

Summary
The sequelae of traumatic brain injury, including posttraumatic epilepsy, represent a major societal problem. Significant resources are required to develop a better understanding of the underlying pathophysiologic mechanisms as targets for potential prophylactic therapies. Posttraumatic epilepsy undoubtedly involves numerous pathogenic factors that develop more or less in parallel. We have highlighted two potential “prime movers”: disinhibition and development of new functional excitatory connectivity, which occur in a number of animal models and some forms of epilepsy in humans. Previous experiments have shown that tetrodotoxin (TTX) applied to injured cortex during a critical period early after lesion placement can prevent epileptogenesis in the partial cortical (“undercut”) model of posttraumatic epilepsy. Here we show that such treatment markedly attenuates histologic indices of axonal and terminal sprouting and presumably associated aberrant excitatory connectivity. A second finding in the undercut model is a decrease in spontaneous inhibitory events. Current experiments show that this is accompanied by regressive alterations in fast-spiking γ-aminobutyric acid (GABA)ergic interneurons, including shrinkage of dendrites, marked decreases in axonal length, structural changes in inhibitory boutons, and loss of inhibitory synapses on pyramidal cells. Other data support the hypothesis that these anatomic abnormalities may result from loss of trophic support normally provided to interneurons by brain-derived neurotrophic factor (BDNF).
Approaches that prevent these two pathophysiologic mechanisms may offer avenues for prophylaxis for posttraumatic epilepsy. However, major issues such as the role of these processes in functional recovery from injury and the timing of the critical period(s) for application of potential therapies in humans are critical and need to be resolved.
Keywords: Posttraumatic epilepsy, Interneurons, Fast-spiking, Sprouting, GAP43, Neurofilaments, Laser uncaging, Caged glutamate, Brain-derived neurotrophic factor, TrkB, Cortical isolation, Undercut, Tetrodotoxin
Introduction
Critical issues
The importance of brain trauma as a risk factor for the development of epilepsy is well established (Salazar et al., 1985; Annegers et al., 1998). The high incidence of epileptogenesis following severe injury such as penetrating brain wounds in war time (Salazar et al., 1985) and the expected marked increase due to the current conflict in Iraq, emphasize the importance of developing prophylactic strategies that might be applied during the latent period between injury and the development of seizures. Unfortunately, multiple trials with anticonvulsant drugs have been ineffective (Temkin et al., 2001).
There are several important unknowns with respect to development of such prophylactic therapies that should be recognized as part of this effort. First, we need to know which pathophysiologic processes to target. A large number of mRNAs are activated in brain by trauma or seizures per se (Raghavendra et al., 2003; Lukasiuk & Pitkanen, 2004). Many abnormalities, including alterations in ion channels (Mody, 1998; Chen et al., 2002), transporters (Tanaka et al., 1997; Jin et al., 2005; Huberfeld et al., 2007), transmitter receptors (Chuang et al., 2001; Furtinger et al., 2001); glial cells (Ding et al., 2007), and development of aberrant connectivity (Tauck & Nadler, 1985; Salin et al., 1995; Jacobs et al., 1999; Wuarin & Dudek, 2001; Jin et al., 2006) have been found in various models of epileptogenesis. Therefore, multiple pathophysiologic epileptogenic processes are likely activated simultaneously or sequentially by brain trauma. Will targeting any one of these be adequate to provide antiepileptogenesis, or are there some “prime movers” that should receive the most focused investigative attention, given the urgency of the problem (Garga & Lowenstein, 2006) and finite resources and manpower?
A second important issue is the relationship between the latent period from injury to onset of epilepsy and the critical period, that is, the time over which a treatment must be administered to abort epileptogenesis. It is encouraging that in the only animal model in which prophylaxis of posttraumatic epileptogenesis has been achieved, the critical period is ~3 days, much shorter than the latency at which epileptogenesis can be detected in slices in vitro–10–14 days or longer (Graber & Prince, 1999, 2004). It seems likely that these important time intervals will vary in different animal models, in different cortical areas (e.g., neocortex vs. hippocampus), and with different degrees and types of injury (e.g., cf. Hoffman et al., 1994; Kelly et al., 2001; and D’Ambrosio et al., 2005). In humans, the latent period may be as long as years (Salazar et al., 1985); however, the critical period is unknown. This makes the timing for onset and duration of prophylactic treatment a critical and unknown variable.
In considering prophylactic approaches, a third critical problem emerges. Both adaptive and maladaptive processes are activated in brain by injury. The former can contribute to functional recovery (e.g., Hurwitz et al., 1990; Chollet et al., 1991), whereas the latter may lead to epileptogenesis. Are these similar processes that differ only quantitatively and in their intensity or timing with respect to injury, or are there distinctly different types of plasticity involved in epileptogenesis versus functional recovery? A prophylactic treatment for epileptogenesis that interfered with motor, sensory, or cognitive recovery following brain injury would not be useful. The answers to these questions are critical if we are to make long-term progress to prevent the occurrence of epileptogenesis after head trauma.
Which model(s)?
The model employed should ideally depend entirely on the question being addressed, and the advantages and disadvantages of the available choices (reviewed in Pitkanen et al., 2006). Certain basic features are desirable including the ease with which a traumatic injury can be applied, reproducibility from animal to animal, a latent period to epileptogenesis that is reasonably short, and, if detailed electrophysiologic data are needed, the capacity to retain epileptogenic activity in vitro to facilitate a variety of complex experiments. We have used the partially isolated neocortex (“undercut”) model of posttraumatic epileptogenesis, which has these advantages as well as others including the presence of a relatively focal lesion at a known neocortical site and relative preservation of adjacent cortex and more remote structures (e.g., hippocampus) Graber & Prince, 2006).
The partially isolated neocortical island with intact pial circulation (“undercuts” below) is an established in vivo and in vitro model for development of chronic posttraumatic hyperexcitability and epileptogenesis (Sharpless & Halpern, 1962; Halpern, 1972; Prince & Tseng, 1993; Hoffman et al., 1994; Salin et al., 1995; reviewed in Graber & Prince, 2006). This model provides a high yield of animals with epileptogenic cortex that may be studied in vitro (Hoffman et al., 1994; Graber & Prince, 2006). Previous results show that this lesion results in epileptogenesis in a variety of species (Graber & Prince, 2006). Isolated islands of neocortical gray matter, with neuropathologic evidence of substantial reorganization, are also present in postmortem specimens from epileptic children who developed extensive underlying white matter lesions as infants (Marin-Padilla, 1997). Seizures were also a frequent occurrence in humans subjected to psychosurgery in which cortical areas were partially isolated (Echlin et al., 1952; Scoville, 1960).
Disinhibition, increases in neuronal membrane excitability, and increases in excitatory synaptic coupling have been suggested as potential mechanisms in this chronic epilepsy model (Purpura & Housepian, 1961; Ribak & Reiffenstein, 1982; Prince & Tseng, 1993; Salin et al., 1995; Bush et al., 1999; Prince, 1999). The undercut cortex becomes progressively more epileptogenic over several weeks (Grafstein & Sastry, 1957; Sharpless & Halpern, 1962), and spontaneous interictal discharges can persist at least 1 year in the monkey (Echlin & Battista, 1963). Interictal epileptiform activity can be recorded within partially isolated cortex of anesthetized rats, and c-fos-immunoreactivity (IR) is increased for weeks in the injured cortex, suggesting ongoing abnormal activity (Jacobs et al., 2001). Clinical seizures have been infrequently observed, perhaps because the partially isolated cortex has limited connections with surrounding or subcortical areas. However, electrographic focal ictal episodes originating in the partial isolation of an implanted rat, associated with subtle behavioral alterations, have been observed during video-EEG (electroencephalography) recording (Graber & Prince, 2006). Spontaneous and evoked interictal activity, and more rarely ictal-like discharges, occurs in vitro in rodent neocortical slices cut through chronic isolations (Prince & Tseng, 1993; Hoffman et al., 1994; Salin et al., 1995).
