Google Scholar]
- 4.Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 5.Langrish CL, Chen Y, Blumenschein WM, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–688. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 8.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 9.Komiyama Y, Nakae S, Matsuki T, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 10.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 11.Röhn TA, Jennings GT, Hernandez M, et al. Vaccination against IL-17 suppresses autoimmune arthritis and encephalomyelitis. Eur J Immunol. 2006;36:2857–2867. doi: 10.1002/eji.200636658. [DOI] [PubMed] [Google Scholar]
- 12.Toh ML, Miossec P. The role of T cells in rheumatoid arthritis: new subsets and new targets. Curr Opin Rheumatol. 2007;19:284–288. doi: 10.1097/BOR.0b013e32805e87e0. [DOI] [PubMed] [Google Scholar]
- 13.Dong C. Diversification of T-helper-cell lineages: finding the family root of IL-17-producing cells. Nat Rev Immunol. 2006;6:329–333. doi: 10.1038/nri1807. [DOI] [PubMed] [Google Scholar]
- 14.Korn T, Oukka M, Bettelli E. Th17 cells: Effector T cells with inflammatory properties. Semin Immunol. 2007;19:362–371. doi: 10.1016/j.smim.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 16.Mangan PR, Harrington LE, O'Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 17.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Korn T, Bettelli E, Gao W, et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nurieva R, Yang XO, Martinez G, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 20.Zhou L, Ivanov I, Spolski R, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 21.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 22.Pappu BP, Borodovsky A, Zheng TS, et al. TL1A-DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J Exp Med. 2008;205:1049–1062. doi: 10.1084/jem.20071364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meylan F, Davidson TS, Kahle E, et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity. 2008;29:79–89. doi: 10.1016/j.immuni.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watts TH. Tnf/Tnfr family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 25.Dong J, Zhong H, Hayden MS, Ghosh G. Repression of gene expression by unphosphorylated NF-κB p65 through epigenetic mechanisms. Genes Dev. 2008;22:1159–1173. doi: 10.1101/gad.1657408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shuai K. The STAT family of proteins in cytokine signaling. Prog Biophys Mol Biol. 1999;71:405–422. doi: 10.1016/s0079-6107(98)00051-0. [DOI] [PubMed] [Google Scholar]
- 27.Ivanov I, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 28.Yang XO, Panopoulos AD, Nurieva R, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 29.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 30.Symons A, Beinke S, Ley SC. MAP kinase kinase kinases and innate immunity. Trends Immunol. 2006;27:40–48. doi: 10.1016/j.it.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 31.Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J. 2004;382:393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foehr ED, Bohuslav J, Chen LF, et al. The NF-kappa B-inducing kinase induces PC12 cell differentiation and prevents apoptosis. J Biol Chem. 2000;275:34021–4. doi: 10.1074/jbc.C000507200. [DOI] [PubMed] [Google Scholar]
- 33.Nadiminty N, Chun JY, Hu Y, Dutt S, Lin X, Gao AC. LIGHT, a member of the TNF superfamily, activates Stat3 mediated by NIK pathway. Biochem Biophys Res Commun. 2007;359:379–384. doi: 10.1016/j.bbrc.2007.05.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Y, Sedwick CE, Hu J, Altman A. Role for protein kinase Ctheta (PKCtheta) in TCR/CD28-mediated signaling through the canonical but not the non-canonical pathway for NF-kappaB activation. J Biol Chem. 2005;280:1217–1223. doi: 10.1074/jbc.M409492200. [DOI] [PubMed] [Google Scholar]
- 35.Yin L, Wu L, Wesche H, et al. Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001;291:2162–2165. doi: 10.1126/science.1058453. [DOI] [PubMed] [Google Scholar]
- 36.Naviaux RN, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free high-titer, recombinant retroviruses. J Virol. 1996;70:5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suen WE, Bergman CM, Hjelmström P, Ruddle NH. A critical role for lymphotoxin in experimental allergic encephalomyelitis. J Exp Med. 1997;186:1233–1240. doi: 10.1084/jem.186.8.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aya K, Alhawagri M, Hagen-Stapleton A, Kitaura H, Kanagawa O, Novack DV. NF-(kappa)B-inducing kinase controls lymphocyte and osteoclast activities in inflammatory arthritis. J Clin Invest. 2005;115:1848–1854. doi: 10.1172/JCI23763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med. 2008;205:1535–1541. doi: 10.1084/jem.20080159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Connor RA, Prendergast CT, Sabatos CA, et al. Cutting edge: Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–3754. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kajiura F, Sun S, Nomura T, et al. NF-kappa B-inducing kinase establishes self-tolerance in a thymic stroma-dependent manner. J Immunol. 2004;172:2067–2075. doi: 10.4049/jimmunol.172.4.2067. [DOI] [PubMed] [Google Scholar]
- 42.Lu LF, Gondek DC, Scott ZA, Noelle RJ. NF kappa B-inducing kinase deficiency results in the development of a subset of regulatory T cells, which shows a hyperproliferative activity upon glucocorticoid-induced TNF receptor family-related gene stimulation. J Immunol. 2005;175:1651–1657. doi: 10.4049/jimmunol.175.3.1651. [DOI] [PubMed] [Google Scholar]
- 43.Hauer J, Püschner S, Ramakrishnan P, et al. TNF receptor (TNFR)-associated factor (TRAF) 3 serves as an inhibitor of TRAF2/5-mediated activation of the noncanonical NF-kappaB pathway by TRAF-binding TNFRs. Proc Natl Acad Sci USA. 2005;102:2874–2879. doi: 10.1073/pnas.0500187102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 45.Xiao G, Cvijic ME, Fong A, et al. Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: evidence for the involvement of IKKalpha. EMBO J. 2001;20:6805–6815. doi: 10.1093/emboj/20.23.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerwien J, Nielsen M, Labuda T, et al. Cutting edge: TCR stimulation by antibody and bacterial superantigen induces Stat3 activation in human T cells. J Immunol. 1999;163:1742–1745. [PubMed] [Google Scholar]
- 47.Browning JL. Inhibition of the lymphotoxin pathway as a therapy for autoimmune disease. Immunol Rev. 2008;223:202–220. doi: 10.1111/j.1600-065X.2008.00633.x. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Anders RA, Wang Y, et al. The critical role of LIGHT in promoting intestinal inflammation and Crohn's disease. J Immunol. 2005;174:8173–8182. doi: 10.4049/jimmunol.174.12.8173. [DOI] [PubMed] [Google Scholar]
- 49.Steinberg MW, Turovskaya O, Shaikh RB, et al. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J Exp Med. 2008;205:1463–1476. doi: 10.1084/jem.20071160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang XO, Pappu BP, Nurieva R, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}