

Abstract
Short-chain fatty acids (SCFAs; butyrate and propionate) up-regulate embryonic/fetal globin gene expression through unclear mechanisms. In a murine model of definitive erythropoiesis, SCFAs increased embryonic β-type globin gene expression in primary erythroid fetal liver cells (eFLCs) after 72 hours in culture, from 1.7% (± 1.2%) of total β-globin gene expression at day 0 to 4.9% (± 2.2%) in propionate and 5.4% (± 3.4%) in butyrate; this effect was greater in butyrate plus insulin/erythropoietin (BIE), at 19.5% (± 8.3%) compared with 0.1% (± 0.1%) in ins/EPO alone (P < .05). Fetal γ-globin gene expression was increased in human transgene-containing eFLCs, to 35.9% (± 7.0%) in BIE compared with 4.4% (± 4.2%) in ins/EPO only (P < .05). Embryonic globin gene expression was detectable in 11 of 15 single eFLCs treated with BIE, but in0 of 15 ins/EPO-only treated cells. Butyrate-treated [65.5% (± 9.9%)] and 77.5% (± 4.0%) propionate-treated eFLCs were highly differentiated in culture, compared with 21.5% (± 3.5%) in ins/EPO (P < .005). Importantly, signaling intermediaries, previously implicated in induced embryonic/fetal globin gene expression (STAT5, p42/44, and p38), were not differentially activated by SCFAs in eFLCs; but increased bulk histone (H3) acetylation was seen in SCFA-treated eFLCs. SCFAs induce embryonic globin gene expression in eFLCS, which are a useful short-term and physiologic primary cell model of embryonic/fetal globin gene induction during definitive erythropoiesis.
Introduction
The human β- and α-globin gene loci are developmentally regulated and are arrayed spatially in the order in which they are expressed developmentally, respectively, 5′-ϵ-Gγ-Aγ-β-3′ and 5′-ζ-α-α-3′. Fetal hemoglobin (HbF), an α2 γ2-tetramer, predominates from week 8 of gestation through birth, after which it is gradually superseded by adult hemoglobin (HbA), an α2 β2-tetramer. HbF is detectable, at approximately 1%, during adult life. The molecular mechanisms underlying the γ- to β-globin switch during development are not fully understood, but are of compelling interest because a persistence of HbF in adults, whether genetic or pharmacologic in origin, is ameliorative in adult β-globin gene disorders such as sickle cell anemia or β-thalassemia.1,2
Intermediaries of mammalian metabolism, such as the short-chain fatty acids (SCFAs) butyrate and propionate, are important fuels during fetal life and, when elevated, are implicated in the delayed fetal-to-adult hemoglobin switch in infants of diabetic mothers (butyrate3) and in the elevated HbF levels seen in children with inherited disorders of branched-chain amino acid metabolism (eg, propionic acidemia4 or β-ketothiolase deficiency5). Importantly, therapeutic trials of butyrate in patients with β-globin gene disorders have increased γ-globin gene expression and HbF levels.6–8
Studies of the molecular and cellular effects of SCFAs in erythroid cells have been constrained by the limited number of experimental models currently available. We were interested in finding additional models in which to study the effects of SCFAs on erythropoiesis and on erythroid gene expression. We evaluated the effects of SCFAs on murine erythropoeisis in primitive and definitive erythroid precursor cells from transgenic mice that had endogenous elevations of SCFAs, and in definitive erythroid precursor cells from wt mice and human β-globin gene locus-containing transgenic mice. These studies were undertaken with the expectation that a biologically relevant, primary, definitive erythroid precursor cell model would be an excellent place in which to study the pleiotropic effects of SCFAs on erythropoiesis.
The murine β-globin gene locus, like the human, is developmentally regulated. βH1 expression is detectable early and persists through embryonic day (E) 12 to 13, while ϵy expression is detected later and is present through E13 through 16.9,10 These embryonic β-type globin genes, and the embryonic α-like ζ-globin gene, are expressed in large, slowly enucleating, primitive erythroid (EryP) cells from the murine yolk sac. Adult β-type globin genes, βAdult, comprise the βmaj and βmin genes (from the β-diffuse haplotype found in most strains) or the βS and βT genes (from the β-single haplotype found in the C57/black 6 strain). βAdult and α are expressed primarily in small, rapidly enucleated, definitive erythroid (EryD) cells that arise from the erythroid fetal liver through late gestation and from the adult bone marrow and, in anemic animals, from the adult erythroid spleen. EryD account for 90% of circulating red cells by E16.5.11 There are no published reports of pharmacologic up regulation of endogenous embryonic globin gene expression in primary murine EryD, and embryonic hemoglobin is not detectable in whole blood from adult mice.
There is no direct counterpart to fetal hemoglobin or to the γ-globin genes themselves in murine erythropoiesis. When expressed transgenically, fetal genes in the human β-globin gene locus are typically expressed in the embryonic yolk sac (aberrantly); later, human γ-globin genes are expressed in the murine fetal liver12,13 and not in the adult murine bone marrow or erythroid spleen.
Mechanistically, SCFAs have been reported to affect several intracellular processes and regulatory molecules, including cyclic nucleotides,14–16 cell-signaling kinases, transcription factors,17–19 mediators of apoptosis,20 corepressors, and as inhibitors of histone deacetylases.21–23 SCFA-mediated activation of signaling molecules has been implicated in stress erythropoiesis and in the up-regulation of embryonic/fetal globin gene expression (summarized in Mabaera et al24). However, models of erythropoiesis in which SCFAs have been studied, including chronic myeloid leukemia (CML)–derived human K562 cells, which do not express adult globin genes, and human CD34+ precursor cells, which require extensive in vitro differentiation, have experimental limitations, including nonphysiologic regulation in the former and, in the latter, a requirement for prolonged cytokine-, especially erythropoietin (EPO)−, driven culture and an absence of transgenic models.
Levels of SCFAs (propionate) are abnormally elevated in the inherited metabolic disease propionic acidemia, which results from an absent or deficient propionyl-CoA carboxylase (PCC) enzyme. In a murine model of propionic acidemia, homozygous PCC subunit A knockout (PCCA KO) transgenic embryos are fully PCC-deficient in utero, and propionate levels are elevated in both embryos and adults.25
Here, we found that embryonic globin gene expression was modestly increased in PCCA KO offspring, but only in erythroid tissues in which these genes were already being expressed. We found that the concentrations of propionic acid achieved in the PCCA KO strain in vivo were not able to activate embryonic globin gene expression in murine EryD in vitro. However, SCFAs at higher concentrations did up-regulate embryonic globin, or human transgenic γ-globin, gene expression in murine EryD in vitro. This was a highly penetrant effect, in which embryonic globin gene expression was induced in more than two-thirds of erythroid fetal liver cells (eFLCs) from wt animals after 3 days in culture.
We then examined this primary cell model of EryD for the impact of SCFAs on globin gene expression, apoptosis, and red cell differentiation; the latter especially difficult to address in currently available models. SCFAs are well known to promote differentiation in transformed erythroid and colonic epithelial cell lines,26–29 but their effects on erythroid differentiation in primary cells had not been described. Normal erythroid differentiation in murine development parallels advancing fetal age. That is, early developmental stages (eg, E11.5 eFLCs) are enriched with early erythroid precursors, while later developmental stages (eg, E14.5 eFLCs) include a continuum of early and late erythroid cells.30 We found that SCFAs increase the proportion of late erythroid cells in cultures of primary EryD; this effect is exaggerated by the relative paucity of early erythroid precursors in SCFA-treated cultures.