Methods
Methods for producing partial neocortical isolations have been reviewed in detail recently (Graber & Prince, 2006) and will not be detailed here. Standard techniques for preparing and maintaining in vitro neocortical slices were used (Hoffman et al., 1994; Li & Prince, 2002). About 2–3 weeks after placement of a lesion producing partially isolated cortex, acute slices cut through the injured area and maintained in vitro show abnormal evoked epileptiform potentials consisting of polyphasic field potentials that are all or none in nature, develop with a variable latency following the normal evoked event, and propagate across the cortical structures (Prince & Tseng, 1993; Hoffman et al., 1994; Graber & Prince, 1999). The incidence and duration of spontaneous interictal discharges are markedly increased when slices are perfused with artificial cerebrospinal fluid (ACSF) containing physiologic concentrations of glutamine (Tani et al., 2007). In the experiments using laser scanning photostimulation of caged glutamate described later and illustrated in Fig. 2, glutamate uncaging was accomplished using 300–600-μs UV laser flashes. Cortical layers II–VI were stimulated in grids of 600–650 × 1000~1200 μm with 50-μm spacing. Evoked EPSCs were recorded from layer V pyramidal neurons that were voltage clamped at −70 mV. In these experiments, “composite amplitude” was defined as the sum of the amplitudes of all detected synaptic events during a 200-ms time window beginning 12 ms after the laser flash. “Region-normalized EPSC amplitude” was obtained by dividing the sum of all composite EPSC amplitudes evoked within a given distance from the soma. A “hot spot” was defined as an uncaging spot from which an EPSC was evoked. Immunocytochemical procedures were as described previously (Salin et al., 1995; Rosen et al., 1998; Jacobs et al., 1999).
Figure 2.

Laser scanning photostimulation with caged glutamate. (A) Representative map of EPSCs onto a pyramidal cell (white triangle) in slice from naive cortex. A 500 × 1200 μm photostimulation grid shows color-coded composite amplitude of EPSCs at each uncaging site detected between 12 and 200 ms of uncaging stimulus onset (scale on left). Roman numerals: cortical lamina. Trace at bottom: Single sweep showing an uncaged glutamate-evoked EPSC, in the absence of direct activation of the neuron, in a slice of control neocortex. Red arrow: time of glutamate uncaging (200 μs UV laser flash); vertical line: time point 200 ms after photostimulation. (B, C) Layer-specific enhancement of excitatory synaptic input onto layer V pyramidal neurons. (B) Comparison of region-normalized EPSC amplitude evoked by uncaging stimuli at various cortical depths. 0: Position of somata. Positive along y-axis: toward pial surface; negative toward white matter in B and C. (C) Mean fraction of hot spots/total uncaging spots at various vertical distances from somata. Asterisks: Two way analyses of variance (ANOVAs) p < 0.0001 for all. Open circles: control; black squares: undercut. Modified from Jin et al., 2006 with permission.
Epilepsia © ILAE
Potential epileptogenic processes during the latent period: Targets for prophylaxis
Increases in excitatory connectivity and sprouting
Immunocytochemical experiments show that there is intense immunoreactivity for 68-kDa and 200-kDa neurofilament proteins in the neuropil as early as 3 days following the partial isolation that persists for weeks following the lesion (I. Parada and D.A. Prince, unpublished data). Both cortical interneurons and pyramidal cells show this increased neurofilament expression (Fig. 5 of Graber & Prince, 2006; Fig. 1D,E). These changes can be taken as a proxy for axonal sprouting within the injured area (Yang et al., 1996; King et al., 2001). Sprouting of axonal terminals has also been demonstrated with immunocytochemistry using an antibody for growth associated protein (GAP) 43, which is prominent both in developing axonal terminals and in those that are reactivated and are sprouting following injury (Fig. 1A,B) (Bendotti et al., 1997; McKinney et al., 1997). Previous studies have shown that there are gross changes in axons of layer V excitatory pyramidal cells following partial isolation, including increases in axonal length, numbers of axon collaterals, and boutons that are presumably sites of synapses (Salin et al., 1995). Electrophysiologic data, including measurements of excitatory currents in layer V pyramidal cells of chronically epileptogenic cortex and field potential recordings of evoked events, support the presence of enhanced excitatory inputs and hyperexcitable circuits (Hoffman et al., 1994; Salin et al., 1995; Li & Prince, 2002).
Figure 1.

Immunoreactivity (IR) of axons and terminals in partially isolated neocortex. (A–C) Sections through layer V of rat sensorimotor cortex reacted with growth-associated protein (GAP) 43 antibody. (D–F) Comparable sections from another rat reacted with antibody for 68-kDa neurofilaments. (A, D) Control from layer V of hemisphere contralateral to the undercut. (B, E). GAP43-IR (B) and 68-kDa neurofilament-IR (E) in layer V of undercuts made 3 days earlier, contralateral to A and D, respectively. (C, F). Representative sections from undercuts of two other rats in which Elvax® impregnated with tetrodotoxin was placed subdurally over the undercut area at the time of surgery. Immunocytochemistry was done after 3 days in C and after 3 weeks in F. Tetrodotoxin (TTX) treatment reduced IR for both GAP43 and neurofilament in the undercuts. Calibrations in C and F: 50 μm for A–C and D–F, respectively.
Epilepsia © ILAE
Recently the use of laser scanning photostimulation of caged glutamate has allowed detailed mapping of synaptic connectivity in the undercut and shown increased excitatory connectivity onto pyramidal cells throughout the cortex in the epileptogenic region (Jin et al., 2006). There is a marked increase in the number of sites in layers II, III, and V from which laser stimulation can evoke EPSCs onto individual recorded neurons in layer V (Fig. 2C), and the evoked events have a larger amplitude than controls (Fig. 2B). Therefore, the electrophysiologic data strongly support the conclusion that the sprouting seen with anatomic studies is associated with new, functional, excessive recurrent excitatory circuits.
Assuming that functionally effective sprouting represents a key pathogenetic mechanism in development of posttraumatic epilepsy, the question arises as to whether this process can be altered by potential therapies applied during the latent period. Previous experiments provide a “proof in principle” that prophylaxis of epileptogenesis is possible in this model. Placement of a sheet of slow release resin (ELVAX®) impregnated with tetrodotoxin (TTX) over the cortical isolation at the time of surgery prevents the occurrence of evoked and spontaneous epileptiform discharges in in vitro slices weeks later, after TTX washout (Graber & Prince, 1999). TTX blocks voltage-dependent sodium channels and presumably eliminates all neurotransmission within the injured tissue. The mechanisms by which blockade of activity interferes with epileptogenesis are unknown. Our results are contrary to the proposal that reduced activity in the isolation underlies epileptogenesis by inducing “homeostatic” upscaling of excitatory synaptic activity (Houweling et al., 2005). There is a critical period for prophylaxis by the TTX in the first 3 days after injury (Graber & Prince, 2006). If the treatment with TTX was delayed for more than 3 days after placement of the undercut, slices were epileptogenic at the end of 2–3 weeks.
How might TTX treatment prevent the development of hyperexcitability? From the preceding, one hypothesis might be that blocking activity with TTX limits the sprouting response and the resulting maladaptive excitatory connectivity. Activity-related wiring of intracortical and interhemispheric connections is known to play a role in cortical ontogenesis and plastic changes in connections in the mature brain following injury (Carmichael & Chesselet, 2002; Mizuno et al., 2007; Wang et al., 2007). We, therefore, tested the hypothesis that there is an activity-dependent link between cortical injury and the axonal sprouting response that would be blocked by TTX. Both the upregulation of 68-kDa and 200-kDa neurofilament and GAP43 immunoreactivity in the undercut, indices of axonal and terminal sprouting, were significantly reduced by the TTX treatment (cf. Fig. 1B with C and E with F). Therefore, there is an as yet undefined activity link between injury and the development of new connectivity within injured cortex that may potentially be a target for prophylactic therapies. It remains to be seen whether therapies that curtail sprouting and epileptogenesis will affect recovery of other functions.
Alterations in GABAergic interneurons: A second potential target for prophylactic strategies
γ-aminobutyric acid (GABA)A receptor-mediated postsynaptic inhibition (termed “inhibition” subsequently) has important roles in normal cortical function (Sillito, 1977; McBain & Fisahn, 2001; Freund, 2003) and in controlling events implicated in epileptogenesis (Prince & Wilder, 1967; Wong & Prince, 1979; Miles & Wong, 1987; Chagnac-Amitai & Connors, 1989; Luhmann & Prince, 1992). Anatomic (Sloper et al., 1980; Ribak et al., 1982; Houser et al., 1986; De Lanerolle et al., 1989; Marco et al., 1996; Rosen et al., 1998; Spreafico et al., 1998; DeFelipe, 1999; Andre et al., 2001) and/or electrophysiologic data (Franck & Schwartzkroin, 1984; Ashwood & Wheal, 1986; Franck et al., 1988; Neumann-Haefelin et al., 1995; Williamson et al., 1999; Zhu & Roper, 2000; Sayin et al., 2003) document decreases in numbers of interneurons and/or postsynaptic inhibition in epileptogenic hippocampus and neocortex. However, other data emphasize the preservation of GABAergic neurons after various types of injury, and the potential for sprouting new inhibitory connections (Nieoullon & Dusticier, 1981; Goldowitz et al., 1982; Westenbroek et al., 1988; Babb et al., 1989a,b; Davenport et al., 1990; Seil et al., 1994; Magloczky & Freund, 2005). Both enhanced inhibitory input (e.g., Gulyas & Freund, 1996; Tamas et al., 1998; Bacci et al., 2003) and decreased excitatory drive onto interneurons (Sloviter, 1991; Lothman et al., 1996; Doherty & Dingledine, 2001) have been proposed as potential epileptogenic mechanisms, (but see Bernard et al., 1998; Jacobs & Prince, 2005). Electrophysiologic data can indicate a reduction in GABAergic inhibition when anatomic indices in the same model do not, presumably because of the limited sensitivity of some techniques for detecting subtle abnormalities (Franck et al., 1988 10636). For example, PV-containing FS cells in the partial cortical isolation are not decreased in number (Graber et al., 1999) and there is also no obvious reduction in calbindin or GAD-containing interneurons (Prince & Jacobs, 1998). However, mIPSC frequency is decreased in layer V P cells of undercut cortex (Li & Prince, 2002), perhaps because of anatomic and electrophysiologic axonal abnormalities not detectable without a more detailed analysis of filled cells. We, therefore, tested the hypothesis that there might be structural abnormalities in GABAergic interneurons in chronic neocortical isolations, using immunocytochemical techniques applied to cortical sections and biocytin-filled layer V FS cells.
Whole cell recordings were obtained from FS interneurons in control cortex and within the partially isolated epileptogenic cortex with biocytin-containing patch pipettes. After processing, a total of 13 cells in control cortex and 11 from the partially isolated cortex were found to have well-filled axonal and dendritic arbors suitable for analysis. These neurons had anatomic characteristics of basket cells (Somogyi et al., 1983), and several in each group that were tested were immunopositive for parvalbumin. Measurements from confocal stacks with Neurolucida software (Methods) showed that there was a very significant reduction in axonal length in the cells of undercut versus control (Fig. 3; 3,429.8 968.1 μm in control, n = 6; 726.9 ± 325.1 μm in undercut, n = 5; p < 0.001). The dendrites of undercut FS interneurons were thinner than in controls, and measurements using Volocity software (Methods) confirmed that they were decreased in volume (~1.3 0 ± 0.0.15 μm3/1 μm length in control and 0.6 ± 0.39 μm3/1 μm length in undercut, p < 0.05).
Figure 3.
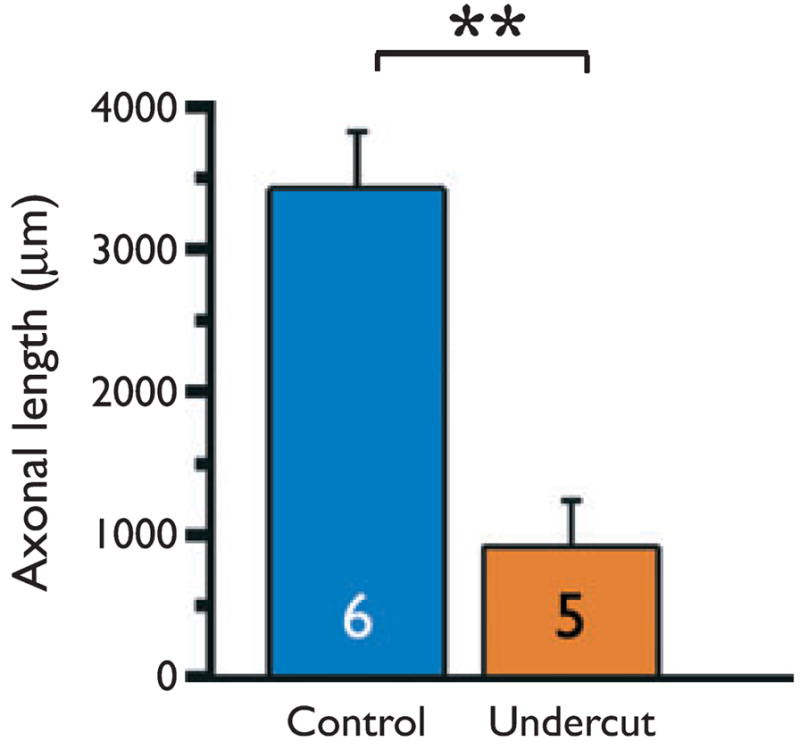
Graph of axonal lengths for six biocytin-filled FS inter-neurons in control and five in undercut cortex. Measurements obtained from stacks of confocal images. Mean ± SD for control: 3429.8 ± 968.1 μm and for undercut: 726.9 ± 325.1 μm. **p < 0.001
Epilepsia © ILAE
When axonal segments of these biocytin-filled inhibitory interneurons were examined in confocal images, several abnormalities were noted. Swellings along segments of axons in both control and undercut were identified as presynaptic boutons, as they were often immunoreactive for the vesicular GABA transporter VGAT (Fig. 4B) and could be in close apposition to post-synaptic gephyrin clusters that presumably marked the location of clusters of postsynaptic GABAA receptors (not shown). The total number of boutons per micrometer of axonal length was not significantly different in FS cells from undercut versus control. However when boutons were classified by size as “big” (>1 μm in diameter; arrows in Fig. 4) and “small” (<1 μm; arrowhead in Fig. 4), a significant reduction in big, and increase in small boutons as a percentage of the total over comparable axonal lengths was found in FS cells from the undercut (Fig. 5). There was also a marked reduction in the percentage of small boutons closely apposed to postsynaptic gephyrin-IR clusters in the undercut (not shown).