We also examined bulk-histone (H3) acetylation and phosphorylation of those signaling intermediaries that had previously been associated with embryonic/fetal β-type globin gene up-regulation during definitive erythropoiesis,18,19,31,32 namely STAT5 and p38 (by SCFAs18,19) and p42/44 (by stem cell factor, SCF33). In our experiments, changes in histone acetylation, as described elsewhere,34 but not changes in p38-, p42/44-, or STAT5-phosphorylation, were specifically associated with SCFA treatment.
We describe a short-term, primary definitive erythroid cell model of embryonic/fetal globin gene activation by SCFAs that allows the investigation of underlying molecular mechanisms in a physiologic milieu.
Methods
Propionate levels
Propionate levels in peripheral blood and homogenized fetal liver were measured in a Hewlett Packard 5890 gas chromatograph (61 × 2 mm glass column with SP-1200/1% H3PO4 on 80/100 Chromosorb W AW; Supelco, Bellefonte, PA). Authentic retention times were confirmed using commercial SCFA standards (NuChek Prep, Elysian, MN).
Murine models
Mouse strains examined in this report included C57/black 6 (designated as wild type, “wt”), CD1, PCCA KO25 mice, and mice transgenic for a 150-kb human β-locus transgene35 (a kind gift of Keiji Tanimoto, Institute of Biochemistry, University of Tsukuba, Tsukuba, Japan). Developmental stages of dissected embryos were confirmed by inspection and by reference to an atlas of murine development.36 All evaluations of murine β-type globin gene expression, adult or embryonic, were made using primers and probes designed to detect transcripts from both β-diffuse and β-single haplotypes. All animal experiments were done with approval from the Albert Einstein College of Medicine Institutional Animal Care and Use Committee (IACUC).
Globin gene expression
RNA was isolated (TelTest, Friendswood, TX) from individual yolk sacs at E10.5, E12.5, and E14.5, from individual erythroid fetal livers at E12.5 and E14.5 (PCCA KO and wt), and from induced cultures of pooled eFLCs at E13.5 and E14.5. DNAse-treated RNA (5 μg) were reverse-transcribed (Oligo dT primers, Superscript II; Invitrogen, Carlsbad, CA). cDNA was quantified fluorimetrically (PicoGreen; Molecular Probes, Invitrogen), and 0.5 ng was analyzed in duplicate on an ABI 7700 instrument (Applied Biosystems, Foster City, CA). Murine globin gene–specific TAMRA-labeled probes (Pr) and sense (S) and antisense (AS) primers, mentioned below, were designed using Primer Express (Applied Biosystems).
Murine globin: ζ: 5′-CGTGTGGATCCGGTCAACTT-3′ (S), 5′-AGCGTGCGGCCATTGT-3′ (AS), and 5′-AAGCTCCTGTCCCACTGTCTGCTGGT-3′ (Pr); α-1 and α-2: 5′-GGTGCTCTCTGGGGAAGACA-3′ (S), 5′-CGGTACTTGGAGGTCAGCACGGTGC-3′ (AS), and 5′-TTCCCCACCACCAAGACCTACTTCCC-3′ (PR).
ϵy: 5′-GAGACTCCCTGTGCTCATCAGAG-3′ (S), 5′-GCCTTCCATGGCGTCTACGT-3′ (AS), and 5′-TCCTGTGTGTCCGCTATGCCTCCTC-3′ (Pr); βH1: 5′-CTCTGGGAAGGTAAGGAATGGAGGG-3′ (S), 5′-GCCAATCACAGTCTCAACTCCCAG-3′ (AS), and 5′-ACTTTCTTGCCATGGGCTCTAATCCGG-3′ (Pr); βAdult: 5′-CCATATTCCCACAGCTCCTGGGC-3′ (S), 5′-CCCCAAGGTCAGACCTGAACCC-3′ (AS), and 5′-CTCTCTTGGGAACAATTAACCATTGTTCACAG-3′ (Pr).
Human globin: ϵ: 5′-GCTGCATGTGGATCCTGAGA-3′ (S), 5′-TGCCAAAGTGAGTAGCCAGAATAA-3′ (AS), and 5′-CTTCAAGCTCCTGGGTAACGTGATGGTG-3′ (PR); G+Aγ: 5′-TGGAAGATGCTGGAGGAGAAA-3′ (S), 5′-TGCCAAAGCTGTCAAAGAACCT-3′ (AS), and 5′-CCTGGGAAGCTCCTGGTTGTCTACCC-3′ (PR); Gγ: 5′-CAGTGCCCTGTCCTCCAGAT-3′ (S), 5′-TTGCAGAATAAAGCCTATCCTTGA-3′ (AS), and 5′-AGCTCACTGCCCATGATGCAGAGCT-3′ (Pr); Aγ: 5′-CAGTGCCCTGTCCTCCAGAT-3′ (S), 5′-TTGCAGAATAAAGCCTATCCTTGA-3′ (AS), and 5′-CCACTGAGCCTCTTGCCCATGATTCA-3′ (Pr); β: 5′-CACGTGGATCCTGAGAACTTCAG-3′ (S), 5′-GGTGAATTCTTTGCCAAAGTGAT-3′ (AS), and 5′-CGTGCTGGTCTGTGTGCTGGCC-3′ (Pr).
Cycle thresholds (Cts) were acceptable only if they differed by at least 5 Cts from a “no reverse transcriptase” control, and had less than 0.3 Ct difference between duplicates. Normalized expression was 2−n, where (Ctglobin − Ct18S = n). Each individual globin gene was corrected to 18S before calculations of embryonic globin gene expression.
Ribosomal 18S primers and probes (internal control) were from a premade kit (TaqMan ribosomal RNA control reagents, no. 4308329; Applied Biosystems).
Flanking primers for single-cell polymerase chain reaction (PCR):
Murine: βH1: 5′-CACTCGAGATCATCTCCAAGC-3′ (S) and 5′-TAACCCCCAAGCCCAAGGATG-3′ (AS); α: 5′-GCTGCCTGGGGGAAGATTGG-3′ (S) and 5′-AGTCAGCACCTTCTTGCC-3′ (AS).
Human: β: 5′-TACCCTTGGACCCAGAGGTTCTTTGA-3′ (S), 5′-ATTAGCCACACCAGCCACCACTTT-3′ (AS); γA+G: 5′-TGGGTCATTTCACAGAGGAGGACA-3′ (S), and 5′-ATGCAGCTTGTCACAGTGCAGTTC-3′ (AS).
Statistical significance of differences in globin gene expression was calculated by a Student t test.
Tissue culture
Ten to 20 million E14.5 wt or transgenic eFLCs were cultured on tissue- culture plates in basal “no cytokine” media, (Iscove modified Dulbecco media [IMDM] with 10% fetal calf serum [FCS], penicillin/streptomycin [PCN/Strep], and 200 μg/mL transferrin), or basal media plus either 10 μg/mL recombinant human insulin and 4 U/mL recombinant human erythropoietin (ins/EPO), or 5 mM propionate (except where noted), pH 7.0, or 1 to 2 mM butyrate, pH 7.0, or both butyrate and ins/EPO (BIE). At 48 hours, media was changed using the same media as at time 0, except without ins/EPO. Cell counts were obtained at 72 hours, with a subset tested at 48 hours before media change. RNA was harvested at times indicated.
Apoptosis
Apoptosis in cultured eFLCs from wt, human transgenic, and CD1 strains was analyzed as reflected by nicked DNA. Cells were incubated with fluorescently labeled dUTPs and terminal deoxynucleotidyl transferase, per protocol (ApoAlert DNA fragmentation assay; Clontech, Mountain View, CA), in which fragmented DNA in apoptotic cells incorporates labeled-dUTP. Fluorescence intensity is proportional to the degree of incorporation of labeled-dUTP and, therefore, to the extent of nicked DNA.