Figure 4.

Confocal images of an axonal segment of biocytin-filled FS interneuron. (A) Axonal segment of control layer V FS cell filled with Texas red-biocytin. Arrows point to large (>1 μm) boutons and arrowhead points to a small (<1 μm) bouton. (B) Same section double-labeled with Texas red and vesicular γ-aminobutyric acid (GABA) transporter (VGAT) antibody (green). Larger swellings along axon (arrows) contain VGAT in merged image (yellow) and are presumed presynaptic GABAergic boutons. Optical sections in A, B: 0.5 μm
Epilepsia © ILAE
Figure 5.

Graphs of bouton sizes for axons of nine control and seven undercut layer V biocytin-filled FS interneurons. Left graph: big boutons (>1 μm diameter) from control (black) and undercut cortex (gray) as a percentage of total boutons counted. Right graph: Small boutons (<1 μm diameter) as a percentage of total boutons in control and undercut. The total number of boutons in control versus undercut cortex is not significantly different (not shown); however, there are significantly fewer big and many smaller boutons in axons of the undercut FS cells. *p < 0.05. Total axon length analyzed from stacks of confocal images was 3,321 μm for nine control cells and 2,528 μm in seven undercut FS cells.
Epilepsia © ILAE
The preceding findings suggested that synaptic contacts between FS and P cells in the undercut might be abnormal. Synapses from FS interneurons are predominantly targeted to somata of P cells (Somogyi et al., 1983) and so a quantitative EM analysis of symmetrical (inhibitory) synapses onto layer V pyramidal cell bodies in control and undercut cortex was performed. (J. Wenzel, P. A. Schwartzkroin, and D. A. Prince, unpublished). Results showed that there was a significant decrease in inhibitory synapses on somata of pyramidal cells in undercut cortex compared to naive or contralateral cortex. The numbers of axonal terminals that were in close approximation to somata, were not different in undercut versus control images. Serial EM was not done, so data regarding bouton sizes or vesicular content are not available. These findings indicate that structural alterations occur in FS interneurons of the partially isolated, epileptogenic cortex that would make GABAergic neurotransmission less effective. Selective loss of inhibitory synapses has been previously reported on layer V/VI P neurons at the margins of chronic cortical isolations in cat (Ribak & Reiffenstein, 1982). The loss of inhibitory synapses in the axotomized pyramidal cells of layer V in the undercut cortex is in some respects similar to that found in motoneurons following axotomy where there is “stripping” of inhibitory, more than excitatory synapses (Sumner & Sutherland, 1973; Takata, 1981; Mendell, 1984) and decreased postsynaptic inhibition. This mechanism may also explain the loss of functional inhibition in kainate-induced hippocampal epileptogenesis (Franck et al., 1988). The prevalence of small boutons perisomatically on P cells in the undercut may also be associated with other pre- and postsynaptic alterations that would make inhibitory transmission less effective (Pierce & Lewin, 1994; Harris & Sultan, 1995).
What mechanisms might underlie the presumed atrophic changes in GABAergic interneurons? We speculate that the changes that we see are a reversion to an earlier developmental stage. Compared to interneurons in mature cortex, those examined early in development have reduced axonal arbors, decreased dendritic volumes and lengths, make fewer synapses on P cells, and would be are less effective in releasing GABA and inducing inhibitory events (Bahr & Wolff, 1985; Seress & Ribak, 1990; De Felipe et al., 1997; Jin et al., 2003). One critical factor for growth and development of interneurons in cortex is the availability of brain-derived neurotrophic factor (BDNF) (McAllister et al., 1995; Marty et al., 1997; Aguado et al., 2003; Jin et al., 2003). Inhibitory contacts and the frequency of mIPSCs onto pyramidal cells genetically altered to limit their capacity to produce BDNF are significantly reduced compared to adjacent nonaltered pyramidal cells (Kohara et al., 2003). We speculate that reductions in the numbers of layer V pyramidal cells (Graber et al., 1999) and alterations in surviving ones indicated by decreased somatic areas and abnormal intrinsic membrane properties (Prince & Tseng, 1993), result in reduced availability of BDNF as a retrograde trophic factor for presynaptic interneurons. This may, in turn, result in the observed functional and structural abnormalities in the epileptogenic cortex. Recent results from gene array experiments show that mRNA for both BDNF and its receptor, TrkB, are significantly decreased 3 days after the partial cortical isolation is placed and that BDNF mRNA remains down at 3 weeks while TrkB recovers (K. Graber and D. A. Prince, unpublished). Immunocytochemical experiments in undercuts show that BDNF protein is also significantly downregulated in pyramidal cells 3 and 7 days after the undercut (not shown), and that there is also a significant reduction in immunoreactivity for the BDNF receptor, Trk B, on the parvalbumin-containing interneurons and P cells (cf. Fig. 6A,B with C,D).
Figure 6.
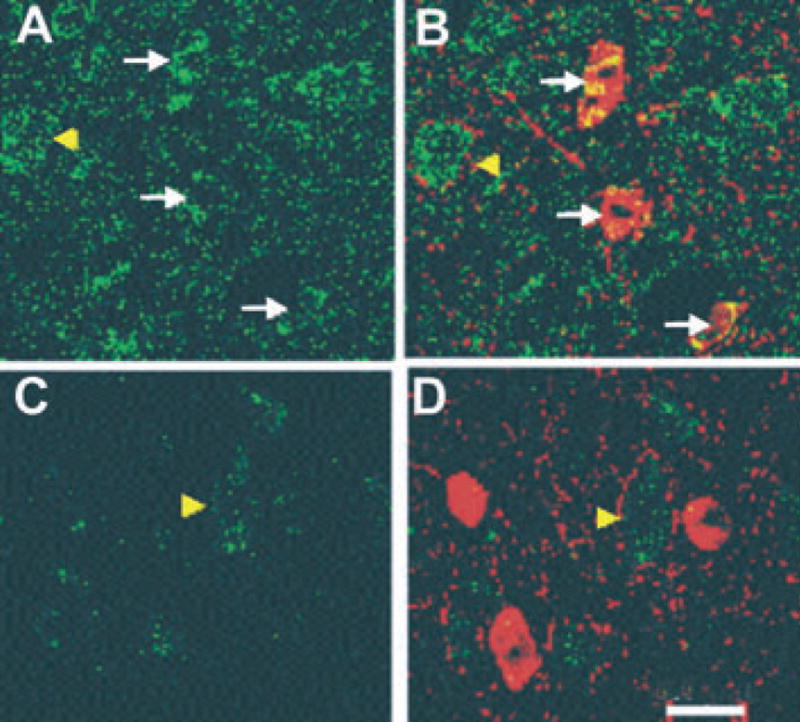
Decreased TrkB-IR (immunoreactivity) in undercut cortex. (A, B). Confocal images of same layer V control section contralateral to undercut, reacted for TrkB-IR (green) (A) and double labeled with TrkB and parvalbumin antibodies (red) (B). TrkB-IR is yellow on the interneurons in merged image. Arrows point to three parvalbumin-containing interneurons. Arrowhead points to probable pyramidal cell also containing TrkB-IR pixels. (C, D) Section from undercut cortex contralateral to A, B, reacted with TrkB antibody (C) and double labeled with TrkB + parvalbumin antibodies (D). Arrowhead in C, D points to a presumed pyramidal cell. TrkB-IR is decreased on pyramidal cell and interneurons in C,D compared to A, B control. Animal perfused 3 days after the partial cortical isolation. Calibration in D: 20 μm for all segments.