Single-cell real-time PCR
Pooled wt E14.5 eFLCs were cultured in ins/EPO or BIE for 72 hours, resuspended in fluorescence-activated cell sorting (FACS) buffer, and sorted (Dako Cytomation MoFlo machine; Dako, Carpinteria, CA) as single cells into 5 μL lysis buffer.
RNA from each well was reverse-transcribed and amplified for 13 cycles, using SuperScript One Step RT-PCR with Platinum Taq (Invitrogen) as per Bender et al,37 and gene-specific primers for mouse α and βH1 and human adult β and fetal γA+G. These primers were designed to amplify sequence that included those regions detected by the real-time PCR primer and probe sets described above. Five microliters (one-tenth volume) of the first amplification reaction was then analyzed by quantitative real-time PCR. Analyses of human globin gene transcription in transgenic murine eFLCS were performed on RNA from 2 single-cell wells, pooled due to otherwise insufficient γ-globin signal.
FACS
E13.5 wt eFLCs were cultured in media with SCFAs and/or ins/EPO. At 72 hours, 2.5 to 5.0 × 106 cells/sample were incubated for 30 minutes at room temperature with fluorochrome-labeled TER-119 and CD71 antibodies, or nonfluorochrome-labeled isotype controls, serially washed, and then analyzed on a FACSCalibur, as per published protocol.30 Cells in regions R1-R5, containing progressively differentiated erythroid cells, and R0, in which binding by neither antibody was detected (containing lineage-negative and nonerythroid cells), were quantitated.
Western blot analysis
Cells per condition (3.0 × 106) were serum-starved overnight, for p42/44 and STAT5, and induced for 10 minutes with SCFAs, FCS, ins/EPO, or SCFAs and ins/EPO, as indicated. Neither serum starvation nor continuous culture eliminated constitutive phospho-p38 from eFLCs. Therefore, Western blot analyses for phospho-p38 were performed on extracts harvested after routine culture for 24 and 32 hours. Cells were lysed (in 20 mM NaPO4, 300 mM NaCl,4 mM ethylenediaminetetraacetic acid [EDTA], 2% sodium deoxycholate, 2% Igepal CA-630, 2% sodium dodecyl sulfate [SDS], 100 mM NaF, 0.2% β-mercaptoethanol [BME], and Complete Protease Inhibitor [Roche, Indianapolis, IN]), on ice for 30 minutes.
Cells for histone Western blot analyses were isolated on E13.5 or cultured for 24 hours in ins/EPO, butyrate, or BIE. eFLCs were lysed, and acid-stable histones were extracted in H2SO4 and serially dialyzed in neutral buffers, per protocol (Upstate Biotechnology, Waltham, MA).
Protein and prestained standards (Bio-Rad Laboratories, Hercules, CA) were separated by electrophoresis in 10% SDS-polyacrylamide gels, transferred onto nitrocellulose membranes, and incubated overnight with primary antibody (1:1000 in 5% milk) and for 1 hour in secondary antibody (1:3800; Santa Cruz Biotechnology, Santa Cruz, CA). Western blot analysis detection reagents (SuperSignal West Pico Luminol Enhancer; Pierce, Thermo Fisher Scientific, Rockford, IL) were added before exposure to x-ray film.
Primary antibodies (from Cell Signaling, Boston, MA) were: phospho-p38 MAP kinase (no. 9211), phospho-p42/44 MAP kinase (no. 9101), p42/44 MAP kinase (no. 9102), and phospho-STAT5 (no. 9351); anti-acetylated histone H3 (no. 06-599; from Upstate Biotechnology), anti-histone H3 (no. 1791; from AbCam, Cambridge, MA) and β-actin (no. 7250; from Santa Cruz Biotechnology). Loading controls for p38 and STAT5 Westerns were β-actin. Loading controls for phosphorylated p42/44 and acetylated histone H3, after incubation in stripping buffer, per protocol (Thermo Fisher Scientific) were, respectively, nonphosphorylated p42/44 and total histone H3.
Results
Endogenous elevations in SCFAs increase embryonic globin gene expression in the murine yolk sac, but not in the fetal liver
Endogenous propionate levels were 2.5 and 3.6 μg/mL in plasma (0.033 and 0.067 mM, respectively) from 2 adult PCCA KO mice and undetectable in 2 non-PCCA KO mice. Propionate levels in E14.5-16.5 fetal livers were 2.3 and 5.0 μg/100 mg tissue in 2 PCCA KO embryos (0.07 and 0.15 mM, respectively, assuming a relative density of 1.045 g/mL38) and undetectable in 2 non-PCCA KO embryos. “Non-PCCA KO” refers to both wt and heterozygous PCCB KO mice.39 Homozygous PCCB KO mice, in which PCC subunit B has been deleted, were studied by us initially. However, they were embryonically lethal and were not examined further.
The globin gene expression profile in wt yolk sac (Figure 1A) was as described by Whitelaw9 and Kingsley,10 in which βH1 transcription and then ϵy transcription from EryP, together comprising greater than 50% of β-type globin gene expression through E12.5, were supplanted by adult βAdult expression from yolk sac–derived EryP and circulating fetal liver–derived EryD. Both ζ- and α-globin gene expression were equal in E10.5 yolk sac, and ζ diminishes as α rises thereafter.
Figure 1.

Globin gene expression in murine yolk sacs; wt and PCCA KO. (A) Proportional β-type ([(x)/(βH1 + ϵy + βAdult)] × 100), gray lines; and α-type globin gene expression ([x/(ζ + α)] × 100), black lines; adult or embryonic (solid or dashed lines, respectively), in wt embryos at times indicated. (B) Fold-globin gene expression in PCCA KO, relative to wt (set equal to 1) in E10.5 yolk sac. (C) Percent embryonic β-type globin gene expression, ([(βH1 + ϵY)/(βH1 + ϵY + βAdult)] × 100), in yolk sacs from E12.5 and E14.5 PCCA KO [dashed line, 74.3% (± 3.3%), n = 4, and 41.6% (± 12.6%), n = 6, respectively] compared with E12.5 and E14.5 wt [solid line, 77.0% (± 6.2%), n = 9, P = n.s., and 20.5% (± 3.0%), n = 6, P < .05, respectively].
Embryonic globin gene expression was modestly increased in the PCCA KO yolk sac; α, βH1, and ζ were expressed in E10.5 PCCA KO yolk sac at, respectively, 2.5-, 3.4-, and 2.4-fold relative to wt (Figure 1B). Control 18S levels did not differ between wt and PCCA KO yolk sac (not shown). It is possible that this result reflects enhanced erythropoiesis in PCCA KO embryos overall at this stage. However, no difference in erythropoiesis was seen in eFLCs at later time points (wt and PCCA KO FACS at E12.5, not shown).
The onset of adult α-type globin gene expression was modestly delayed in the E12.5 and E14.5 PCCA KO yolk sac, compared with wt (P < .05, not shown). Embryonic β-type globin gene expression was equivalent, and high, in both the E12.5 PCCA KO [Figure 1C dashed line, 74.3% (± 3.3%)] and wt E12.5 yolk sacs [Figure 1C solid line, 77.0% (± 6.2%)]. However, at E14.5 embryonic β-type globin gene expression had dropped overall, but remained higher in the PCCA KO yolk sacs [Figure 1C dashed line, sac, 41.6% (± 12.6%)] relative to wt [Figure 1C solid line, 20.5% (± 3.0%)].
Embryonic β-type globin gene expression in E12.5 and 14.5 fetal liver did not differ between PCCA KO and wt, but adult α- and β-globin gene expression were significantly depressed, at 0.82 and 0.59, respectively, relative to wt, in PCCA KO embryos (P < .05, not shown).
Adult male PCCA KO mice had normal hemoglobin levels, as did wt controls (15-16.5 g/dL, n = 2 and 3, respectively). Adult bone marrow from PCCA and wt mice all showed less than .005% embryonic β-type globin gene expression (not shown). Embryonic hemoglobin was not detectable in adult PCCA KO mice by Triton-acid-urea gel electrophoresis (n = 2 PCCA KO, n = 4 non-PCCA KO, not shown; kindness of J. Eric Russell, Department of Medicine and Pediatrics, School of Medicine, University of Pennsylvania).
SCFAs increase embryonic globin gene expression in murine fetal liver–derived EryD in vitro
We reasoned that either SCFAs could not reactivate embryonic globin gene expression in murine fetal livers, or that SCFA concentrations achievable in vivo were insufficient, therefore we tested dose-dependent SCFA-responsiveness in fetal liver–derived EryD in vitro. Globin gene expression was measured in wt and PCCA KO eFLCs at baseline and at 72 hours in culture at a range of propionate concentrations. Maximal induction of murine embryonic globin gene expression was seen at 5 mM propionate (∼ 30-fold the concentration measured in PCCA KO fetal liver and > 50-fold the concentration measured in PCCA KO adult plasma), with lesser induction at 2.5 mM, and only basal expression at less than or equal to 1.25 mM propionate (Figure 2A).
Figure 2.

Embryonic/fetal globin gene expression in SCFA-induced eFLCs. (A) Percent embryonic β-type globin gene expression ([(βH1 + ϵY)/(βH1 + ϵy + βAdult)] × 100), in a representative experiment from E13.5 to 14.5 wt (n = 2) and PCCA KO (n = 2) eFLCs (which acted identically), after 72 hours of culture, in “no cytokines,” ins/EPO, or propionate, at from 0.025 to 5 mM. The arrow indicates the maximum endogenous propionate concentrations measured in fetal livers from PCCA KO. (B) Percent embryonic β-type globin gene expression at day 0 [1.7% (± 1.2%)] and at 72 hours in ins/EPO [0.1% (± 0.1%)], propionate [4.9% (± 2.2%)], butyrate [5.4% (± 3.4%)], or BIE [19.5% (± 8.3%), n = 6, 7, 4, 5, and 3 wt litters, respectively; *P < .05, for all relative to ins/EPO or day 0]. (C) Human fetal globin gene expression ([(G+Aγ)/(G+Aγ + β)] × 100) in eFLCs containing the 100 Kb human locus, at day 0 [11.2%(± 7.9%), n = 5], or after 72 hours in butyrate [32.6% (± 5.3%)], BIE [36.0% (± 7.0%)], or ins/EPO [3.9% (± 3.6%)] as shown (n = 4; *P < .05 relative to ins/EPO or day 0). (D) Percent human fetal β-type globin gene expression in E13.5 murine eFLCs, transgenic for the human β-globin gene locus, in 2 experiments after 72 hours in ins/EPO, or propionate, at from 0.34 to 10 mM. As in panel A, the arrow indicates maximum endogenous propionate concentrations in fetal livers from PCCA KO animals.
These experiments were repeated numerous times, with a consistent robust induction by SCFAs and ins/EPO in vitro. There was some intrastrain variability in SCFA-responsiveness (wt, human transgene–containing and PCCA KO at 1 mM and CD-1 at 2 mM). However, all strains showed enhanced embryonic/fetal globin gene expression when exposed to SCFAs.
Propionate and butyrate, with or without insulin and erythropoietin, were then tested. Percent embryonic β-type globin gene expression in eFLCs was 1.7% (± 1.2%) at baseline and, after 72 hours in culture, 4.9% (± 2.2%) in propionate, 5.4% (± 3.4%) in butyrate, and 19.5% (± 8.3%) in BIE compared with 0.1% (± 0.1%) in ins/EPO (Figure 2B). SCFA-mediated induction of embryonic globin gene expression was detectable by 24 hours (not shown).
We calculated βH1 globin gene expression, and it accounted for most of the embryonic globin gene induction in SCFA-only treated eFLCs (not shown). The ratio of βH1 to ϵY, each normalized to 18S, was 2.0 (± 1.1) (n = 5) at baseline, 10.8 (± 6.8) in propionate (n = 6, P < .05), and 7.4 (± 3.9) in butyrate (n = 8, P < .05). This predisposition was lost when cells were cultured in both SCFAs and ins/EPO, in which the βH1 to ϵY ratio was 2.7 (± 1.1) (n = 4, P = not significant [n.s.] relative to baseline).
Murine ζ-globin gene expression was unchanged in culture, 0.37 (± 0.30) at baseline (n = 6 pooled litters) and 0.68 (± 0.66) after 72 hours in butyrate (n = 6 pooled litters, P = n.s.), and was not examined further.
Expression from human fetal β-type globin genes, γA and γG, was also up-regulated by SCFAs in cultured eFLCs from human β-locus transgenic mice, from 11.2% (± 7.9%) of total human β-type globin gene expression at day 0 to, after 72 hours, 32.6% (± 5.3%) in butyrate-only and 36.0% (± 7.0%) in BIE compared with 3.9% (± 3.6%) in ins/EPO-only (Figure 2C).
We calculated Gγ- and Aγ-globin gene expression, and Gγ accounted for the bulk of SCFA-induced fetal globin gene expression in eFLCs, with a 0.2 (± 0.1) Gγ:Aγ ratio at day 0 (n = 5) compared with 0.9 (± 0.3) Gγ:Aγ after 72 hours in butyrate (n = 5, P < .01, not shown). Again, this predilection was lost when ins/EPO was added to butyrate, with a Gγ:Aγ ratio at 72 hours of 0.1 (± 0.1) in BIE (n = 3, P = n.s. relative to day 0).
eFLCs from human β-globin locus transgenic animals also showed a dose-responsiveness to propionate, with maximal fetal globin gene induction at 10 mM propionate, less at 5.0 mM, and at baseline levels at less than or equal to 2.5 mM (Figure 2D). Only minimal human embryonic β-type globin gene expression, ϵ, was detected in these cultures.
SCFAs accelerate erythroid differentiation in EryD
Prominent induction of differentiation was seen in SCFA-treated eFLCs. FACS analysis of erythroid differentiation at 72 hours (Figure 3A), using coimmunostaining by CD71 and TER119 antibodies per Zhang et al30 showed that 65.5% (± 9.9%) of butyrate-only treated and 77.5% (± 4.0%) of propionate-only treated eFLCs were highly differentiated at 72 hours, in regions R4-5, compared with 21.5% (± 3.5%) of ins/EPO-treated eFLCs (Figure 3B). BIE-treatment showed an intermediate phenotype, with 57.2% (± 9.0%) highly differentiated cells. Cytospins of sorted, cultured eFLCs (Figure 3C) confirmed progressive differentiation in FACS defined regions. Cytospins confirmed the presence of prominent erythroid differentiation in SCFA-treated eFLCs, and of prominent proliferation of erythroid precursors in ins/EPO-only treated eFLCs (Figure 3C). Manual cell counts of SCFA-only, ins/EPO-only, and SCFA and ins/EPO cytospins (Figure 3D) confirmed a relative preponderance of early erythroid precursors in ins/EPO-only treated eFLCs and a relative preponderance of more differentiated erythroid precursors in SCFA-treated cells with or without ins/EPO.
Figure 3.