Epilepsia © ILAE
Discussion
There are many processes initiated by cortical injury that are ongoing in parallel with the ones described earlier. Each of them, or combinations of several, might well be important contributors to epileptogenesis and targets for prophylaxis. As mentioned previously, even if there are multiple mechanisms involved, it may be that intervention to affect particularly critical ones would be sufficient to prevent or markedly reduce subsequent hyperexcitability. However, at least in experimental models and likely also in humans, the timing of the lesion during development, the latency between the occurrence of a lesion and the onset of prophylactic therapy, the structures involved, the genetic background, and other factors will influence the results. Although this is a daunting proposition, it is critically important to begin to address the issue of prophylaxis, particularly because of the specter of increasing numbers of individuals who have sustained significant brain injury in military service (Garga & Lowenstein, 2006).
Here we have highlighted two potentially epileptogenic processes in the chronically injured cortex. The first, development of maladaptive axonal sprouting of excitatory connectivity, appears to depend at least in part on the level of activity within the circuit and may be altered by “ quieting” the involved cells. A similar result has been reported in thermal ischemic lesions of somatosensory cortex (Carmichael & Chesselet, 2002). Neural activity is well established as an important element in normal development of connectivity in cortical and other structures. For example, in neocortex, activity of layer II/III pyramidal cells is important in development of intrinsic and callosal connections (Mizuno et al., 2007; Wang et al., 2007). The molecular link between activity and sprouting in the above-noted experiments is not known.
One major challenge will be to develop experimental protocols that differentially affect maladaptive (i.e., epileptogenic) versus adaptive, or functionally restorative sprouting. It may be possible to accomplish this if the two processes have different time courses or critical periods, or have different cascades of underlying molecular events. A second avenue for investigation will be a search for agents that are more selective and less toxic than TTX, and can be applied directly to injured cortex or administered parenterally to target the events underlying aberrant circuit rewiring.
Structural and functional alterations in interneurons leading to defects in GABAergic neurotransmission represent a second set of abnormalities potentially approachable with new prophylactic strategies. It will be important to determine whether there is also an activity-related link between injury and the events leading to these interneuronal alterations, as suggested for the maladaptive sprouting. If the atrophic changes in FS interneurons are related to de-afferentation and a decrease in BDNF signaling, silencing the cortex would be expected to make matters worse, as both BDNF release and maintenance of interneuronal structure/function are activity dependent (see Marty et al., 1997 for review). Decreased activity as a result of the de-afferentation in the partial isolation and the TTX treatment should decrease BDNF release. So block of activity might have opposite effects on sprouting versus maintenance of inhibitory functionality.
If the decreases in BDNF and TrkB in the undercut cortex underlie the anatomic alterations in FS cells, would supplying exogenous BDNF rescue these cells from their presumed reversion to an early developmental stage? Experiments focused on this question are underway, with the caveat that BDNF signaling may also enhance excitatory neurotransmission (Kang & Schuman, 1995; Carmignoto et al., 1997; but see Frerking et al., 1998), and under some circumstances, promote epileptogenesis (Kokaia et al., 1995; Binder et al., 1999). Further information regarding the time course of adaptive versus aberrant sprouting and alterations in interneurons in relation to injury will be important in developing effective prophylactic strategies.
Acknowledgments
Supported by NIH grants NS12151 and NS39579 from the NINDS.
Footnotes
Disclosure: The authors declare no conflicts of interest.
References
- Aguado F, Carmona MA, Pozas E, Aguilo A, Martinez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibanez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl− co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Andre V, Marescaux C, Nehlig A, Fritschy JM. Alterations of hippocampal GABAergic system contribute to development of spontaneous recurrent seizures in the rat lithium-pilocarpine model of temporal lobe epilepsy. Hippocampus. 2001;11:452–468. doi: 10.1002/hipo.1060. [DOI] [PubMed] [Google Scholar]
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- Ashwood TJ, Wheal HV. Loss of inhibition in the CA1 region of the kainic acid lesioned hippocampus is not associated with changes in postsynaptic responses to GABA. Brain Res. 1986;367:390–394. doi: 10.1016/0006-8993(86)91625-2. [DOI] [PubMed] [Google Scholar]
- Babb TL, Pretorius JK, Kupfer WR, Crandall PH. Glutamate decarboxylase-immunoreactive neurons are preserved in human epileptic hippocampus. J Neurosci. 1989a;9:2562–2574. doi: 10.1523/JNEUROSCI.09-07-02562.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babb TL, Pretorius JK, Kupfer WR, Feldblum S. Recovery of decreased glutamate decarboxylase immunoreactivity after rat hippocampal kindling. Epilepsy Res. 1989b;3:18–30. doi: 10.1016/0920-1211(89)90064-8. [DOI] [PubMed] [Google Scholar]
- Bacci A, Rudolph U, Huguenard JR, Prince DA. Major differences in inhibitory synaptic transmission onto two neocortical inter-neuron subclasses. J Neurosci. 2003;23:9664–9674. doi: 10.1523/JNEUROSCI.23-29-09664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr S, Wolff JR. Postnatal development of axosomatic synapses in the rat visual cortex: morphogenesis and quantitative evaluation. J Comp Neurol. 1985;233:405–420. doi: 10.1002/cne.902330309. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Baldessari S, Pende M, Southgate T, Guglielmetti F, Samanin R. Relationship between GAP-43 expression in the dentate gyrus and synaptic reorganization of hippocampal mossy fibres in rats treated with kainic acid. Eur J Neurosci. 1997;9:93–101. doi: 10.1111/j.1460-9568.1997.tb01357.x. [DOI] [PubMed] [Google Scholar]
- Bernard C, Esclapez M, Hirsch JC, Ben-Ari Y. Interneurones are not so dormant in temporal lobe epilepsy: a critical reappraisal of the dormant basket cell hypothesis. Epilepsy Res. 1998;32:93–103. doi: 10.1016/s0920-1211(98)00043-6. [DOI] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, Ryan TE, Yancopoulos GD, McNamara JO. Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J Neurosci. 1999;19:1424–1436. doi: 10.1523/JNEUROSCI.19-04-01424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush PC, Prince DA, Miller KD. Increased pyramidal excitability and NMDA conductance can explain posttraumatic epileptogenesis without disinhibition: a model. J Neurophysiol. 1999;82:1748–1758. doi: 10.1152/jn.1999.82.4.1748. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Chesselet MF. Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J Neurosci. 2002;22:6062–6070. doi: 10.1523/JNEUROSCI.22-14-06062.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagnac-Amitai Y, Connors BW. Horizontal spread of synchronized activity in neocortex and its control by GABA-mediated inhibition. J Neurophysiol. 1989;61:747–758. doi: 10.1152/jn.1989.61.4.747. [DOI] [PubMed] [Google Scholar]
- Chen K, Aradi I, Santhakumar V, Soltesz I. H-channels in epilepsy: new targets for seizure control? Trends Pharmacol Sci. 2002;23:552–557. doi: 10.1016/s0165-6147(02)02110-7. [DOI] [PubMed] [Google Scholar]
- Chollet F, DiPiero V, Wise RJ, Brooks DJ, Dolan RJ, Frackowiak RS. The functional anatomy of motor recovery after stroke in humans: a study with positron emission tomography. Ann Neurol. 1991;29:63–71. doi: 10.1002/ana.410290112. [DOI] [PubMed] [Google Scholar]
- Chuang SC, Bianchi R, Kim D, Shin HS, Wong RK. Group I metabotropic glutamate receptors elicit epileptiform discharges in the hippocampus through PLCbeta1 signaling. J Neurosci. 2001;21:6387–6394. doi: 10.1523/JNEUROSCI.21-16-06387.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Fender JS, Fairbanks JP, Simon EA, Born DE, Doyle DL, Miller JW. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain. 2005;128:174–188. doi: 10.1093/brain/awh337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport CJ, Brown WJ, Babb TL. Sprouting of GABAergic and mossy fiber axons in dentate gyrus following intrahippocampal kainate in the rat. Exp Neurol. 1990;109:180–190. doi: 10.1016/0014-4886(90)90072-z. [DOI] [PubMed] [Google Scholar]
- De Felipe J, Marco P, Fairen A, Jones EG. Inhibitory synaptogenesis in mouse somatosensory cortex. Cereb Cortex. 1997;7:619–634. doi: 10.1093/cercor/7.7.619. [DOI] [PubMed] [Google Scholar]
- De Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- DeFelipe J. Chandelier cells and epilepsy. Brain. 1999;122:1807–1822. doi: 10.1093/brain/122.10.1807. [DOI] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2 + signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27:10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Dingledine R. Reduced excitatory drive onto interneurons in the dentate gyrus after status epilepticus. J Neurosci. 2001;21:2048–2057. doi: 10.1523/JNEUROSCI.21-06-02048.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echlin FA, Battista A. Epileptiform seizures from chronic isolated cortex. Arch Neurol. 1963;9:154–170. doi: 10.1001/archneur.1963.00460080064009. [DOI] [PubMed] [Google Scholar]
- Echlin FA, Arnett V, Zoll J. Paroxysmal high voltage discharges from isolated and partially isolated human and animal cerebral cortex. Electroencephalogr Clin Neurophysiol. 1952;4:147–164. doi: 10.1016/0013-4694(52)90004-7. [DOI] [PubMed] [Google Scholar]
- Franck JE, Schwartzkroin PA. Immature rabbit hippocampus is damaged by systemic but not intraventricular kainic acid. Brain Res Dev Brain Res. 1984;13:219–227. doi: 10.1016/0165-3806(84)90156-1. [DOI] [PubMed] [Google Scholar]
- Franck JE, Kunkel DD, Baskin DG, Schwartzkroin PA. Inhibition in kainate-lesioned hyperexcitable hippocampi: physiologic, autoradiographic, and immunocytochemical observations. J Neurosci. 1988;8:1991–2002. doi: 10.1523/JNEUROSCI.08-06-01991.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- Freund TF. Interneuron diversity series: rhythm and mood in peri-somatic inhibition. Trends Neurosci. 2003;26:489–495. doi: 10.1016/S0166-2236(03)00227-3. [DOI] [PubMed] [Google Scholar]
- Furtinger S, Pirker S, Czech T, Baumgartner C, Ransmayr G, Sperk G. Plasticity of Y1 and Y2 receptors and neuropeptide Y fibers in patients with temporal lobe epilepsy. J Neurosci. 2001;21:5804–5812. doi: 10.1523/JNEUROSCI.21-15-05804.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garga N, Lowenstein DH. Posttraumatic epilepsy: a major problem in desperate need of major advances. Epilepsy Curr. 2006;6:1–5. doi: 10.1111/j.1535-7511.2005.00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldowitz D, Vincent SR, Wu JY, Hokfelt T. Immunohistochemical demonstration of plasticity in GABA neurons of the adult rat dentate gyrus. Brain Res. 1982;238:413–420. doi: 10.1016/0006-8993(82)90116-0. [DOI] [PubMed] [Google Scholar]
- Graber K, Prince DA. Tetrodotoxin prevents post-traumatic epileptogenesis in rats. Ann Neurol. 1999;46:234–242. doi: 10.1002/1531-8249(199908)46:2<234::aid-ana13>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA. A critical period for prevention of neocortical post-traumatic epileptogenesis in rats. Ann Neurol. 2004;55:860–870. doi: 10.1002/ana.20124. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA. Chronic partial cortical isolation. In: Pitkanen A, Schwartzkroin P, Moshe S, editors. Models of Seizures and Epilepsy. Elsevier Academic Press; San Diego: 2006. pp. 477–493. [Google Scholar]
- Graber KD, Kharazia VN, Parada I, Prince DA. Parvalbumin-containing interneurons are spared in the undercut model of posttraumatic epileptogenesis. Epilepsia. 1999;40:31. [Google Scholar]
- Grafstein B, Sastry P. Some preliminary electrophysiological studies on chronically neuronally isolated cerebral cortex. Electroencephalogr Clin Neurophysiol. 1957;9:723–725. doi: 10.1016/0013-4694(57)90096-2. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Freund TF. Pyramidal cell dendrites are the primary targets of calbindin D28k-immunoreactive interneurons in the hippocampus. Hippocampus. 1996;6:525–534. doi: 10.1002/(SICI)1098-1063(1996)6:5<525::AID-HIPO5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Halpern LM. Chronically isolated aggregates of mammalian cortical neurons studied in situ. In: Purpura DP, Penry JK, Tower D, Woodbury DM, Walter R, editors. Experimental Models of Epilepsy. Raven; New York: 1972. pp. 197–221. [Google Scholar]
- Harris KM, Sultan P. Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses. Can J Physiol Pharmacol. 1995;34:1387–1395. doi: 10.1016/0028-3908(95)00142-s. [DOI] [PubMed] [Google Scholar]
- Hoffman SN, Salin PA, Prince DA. Chronic neocortical epileptogenesis in vitro. J Neurophysiol. 1994;71:1762–1773. doi: 10.1152/jn.1994.71.5.1762. [DOI] [PubMed] [Google Scholar]
- Houser CR, Harris AB, Vaughn JE. Time course of the reduction of GABA terminals in a model of focal epilepsy: a glutamic acid decarboxylase immunocytochemical study. Brain Res. 1986;383:129–145. doi: 10.1016/0006-8993(86)90014-4. [DOI] [PubMed] [Google Scholar]