Erythroid differentiation of SCFA-treated eFLCs. (A) FACS analysis of fresh uncultured E13.5 eFLCs, immunostained with antibodies against CD71 and TER119, is shown. Gates for regions of progressive erythroid differentiation, as published by Zhang et al30 are R0 (nonerythroid and lineage negative cells), R1 through R3 (progenitor cells, proerythroblast, and early basophilic erythroid precursors), and R4 and R5 (chromatophilic and orthochromatophilic erythroid precursors, plus reticulocytes). (B) Wright-Giemsa–stained cytospins from FACS-sorted E13.5 wt eFLCs (40×, Nikon Eclipse TE 2000-5 with a Nikon LUD lens; Nikon, Melville, NY) that had been cultured in ins/EPO are shown for regions R3 (basophilic and orthochromatic erythroid precursors) and R4 (orthochromatophilic erythroid precursors and reticulocytes). Shown also is a graph of quantitative analyses of R0-R5 for fresh (n = 2) and cultured E13.5 eFLCs (n = 4 pooled eFLCs in butyrate, propionate, or ins/EPO; n = 2 butyrate and ins/EPO). SCFA-treated eFLCs had a relative increase in more differentiated erythroid cells, in regions R4 and 5, compared with ins/EPO-only treated cells, 77.5% (± 4.0%) in propionate-treated or 65.5% (± 9.9%) in butyrate-treated eFLCs compared with 21.5% (± 3.5%) in ins/EPO-treated eFLCs (P < .05). BIE-treated eFLCS had an intermediate differentiation pattern, at 57.2% (± 9.0%) (P = n.s. to SCFAs alone, P = .08 to ins/EPO alone). (C) Wrights-Giemsa–stained cytospins of eFLCs (40×, Nikon Eclipse TE 2000-5 with a Nikon LUD lens) that had been cultured for 72 hours in ins/EPO, butyrate, or both BIE. Early erythroid precursors (arrowhead ▷) are prominent ins/EPO-only treated cells, while mature erythroid cells (brackets) predominate in butyrate-containing media, with or without ins/EPO. (D) Median differential cell count of erythroid precursors (%) from eFLCs that had been cultured in ins/EPO, butyrate, or BIE (200 cell counts from 2 independent experiments).
Cell growth was seen in ins/EPO-treated eFLCs, but not in SCFA-treated eFLCs (not shown). At 72 hours, baseline-normalized cell counts were 1.3 (± 0.4)–fold (n = 9) in ins/EPO, 0.8 (± 0.1)–fold in propionate (n = 9), 0.8 (± 0.1)–fold in butyrate alone, and 0.7 (± 0.2)–fold in “no cytokine” cultures (n = 11, P < .05 for either SCFA or “no cytokine” compared with ins/EPO alone); ins/EPO, when added to butyrate (BIE), marginally preserved cell counts, at 0.96 (± 0.04)–fold (n = 4, P < .05 relative to ins/EPO alone and P = 0.055 relative to butyrate alone). Absolute cell growth was underestimated slightly, but proportionately, in these experiments due to modest cell loss during media change at 48 hours.
Apoptosis was not prominent in eFLCS, whatever the culture conditions. At 24 hours, rates of apoptosis were similar among ins/EPO-, butyrate, or BIE-treated cultures (Table 1).
Table 1.
Apoptosis in cultured eFLCs
| Apoptosis in eFLCs | Butyrate | Ins/EPO | BIE |
|---|---|---|---|
| Mean (± SD) | 9.9% (± 0.7%) | 10.1% (± 0.7%) | 8.9% (± 5.3%) |
| P, relative to other conditions | ns | ns | ns |
Mean apoptosis (± SD) for 3 independent analyses in eFLCs, cultured as shown.
eFLC indicates erythroid fetal liver cell; Ins, insulin; EPO, erythropoietin; BIE, butyrate plus ins/EPO; and ns, not significant.
Embryonic globin gene expression is detectable in SCFA-induced EryD in vitro
wt E14.5 eFLCs cultured in ins/EPO or in BIE were sorted as single cells onto a 96-well plate. All cells had detectable α-globin gene expression (15/15 from each culture, Table 2). The range of Cts for α cDNA was wider in ins/EPO-treated, compared with BIE-treated, eFLCs, consistent with the more heterogeneous cellular population seen by FACS and cytospin in ins/EPO cultures. SCFAs induce embryonic globin genes in the majority of erythroid cells, with BIE-treated eFLCs having detectable βH1-globin gene expression in 11 of 15 individual eFLCs, compared with 0 of 15 individual eFLCs cultured in ins/EPO.
Table 2.
Single-cell PCR for adult and embryonic globin genes
| Number of individual FLCs that contain detectable globin mRNA, and Cts for each analysis | ||||
|---|---|---|---|---|
| Culture conditions | α: P = ns | α: Ct mean (± SD), range | βH1: P < .005 | βH1: Ct mean (± SD), range |
| Ins/EPO | 15/15 | 24.4 (± 5.3), 19.5-35.6 | 0/15 | 40.0 (± 0), 40.0 |
| BIE | 15/15 | 23.4 (± 2.2), 20.5-28.4 | 11/15 | 36.3 (± 3.0), 32.4-40 |
Individual eFLCs were tested for both α and βH1 globin gene expression. α-globin gene expression was detectable in all cells; βH1 was detectable in most (11/15) eFLCs that had been treated with ins/EPO and SCFAs, but not (0/15) in eFLCs treated only with ins/EPO (P < .005). Mean cycle threshold (Ct) for α- and βH1-globin gene analysis of cDNA real-time polymerase chain reaction (PCR) are also shown.
FLC indicates fetal liver cell; Ins, insulin; EPO, erythropoietin; BIE, butyrate plus ins/EPO; and eFLC, erythroid fetal liver cell.
Only adult human β-globin gene expression was detectable in single cells isolated from cultured eFLCs transgenic for the human β-globin gene locus. However, γ-globin gene expression was detectable in 10 of 15 BIE-treated 2-cell pools compared with 0 of 15 ins/EPO-treated 2-cell pools. Statistically, this indicates that there is SCFA-induced globin gene expression in at least 35% of cells in SCFA and ins/EPO-treated human-globin eFLCs, versus 0% in ins/EPO-only treated eFLCs.
SCFAs cause bulk histone acetylation, but do not change the phosphorylation status of p38, p42/44, or STAT5 in eFLCs
We examined the acetylation status of bulk histone H3 in eFLCs and found that histone H3 was hyperacetylated only in eFLCs that had been exposed to SCFAs, with or without ins/EPO; ins/EPO alone did not change bulk histone H3 acetlyation (Figure 4).
Figure 4.
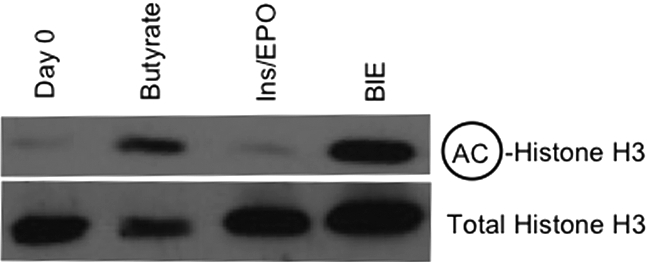
Bulk histone acetylation in eFLCs. Shown are Western blot analyses for total acetylated histone H3 with total histone H3 as a loading control, from E14.5 eFLCs harvested at day 0 or after 24 hours in culture, as indicated.
We investigated whether changes in signaling molecules, described in other models of SCFA- or cytokine-mediated up-regulation of fetal globin gene expression, were detectable in eFLCs. Both p3818,40 and STAT5 phosphorylation19 have been seen during SCFA-mediated up-regulation of embryonic globin gene expression. Changes in p42/44 phosphorylation with embryonic/fetal globin gene induction have also been described, whether diminished in butyrate-treated K562 cells41 or increased in stem cell factor and EPO-treated human CD34+-derived erythroid progenitors.32
In eFLCs, p42/44 and STAT5 were phosphorylated by ins/EPO, with or without SCFAs, but not by SCFAs alone (Figure 5). Phosphorylation of p38 was constitutive on day 0 and persisted in spite of overnight serum starvation. Thereafter, p38 phosphorylation remained detectable in continuous culture in all conditions at 24 hours; but, by 32 hours, was least detectable in SCFA-only or untreated eFLCs (Figure 5).