- Houweling AR, Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Homeostatic synaptic plasticity can explain post-traumatic epileptogenesis in chronically isolated neocortex. Cereb Cortex. 2005;15:834–845. doi: 10.1093/cercor/bhh184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz BE, Dietrich WD, McCabe PM, Watson BD, Ginsberg MD, Schneiderman N. Sensory-motor deficit and recovery from thrombotic infarction of the vibrissal barrel-field cortex. Brain Res. 1990;512:210–220. doi: 10.1016/0006-8993(90)90628-o. [DOI] [PubMed] [Google Scholar]
- Jacobs KM, Prince DA. Excitatory and inhibitory postsynaptic currents in a rat model of epileptogenic microgyria. J Neurophysiol. 2005;93:687–696. doi: 10.1152/jn.00288.2004. [DOI] [PubMed] [Google Scholar]
- Jacobs KM, Mogensen M, Warren E, Prince DA. Experimental microgyri disrupt the barrel field pattern in rat somatosensory cortex. Cereb Cortex. 1999;9:733–744. doi: 10.1093/cercor/9.7.733. [DOI] [PubMed] [Google Scholar]
- Jacobs KM, Parada I, Prince DA. Enhanced c-fos staining in two post-lesional models of cortical hyperexcitability: neonatal freeze lesions and partial cortical isolations. Epilepsia. 2001;42(Suppl 7):221. [Google Scholar]
- Jin X, Hu H, Mathers PH, Agmon A. Brain-derived neurotrophic factor mediates activity-dependent dendritic growth in nonpyramidal neocortical interneurons in developing organotypic cultures. J Neurosci. 2003;23:5662–5673. doi: 10.1523/JNEUROSCI.23-13-05662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Impaired CL- extrusion in layer V pyramidal neurons of chronically injured epileptogenic cortex. J Neurophysiol. 2005;93:2117–2126. doi: 10.1152/jn.00728.2004. [DOI] [PubMed] [Google Scholar]
- Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer V pyramidal neurons of chronically injured epileptogenic neocortex in rats. J Neurosci. 2006;26:4891–4900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Kelly KM, Kharlamov A, Hentosz TM, Kharlamova EA, Williamson JM, Bertram EH, III, Kapur J, Armstrong DM. Photothrombotic brain infarction results in seizure activity in aging Fischer 344 and Sprague Dawley rats. Epilepsy Res. 2001;47:189–203. doi: 10.1016/s0920-1211(01)00294-7. [DOI] [PubMed] [Google Scholar]
- King CE, Canty AJ, Vickers JC. Alterations in neurofilaments associated with reactive brain changes and axonal sprouting following acute physical injury to the rat neocortex. Neuropathol Appl Neurobiol. 2001;27:115–126. doi: 10.1046/j.1365-2990.2001.00317.x. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Adachi N, Nishida M, Itami C, Nakamura S, Tsumoto T. Inhibitory but not excitatory cortical neurons require presynaptic brain-derived neurotrophic factor for dendritic development, as revealed by chimera cell culture. J Neurosci. 2003;23:6123–6131. doi: 10.1523/JNEUROSCI.23-14-06123.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia M, Ernfors P, Kokaia Z, Elmér E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol. 1995;133:215–224. doi: 10.1006/exnr.1995.1024. [DOI] [PubMed] [Google Scholar]
- Li H, Prince DA. Synaptic activity in chronically injured, epileptogenic sensory-motor neocortex. J Neurophysiol. 2002;88:2–12. doi: 10.1152/jn.00507.2001. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Bertram EH, III, Kapur J, Bekenstein JW. Temporal lobe epilepsy: studies in a rat model showing dormancy of GAB-Aergic inhibitory interneurons. Epilepsy Res Suppl. 1996;12:145–156. [PubMed] [Google Scholar]
- Luhmann HJ, Prince DA. Ontogenetic modifications in neocortical inhibition and functional consequences for epileptiform discharge. In: Speckmann E-J, Gutnick MJ, editors. Epilepsy and Inhibition. Urban and Schwarzenbeck; Munich: 1992. pp. 131–141. [Google Scholar]
- Lukasiuk K, Pitkanen A. Large-scale analysis of gene expression in epilepsy research: is synthesis already possible? Neurochem Res. 2004;29:1169–1178. doi: 10.1023/b:nere.0000023604.91584.6c. [DOI] [PubMed] [Google Scholar]
- Magloczky Z, Freund TF. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci. 2005;28:334–340. doi: 10.1016/j.tins.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Marco P, Sola RG, Pulido P, Alijarde MT, Sanchez A, Ramon CS, DeFelipe J. Inhibitory neurons in the human epileptogenic temporal neocortex. An immunocytochemical study. Brain. 1996;119:1327–1347. doi: 10.1093/brain/119.4.1327. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex. J Neuropathol Exp Neurol. 1997;56:219–235. doi: 10.1097/00005072-199703000-00001. [DOI] [PubMed] [Google Scholar]
- Marty S, Berzaghi M, Berninger B. Neurotrophins and activity-dependent plasticity of cortical interneurons. Trends Neurosci. 1997;20:198–202. doi: 10.1016/s0166-2236(96)01026-0. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. J Physiol. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Fisahn A. Interneurons unbound. Nat Rev Neurosci. 2001;2:11–23. doi: 10.1038/35049047. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat Med. 1997;3:990–996. doi: 10.1038/nm0997-990. [DOI] [PubMed] [Google Scholar]
- Mendell LM. Modifiability of spinal synapses. Physiol Rev. 1984;64:260–324. doi: 10.1152/physrev.1984.64.1.260. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RK. Latent synaptic pathways revealed after tetanic stimulation in the hippocampus. Nature. 1987;329:724–726. doi: 10.1038/329724a0. [DOI] [PubMed] [Google Scholar]
- Mizuno H, Hirano T, Tagawa Y. Evidence for activity-dependent cortical wiring: formation of interhemispheric connections in neonatal mouse visual cortex requires projection neuron activity. J Neurosci. 2007;27:6760–6770. doi: 10.1523/JNEUROSCI.1215-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I. Ion channels in epilepsy. Int Rev Neurobiol. 1998;42:199–226. doi: 10.1016/s0074-7742(08)60611-x. [DOI] [PubMed] [Google Scholar]
- Neumann-Haefelin T, Hagemann G, Witte OW. Cellular correlates of neuronal hyperexcitability in the vicinity of photochemically induced cortical infarcts in rats in vitro. Neurosci Lett. 1995;193:101–104. doi: 10.1016/0304-3940(95)11677-o. [DOI] [PubMed] [Google Scholar]
- Nieoullon A, Dusticier N. Increased glutamate decarboxylase activity in the red nucleus of the adult cat after cerebellar lesions. Brain Res. 1981;224:129–139. doi: 10.1016/0006-8993(81)91122-7. [DOI] [PubMed] [Google Scholar]
- Pierce JP, Lewin GR. An ultrastructural size principle. Neuroscience. 1994;58:441–446. doi: 10.1016/0306-4522(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Schwartzkroin P, Moshe S, editors. Models of Seizures and Epilepsy. Elsevier Academic Press; San Diego: 2006. pp. 477–493. [Google Scholar]
- Prince DA. Epileptic neurons and circuits. In: Delgado-Escueta A, Wilson W, Olsen R, Porter R, editors. Third edition of basic mechanisms of the epilepsies. Vol. 79. Lippincott, Raven; Philadelphia, PA: 1999. pp. 665–684. [Google Scholar]
- Prince DA, Jacobs K. Inhibitory function in two models of chronic epileptogenesis. Epilepsy Res. 1998;32:83–92. doi: 10.1016/s0920-1211(98)00042-4. [DOI] [PubMed] [Google Scholar]
- Prince DA, Tseng G-F. Epileptogenesis in chronically injured cortex: in vitro studies. J Neurophysiol. 1993;69:1276–1291. doi: 10.1152/jn.1993.69.4.1276. [DOI] [PubMed] [Google Scholar]