Figure 5.
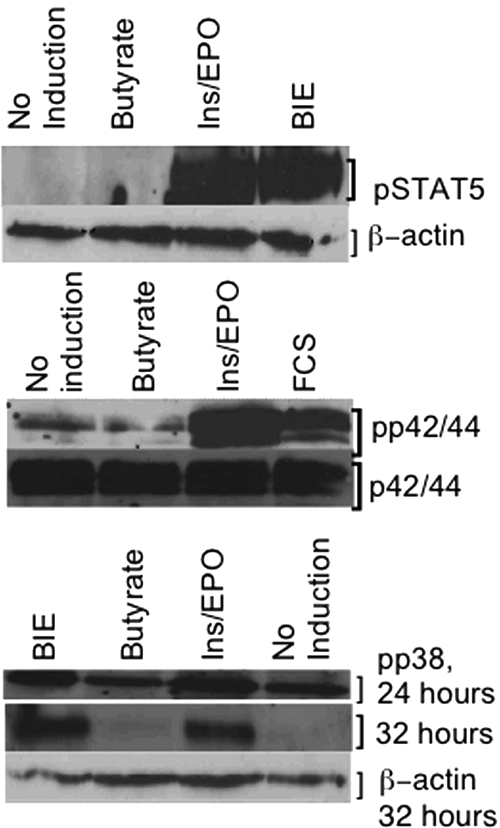
Cell-signaling intermediaries in eFLCs. Shown are Western blot analyses for (A) phospho-STAT5 (pSTAT5) with β-actin as a loading control, cultured as indicated, and (B) phospho-p42/44 (pp42/44), and unphosphorylated p42/44 as a loading control (reprobed), cultured as indicated. Both panels A and B were induced for 10 minutes, after overnight serum starvation. (C) Phospho-p38 (pp38), after 24 and 32 hours in culture, plus β-actin loading control at 32 hours, run in parallel.
Discussion
Here we show SCFAs induce the expression of embryonic/fetal β-type globin genes in EryD, a key therapeutic goal in β-globin gene disorders. This finding offers the opportunity to study SCFAs in a model of definitive erythropoiesis that is responsive to SCFAs, nontransformed, and accessible to transgenic manipulation.
The effects of endogenous elevations of SCFAs on globin gene expression in erythroid precursors were examined during development, in a transgenic model of perturbed intermediary metabolism. In PCCA KO mice, propionate levels were elevated in the dam throughout pregnancy and in the pups throughout gestation. This allowed us to minimize the complications of, and unclear dosing to embryos from, exogenous SCFAs given intraperitoneally or subcutaneously to wt dams. We found that elevated levels of SCFAs in utero modestly increased embryonic globin gene expression during primitive erythropoiesis in the yolk sac, but not during definitive erythropoiesis in the fetal liver.
There have been no examples of murine embryonic globin genes being induced in primary EryD, and murine embryonic hemoglobin, unlike human fetal hemoglobin, is not detectable in adult animals. Further, DNA around the murine embryonic β-type globin genes, and not the adult globin genes, becomes epigenetically modified by DNA methylation during development.42 Therefore, it was plausible that the embryonic β-globin genes in EryD were packaged in a chromosomally inaccessible state and could not be reactivated by SCFAs alone.
We tested SCFAs in vitro, at concentrations of propionate not achievable in PCCA KO mice in vivo, for their ability to up-regulate embryonic/fetal globin gene expression in EryD from wt or human β-locus transgene-containing eFLCs. Marked augmentation of murine embryonic and human fetal β-type globin gene expression was seen. This finding implied that embryonic globin genes could be induced in primary EryD by SCFAs alone.
SCFA-mediated up-regulation of globin gene expression in vitro was detectable in most cells and, in the absence of ins/EPO, affected primarily βH1 in the murine and Gγ in the human loci. Gγ, but not Aγ, is up-regulated by hydroxyurea or butyrate in K562 cells.22,43 Sangeman et al22 described a cAMP-response element (CRE) at an ATF/CREB binding site in the Gγ promoter, absent from the Aγ promoter, through which butyrate acts in K562s. Similarly, in our work, βH1 or Gγ expression is seen after SCFA-only treatment in wt and transgenic eFLCs, respectively, although the βH1 promoter does not contain an obvious ATF/CREB binding site per “TESS” analysis (http://www.cbil.upenn.edu/tess).44
EPO alone in our model, when apart from the complex physiology of stress erythropoiesis in vivo, did not increase embryonic/fetal globin gene expression in EryD in vitro. However, EPO abrogated differences in induction between βH1 and ϵY in murine EryD and between Gγ and Aγ in the human β-globin loci of transgenic EryD (SCFAs + ins/EPO in our experiments) and in human erythroid precursors in vitro (SCFAs + EPO45 or hydroxyurea + EPO46).
SCFAs may have a variable effect on individual globin gene alleles (βH1 or ϵY and Gγ and Aγ) depending upon the differentiation state of the erythroid precursor upon which they act. Here, SCFAs and ins/EPO-treated eFLCs (ie, BIE) proliferate more than SCFA-only treated cells, and FACS analysis suggests slightly increased numbers of early erythroid precursors by cell-surface expression in SCFA and ins/EPO-treated cells. Others have found that transcription factor levels and histone acetylation patterns at the α-globin loci change during red cell differentiation;47 these differences, between early and late erythroid precursors, may alter the susceptibility of specific embryonic or fetal β-type globin genes to pharmacologic manipulation. This could explain the difference in induced human fetal (γA vs γG) or induced murine embryonic (βH1 versus ϵY) globin gene expression that is seen with SCFAs alone compared with SCFAs and ins/EPO.
SCFAs did not trigger apoptosis in fetal liver–derived definitive erythroid cells, although enhanced SCFA-mediated erythroid differentiation was seen. This parallels the gastrointestinal cell differentiation that is seen in colonic cells after exposure to SCFAs.27,48
In our experiments, those conditions in which embryonic/fetal globin gene expression was induced (eg, SCFAs with or without ins/EPO), resulted in net histone acetylation, as described previously,22,23,34,40,49 while ins/EPO alone, in which embryonic globin gene expression was not induced, did not. In SCFA-treated eFLCs, we did not detect the changes in cell-signaling intermediaries that had been seen, for p38 and p42/44,18,32 with induced embryonic/fetal globin gene expression and, for STAT5, with SCFA-associated rescue of cytokine-dependent hematopoiesis.19 p42/44 and STAT5 were activated by ins/EPO treatment in eFLCs, with or without SCFAs, but not by SCFAs alone. p38 was phosphorylated in eFLCs at baseline, at 24 hours, and in all culture conditions, without any enhancement by SCFAs. Importantly, p38 phosphorylation, associated with red cell differentiation,50 may be essential to SCFA-mediated reactivation of embryonic globin gene expression without being a unique mediator of this induction.
EryD normally express only adult globin genes. Paired cellular events, such as HDAC inhibition and an additional activating molecular signal, have been found to play a role in the pathologic reactivation of fetal molecular programs during cardiac hypertrophy51 and have been implicated in SCFA- or thalidomide-mediated fetal globin gene induction in erythroid precursors.22,52 Our work shows that SCFA induction in EryD is associated with constitutive p38 phosphorylation during erythroid differentiation, paired with a SCFA-specific inhibition of histone deacetylation. This is an important refinement of earlier models.
We expect that eFLCs, as a robust, short-term, primary cell model, will offer distinct advantages for obtaining mechanistic insights into induction of embryonic/fetal globin gene expression by SCFAs, and perhaps other agents, in EryD in a physiologic setting.
Acknowledgments
We acknowledge with gratitude Dr Larry Swift and the Lipid Core, Vanderbilt-National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Mouse Metabolic Phenotyping Center, Nashville, TN (support: U24, DK59637) for propionate measurements; Dr J. Eric Russell, Department of Medicine and Pediatrics, School of Medicine, University of Pennsylvania, for analyzing embryonic hemoglobin protein; Galina Baibakova, Laboratory of Cellular and Developmental Biology (LCDB; NIDDK, National Institutes of Health [NIH]), for expert animal husbandry; and Drs Margaret Baron and Joan Isern, Departments of Medicine, Developmental and Regenerative Biology, Gene and Cell Medicine, and Oncological Sciences, Mt Sinai School of Medicine, for advice and for access to complementary experimental data. Support from the Department of Medicine, Albert Einstein College of Medicine (H.B., A. Dutta, S.K, and J.A.L.), and the Intramural Program, NIDDK, NIH (J.L.H., L.S.S., A. Dean, and J.A.L.) is gratefully acknowledged. We thank our colleagues in the Pasta and Red Cell Society of New York for critical review of the data.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: H.B., J.L.H., and A. Dutta contributed to the design and analysis of, and carried out, the experiments; S.K. and L.S.S. carried out the experiments; T.M. supplied a critical reagent and expertise; A. Dean contributed to the design and analysis of the experiments; J.A.L. contributed to the design and analysis of and carried out the experiments and prepared the manuscript; and all authors have read the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jane A. Little, Division of Hematology, Department of Medicine, Albert Einstein College of Medicine, Ullman 505, 1300 Morris Park Ave, Bronx, NY 10461; e-mail: jlittle@aecom.yu.edu.
References
- 1.Wood WG, Pembrey ME, Serjeant GR, Perrine RP, Weatherall DJ. Hb F synthesis in sickle cell anaemia: a comparison of Saudi Arab cases with those of African origin. Br J Haematol. 1980;45:431–445. doi: 10.1111/j.1365-2141.1980.tb07163.x. [DOI] [PubMed] [Google Scholar]
- 2.Ottolenghi S, Giglioni B, Taramelli R, Comi P, Gianni AM. δβ-Thalassemia and HPFH. Birth Defects Orig Artic Ser. 1982;18:65–67. [PubMed] [Google Scholar]
- 3.Perrine SP, Greene MF, Faller DV. Delay in the fetal globin switch in infants of diabetic mothers. N Engl J Med. 1985;312:334–338. doi: 10.1056/NEJM198502073120602. [DOI] [PubMed] [Google Scholar]
- 4.Little JA, Dempsey NJ, Tuchman M, Ginder GD. Metabolic persistence of fetal hemoglobin. Blood. 1995;85:1712–1718. [PubMed] [Google Scholar]
- 5.Galanello R, Cao A, Olivieri N. Induction of fetal hemoglobin in the presence of increased 3-hydroxybutyric acid associated with β-ketothiolase deficiency. N Engl J Med. 1994;331:746–747. doi: 10.1056/NEJM199409153311114. [DOI] [PubMed] [Google Scholar]
- 6.Sher GD, Ginder GD, Little J, Yang S, Dover GJ, Olivieri NF. Extended therapy with intravenous arginine butyrate in patients with beta-hemoglobinopathies [see comments]. N Engl J Med. 1995;332:1606–1610. doi: 10.1056/NEJM199506153322404. [DOI] [PubMed] [Google Scholar]
- 7.Perrine SP, Ginder GD, Faller DV, et al. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the β-globin disorders [see comments]. N Engl J Med. 1993;328:81–86. doi: 10.1056/NEJM199301143280202. [DOI] [PubMed] [Google Scholar]
- 8.Atweh GF, Sutton M, Nassif I, et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood. 1999;93:1790–1797. [PMC free article] [PubMed] [Google Scholar]
- 9.Whitelaw E, Tsai S-F, Hogben P, Orkin SH. Regulated expression of globin chains and the erythroid transcription factor GATA-1 during erythropoiesis in the developing mouse. Mol Cell Biol. 1990;10:6596–6606. doi: 10.1128/mcb.10.12.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kingsley PD, Malik J, Emerson RL, et al. “Maturational” globin switching in primary primitive erythroid cells. Blood. 2006;107:1665–1672. doi: 10.1182/blood-2005-08-3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kingsley PD, Malik J, Fantauzzo KA, Palis J. Yolk sac-derived primitive erythroblasts enucleate during mammalian embryogenesis. Blood. 2004;104:19–25. doi: 10.1182/blood-2003-12-4162. [DOI] [PubMed] [Google Scholar]
- 12.Gaensler KM, Kitamura M, Kan YW. Germ-line transmission and developmental regulation of a 150-kb yeast artificial chromosome containing the human β-globin locus in transgenic mice. Proc Natl Acad Sci U S A. 1993;90:11381–11385. doi: 10.1073/pnas.90.23.11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson KR, Clegg CH, Huxley C, et al. Transgenic mice containing a 248-kb yeast artificial chromosome carrying the human β-globin locus display proper developmental control of human globin genes. Proc Natl Acad Sci U S A. 1993;90:7593–7597. doi: 10.1073/pnas.90.16.7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keefer JR, Schneidereith TA, Mays A, Purvis SH, Dover GJ, Smith KD. Role of cyclic nucleotides in fetal hemoglobin induction in cultured CD34+ cells. Exp Hematol. 2006;34:1151–1161. doi: 10.1016/j.exphem.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Inoue A, Kuroyanagi Y, Terui K, Moi P, Ikuta T. Negative regulation of γ-globin gene expression by cyclic AMP-dependent pathway in erythroid cells. Exp Hematol. 2004;32:244–253. doi: 10.1016/j.exphem.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 16.Ikuta T, Ausenda S, Cappellini MD. Mechanism for fetal globin gene expression: role of the soluble guanylate cyclase-cGMP-dependent protein kinase pathway. Proc Natl Acad Sci U S A. 2001;98:1847–1852. doi: 10.1073/pnas.041599798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Asleh R, Marsh S, Shilkrut M, et al. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ Res. 2003;92:1193–1200. doi: 10.1161/01.RES.0000076889.23082.F1. [DOI] [PubMed] [Google Scholar]
- 18.Pace BS, Qian XH, Sangerman J, et al. p38 MAP kinase activation mediates γ-globin gene induction in erythroid progenitors. Exp Hematol. 2003;31:1089–1096. doi: 10.1016/s0301-472x(03)00235-2. [DOI] [PubMed] [Google Scholar]
- 19.Boosalis MS, Bandyopadhyay R, Bresnick EH, et al. Short-chain fatty acid derivatives stimulate cell proliferation and induce STAT-5 activation. Blood. 2001;97:3259–3267. doi: 10.1182/blood.v97.10.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castaneda S, Boosalis MS, Emery D, Thies A, Faller DV, Perrine SP. Enhancement of growth and survival and alterations in Bcl-family proteins in beta-thalassemic erythroid progenitors by novel short-chain fatty acid derivatives. Blood Cells Mol Dis. 2005;35:217–226. doi: 10.1016/j.bcmd.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCaffrey PG, Newsome DA, Fibach E, Yoshida M, Su MS. Induction of gamma-globin by histone deacetylase inhibitors. Blood. 1997;90:2075–2083. [PubMed] [Google Scholar]