- Prince DA, Wilder BJ. Control mechanisms in cortical epileptogenic foci: “Surround” inhibition. Arch Neurol. 1967;16:194–202. doi: 10.1001/archneur.1967.00470200082007. [DOI] [PubMed] [Google Scholar]
- Purpura DP, Housepian EM. Morphological and physiological properties of chronically isolated immature neocortex. Exp Neurol. 1961;4:377–401. doi: 10.1016/0014-4886(61)90025-5. [DOI] [PubMed] [Google Scholar]
- Raghavendra RV, Dhodda VK, Song G, Bowen KK, Dempsey RJ. Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis. J Neurosci Res. 2003;71:208–219. doi: 10.1002/jnr.10486. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Reiffenstein RJ. Selective inhibitory synapse loss in chronic cortical slabs: a morphological basis for epileptic susceptibility. Can J Physiol Pharmacol. 1982;60:864–870. doi: 10.1139/y82-122. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Bradurne RM, Harris AB. A preferential loss of GAB-Aergic, symmetric synapses in epileptic foci: a quantitative ultrastructural analysis of monkey neocortex. J Neurosci. 1982;2:1725–1735. doi: 10.1523/JNEUROSCI.02-12-01725.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen GD, Jacobs KM, Prince DA. Effects of neonatal freeze lesions on expression of parvalbumin in rat neocortex. Cereb Cortex. 1998;8:753–761. doi: 10.1093/cercor/8.8.753. [DOI] [PubMed] [Google Scholar]
- Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985;35:1406–1414. doi: 10.1212/wnl.35.10.1406. [DOI] [PubMed] [Google Scholar]
- Salin PA, Tseng G-F, Hoffman SN, Parada I, Prince DA. Axonal sprouting in layer V pyramidal neurons of chronically injured cerebral cortex. J Neurosci. 1995;15:8234–8245. doi: 10.1523/JNEUROSCI.15-12-08234.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin U, Osting S, Hagen J, Rutecki P, Sutula T. Spontaneous seizures and loss of axo-axonic and axo-somatic inhibition induced by repeated brief seizures in kindled rats. J Neurosci. 2003;23:2759–2768. doi: 10.1523/JNEUROSCI.23-07-02759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville WB. Late results of orbital undercutting. Report of 76 patients undergoing quantitative selective lobotomies. Am J Psychiatry. 1960;117:525–532. doi: 10.1176/ajp.117.6.525. [DOI] [PubMed] [Google Scholar]
- Seil FJ, Drake-Baumann R, Leiman AL, Herndon RM, Tiekotter KL. Morphological correlates of altered neuronal activity in organotypic cerebellar cultures chronically exposed to anti-GABA agents. Brain Res Dev Brain Res. 1994;77:123–132. doi: 10.1016/0165-3806(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Seress L, Ribak CE. Postnatal development of the light and electron microscopic features of basket cells in the hippocampal dentate gyrus of the rat. Anat Embryol (Berl) 1990;181:547–565. doi: 10.1007/BF00174627. [DOI] [PubMed] [Google Scholar]
- Sharpless SK, Halpern LM. The electrical excitability of chronically isolated cortex studied by means of permanently implanted electrodes. Electroencephalogr Clin Neurophysiol. 1962;14:244–255. doi: 10.1016/0013-4694(62)90034-2. [DOI] [PubMed] [Google Scholar]
- Sillito AM. Inhibitory processes underlying the directional specificity of simple, complex and hypercomplex cells in the cat’s visual cortex. J Physiol. 1977;271:669–720. doi: 10.1113/jphysiol.1977.sp012021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloper JJ, Johnson P, Powell TP. Selective degeneration of inter-neurons in the motor cortex of infant monkeys following controlled hypoxia: a possible cause of epilepsy. Brain Res. 1980;198:204–209. doi: 10.1016/0006-8993(80)90356-x. [DOI] [PubMed] [Google Scholar]
- Sloviter RS. Permanently altered hippocampal structure, excitability, and inhibition after experimental status epilepticus in the rat: the “dormant basket cell” hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Kisvarday ZF, Martin KA, Whitteridge D. Synaptic connections of morphologically identified and physiologically characterized large basket cells in the striate cortex of cat. Neuroscience. 1983;10:261–294. doi: 10.1016/0306-4522(83)90133-1. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Battaglia G, Arcelli P, Andermann F, Dubeau F, Palmini A, Olivier A, Villemure JG, Tampieri D, Avanzini G, Avoli M. Cortical dysplasia: an immunocytochemical study of three patients. Neurology. 1998;50:27–36. doi: 10.1212/wnl.50.1.27. [DOI] [PubMed] [Google Scholar]
- Sumner BE, Sutherland FI. Quantitative electron microscopy on the injured hypoglossal nucleus in the rat. J Neurocytol. 1973;2:315–328. doi: 10.1007/BF01104033. [DOI] [PubMed] [Google Scholar]
- Takata M. Lingually induced inhibitory postsynaptic potentials in hypoglossal motoneurons after axotomy. Brain Res. 1981;224:165–169. doi: 10.1016/0006-8993(81)91127-6. [DOI] [PubMed] [Google Scholar]
- Tamas G, Somogyi P, Buhl EH. Differentially interconnected networks of GABAergic interneurons in the visual cortex of the cat. J Neurosci. 1998;18:4255–4270. doi: 10.1523/JNEUROSCI.18-11-04255.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tani H, Bandrowski AE, Parada I, Wynn M, Huguenard JR, Prince DA, Reimer RJ. Modulation of epileptiform activity by glutamine and system A transport in a model of post-traumatic epilepsy. Neurobiol Dis. 2007;25(2):230–238. doi: 10.1016/j.nbd.2006.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR, Jarell AD, Anderson GD. Antiepileptogenic agents: how close are we? Drugs. 2001;61:1045–1055. doi: 10.2165/00003495-200161080-00002. [DOI] [PubMed] [Google Scholar]
- Wang CL, Zhang L, Zhou Y, Zhou J, Yang XJ, Duan SM, Xiong ZQ, Ding YQ. Activity-dependent development of callosal projections in the somatosensory cortex. J Neurosci. 2007;27:11334–11342. doi: 10.1523/JNEUROSCI.3380-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Westrum LE, Hendrickson AE, Wu JY. Ultrastructure of synaptic remodeling in piriform cortex of adult rats after neonatal olfactory bulb removal: an immunocytochemical study. J Comp Neurol. 1988;274:334–346. doi: 10.1002/cne.902740304. [DOI] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Spencer DD. Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol. 1999;45:92–99. doi: 10.1002/1531-8249(199901)45:1<92::aid-art15>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Wong RK, Prince DA. Dendritic mechanisms underlying penicillin-induced epileptiform activity. Science. 1979;204:1228–1231. doi: 10.1126/science.451569. [DOI] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol. 2001;85:1067–1077. doi: 10.1152/jn.2001.85.3.1067. [DOI] [PubMed] [Google Scholar]
- Yang Q, Wang S, Hamberger A, Haglid KG. Plasticity of granule cell-mossy fiber system following kainic acid induced seizures: an immunocytochemical study on neurofilament proteins. Neurosci Res. 1996;26:57–64. doi: 10.1016/0168-0102(96)01077-2. [DOI] [PubMed] [Google Scholar]
- Zhu WJ, Roper SN. Reduced inhibition in an animal model of cortical dysplasia. J Neurosci. 2000;20:8925–8931. doi: 10.1523/JNEUROSCI.20-23-08925.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]