- 22.Sangerman J, Lee MS, Yao X, et al. Mechanism for fetal hemoglobin induction by histone deacetylase inhibitors involves γ-globin activation by CREB1 and ATF-2. Blood. 2006;108:3590–3599. doi: 10.1182/blood-2006-01-023713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mankidy R, Faller DV, Mabaera R, et al. Short-chain fatty acids induce γ-globin gene expression by displacement of a HDAC3-NCoR repressor complex. Blood. 2006;108:3179–3186. doi: 10.1182/blood-2005-12-010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mabaera R, Greene MR, Richardson CA, Conine SJ, Kozul CD, Lowrey CH. Neither DNA hypomethylation nor changes in the kinetics of erythroid differentiation explain 5-azacytidine's ability to induce human fetal hemoglobin. Blood. 2008;111:411–420. doi: 10.1182/blood-2007-06-093948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazaki T, Ohura T, Kobayashi M, et al. Fatal propionic acidemia in mice lacking propionyl-CoA carboxylase and its rescue by postnatal, liver-specific supplementation via a transgene. J Biol Chem. 2001;276:35995–35999. doi: 10.1074/jbc.M105467200. [DOI] [PubMed] [Google Scholar]
- 26.Leder A, Leder P. Butyric acid, a potent inducer of erythroid differentiation in cultured erythroleukemic cells. Cell. 1975;5:319–322. doi: 10.1016/0092-8674(75)90107-5. [DOI] [PubMed] [Google Scholar]
- 27.Heerdt BG, Houston MA, Augenlicht LH. Potentiation by specific short-chain fatty acids of differentiation and apoptosis in human colonic carcinoma cell lines. Cancer Res. 1994;54:3288–3293. [PubMed] [Google Scholar]
- 28.Basson MD, Turowski GA, Rashid Z, Hong F, Madri JA. Regulation of human colonic cell line proliferation and phenotype by sodium butyrate. Dig Dis Sci. 1996;41:1989–1993. doi: 10.1007/BF02093601. [DOI] [PubMed] [Google Scholar]
- 29.Basson MD, Emenaker NJ, Hong F. Differential modulation of human (Caco-2) colon cancer cell line phenotype by short chain fatty acids. Proc Soc Exp Biol Med. 1998;217:476–483. doi: 10.3181/00379727-217-44261. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Socolovsky M, Gross AW, Lodish HF. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood. 2003;102:3938–3946. doi: 10.1182/blood-2003-05-1479. [DOI] [PubMed] [Google Scholar]
- 31.McElveen RL, Lou TF, Reese K, Xia S, Baliga BS, Pace BS. Erk pathway inhibitor U0126 induces γ-globin expression in erythroid cells. Cell Mol Biol (Noisy-le-grand) 2005;51:215–227. [PubMed] [Google Scholar]
- 32.Bhanu NV, Trice TA, Lee YT, Miller JL. A signaling mechanism for growth-related expression of fetal hemoglobin. Blood. 2004;103:1929–1933. doi: 10.1182/blood-2003-05-1624. [DOI] [PubMed] [Google Scholar]
- 33.Miller JL. Signaled expression of fetal hemoglobin during development. Transfusion. 2005;45:1229–1232. doi: 10.1111/j.1537-2995.2005.00182.x. [DOI] [PubMed] [Google Scholar]
- 34.Wilson AJ, Byun DS, Popova N, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem. 2006;281:13548–13558. doi: 10.1074/jbc.M510023200. [DOI] [PubMed] [Google Scholar]
- 35.Tanimoto K, Liu Q, Bungert J, Engel JD. Effects of altered gene order or orientation of the locus control region on human beta-globin gene expression in mice. Nature. 1999;398:344–348. doi: 10.1038/18698. [DOI] [PubMed] [Google Scholar]
- 36.Kaufmann MH. London: Elsevier Academic Press; 2003. The Atlas of Mouse Development. [Google Scholar]
- 37.Bender MA, Reik A, Close J, et al. Description and targeted deletion of 5′ hypersensitive site 5 and 6 of the mouse β-globin locus control region. Blood. 1998;92:4394–4403. [PubMed] [Google Scholar]
- 38.Dick DAT. Growth and function in the foetal liver. J Embrol Exp Morphol. 1956;4:97–109. [Google Scholar]
- 39.Schrick JJ, Lingrel JB. cDNA cloning, mapping and expression of the mouse propionyl CoA carboxylase beta (pccb), the gene for human type II propionic acidaemia. Gene. 2001;264:147–152. doi: 10.1016/s0378-1119(00)00586-2. [DOI] [PubMed] [Google Scholar]
- 40.Witt O, Monkemeyer S, Ronndahl G, et al. Induction of fetal hemoglobin expression by the histone deacetylase inhibitor apicidin. Blood. 2003;101:2001–2007. doi: 10.1182/blood-2002-08-2617. [DOI] [PubMed] [Google Scholar]
- 41.Witt O, Sand K, Pekrun A. Butyrate-induced erythroid differentiation of human K562 leukemia cells involves inhibition of ERK and activation of p38 MAP kinase pathways. Blood. 2000;95:2391–2396. [PubMed] [Google Scholar]
- 42.Hsu M, Mabaera R, Lowrey CH, Martin DI, Fiering S. CpG hypomethylation in a large domain encompassing the embryonic β-like globin genes in primitive erythrocytes. Mol Cell Biol. 2007;27:5047–5054. doi: 10.1128/MCB.02234-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu J, Zimmer DB. Differential regulation of A γ and G γ fetal hemoglobin mRNA levels by hydroxyurea and butyrate. Exp Hematol. 1998;26:265–272. [PubMed] [Google Scholar]
- 44.Schug J, Overton GC. Computational Biology and Informatics Laboratory, School of Medicine, University of Pennsylvania; 1997. TESS: Transcription Element Search Software on the WWW. [Google Scholar]
- 45.Perrine SP, Miller BA, Faller DV, et al. Sodium butyrate enhances fetal globin gene expression in erythroid progenitors of patients with Hb SS and b thalassemia. Blood. 1989;74:454–459. [PubMed] [Google Scholar]
- 46.Watanapokasin R, Sanmund D, Winichagoon P, Muta K, Fucharoen S. Hydroxyurea responses and fetal hemoglobin induction in β-thalassemia/HbE patients' peripheral blood erythroid cell culture. Ann Hematol. 2006;85:164–169. doi: 10.1007/s00277-005-0049-1. [DOI] [PubMed] [Google Scholar]
- 47.Anguita E, Hughes J, Heyworth C, Blobel GA, Wood WG, Higgs DR. Globin gene activation during haemopoiesis is driven by protein complexes nucleated by GATA-1 and GATA-2. EMBO J. 2004;23:2841–2852. doi: 10.1038/sj.emboj.7600274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mariadason JM, Corner GA, Augenlicht LH. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. 2000;60:4561–4572. [PubMed] [Google Scholar]
- 49.Johnson J, Hunter R, McElveen R, Qian XH, Baliga BS, Pace BS. Fetal hemoglobin induction by the histone deacetylase inhibitor, scriptaid. Cell Mol Biol (Noisy-le-grand) 2005;51:229–238. [PubMed] [Google Scholar]
- 50.Rubiolo C, Piazzolla D, Meissl K, et al. A balance between Raf-1 and Fas expression sets the pace of erythroid differentiation. Blood. 2006;108:152–159. doi: 10.1182/blood-2005-09-3866. [DOI] [PubMed] [Google Scholar]
- 51.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aerbajinai W, Zhu J, Gao Z, Chin K, Rodgers GP. Thalidomide induces γ-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood. 2007;110:2864–2871. doi: 10.1182/blood-2007-01-065201. [DOI] [PMC free article] [PubMed] [Google Scholar]