

The involvement of the ubiquitin–proteasome pathway in the degradation of critical intracellular regulatory proteins suggested a few years ago the potential use of proteasome inhibitors as novel therapeutic agents being applicable in many different disease indications, and in particular for cancer therapy. This article reviews recent salient medicinal chemistry achievements in the design, synthesis, and biological characterization of both synthetic and natural peptide-like proteasome inhibitors, updating recent reviews on this class of agents. As shown herein, different compound classes are capable of modulating the subunit-specific proteolytic activities of the 20S proteasome in ways not previously possible, and one of them, bortezomib, has provided proof-of-concept for this therapeutic approach in cancer clinical settings.
KEY WORDS: Anticancer agents, peptidomimetics, drug design, enzyme inhibitors, irreversible inhibitors, reversible inhibitors, suicide inhibitors
Introduction
The 26S proteasome1 is a multifunctional ATP-dependent proteolytic system that is responsible for intracellular protein turnover (Goldberg et al., 1995; Hochstrasser, 1995; Coux et al., 1996; Baumeister and Lupas, 1997; Baumeister et al., 1998; Voges et al., 1999). The enzymatic activity of this protease complex occurs in a 700-kDa barrel-shaped core structure known as the 20S proteasome (Coux et al., 1996). This protein consists of four stacked rings arrayed in an α7β7β7α7 manner (Groll et al., 1997; Tanaka, 1998). Three β-subunits in each β-ring are catalytically active and have an N-terminal threonine as the active nucleophile involved in protein and peptide proteolysis (Loewe et al., 1995). The proteasome has at least three distinct peptidase activities and, by comparison with substrate specificities of known proteases, they are designated as: chymotrypsin-like (β5-subunit), trypsin-like (β2-subunit), and peptidylglutamyl-peptide hydrolytic (PGPH) activities (β1-subunit)2 (Orlowski et al., 1993). The involvement of the 20S proteasome in the ubiquitin (Ub)-dependent and Ub-independent degradation of critical regulatory proteins suggested a few years ago the potential use of proteasome inhibitors as novel therapeutic agents being applicable in many different disease indications (Elliott et al., 1999; Checler et al., 2000; Dou and Nam, 2000; Myung et al., 2001; Adams, 2002). The specific target in the search for novel cytotoxic and antiproliferative agents is the chymotrypsin-like activity of the 20S proteasome. Modulation of this enzymatic activity by β5-subunit-specific inhibitors may convey an anti-tumor effect by induction of cell cycle arrest and apoptosis in tumor cells.3
Early synthetic proteasome inhibitors were relatively nonspecific compounds but proved to be valuable molecular probes for improving our understanding of biological processes associated with the ubiquitin–proteasome pathway (Garcia-Echeverria, 2002). To overcome the inherent drawbacks of these molecular probes, new classes of inhibitors that target this proteolytic enzyme have emerged in the last few years by natural product screening, and by combining traditional drug discovery approaches with new methods to find and optimize lead structures. This article reviews recent salient medicinal chemistry achievements in the design, synthesis, and biological characterization of both synthetic and natural peptide-like proteasome inhibitors, updating recent reviews on this class of agents (Garcia-Echeverria, 2002; Delcros et al., 2003; Groll and Huber, 2004). As shown herein, different compound classes are capable of modulating the subunit-specific proteolytic activities of the 20S proteasome in ways not previously possible, and one of them, bortezomib, has provided proof-of-concept for this therapeutic approach in cancer clinical settings (Adams, 2001; Chauhan et al., 2005; Mendoza et al., 2005; Mitsiades et al., 2005; Zavrski et al., 2005).
The 20S proteasome modulators covered in this manuscript are categorized in two broad groups, covalent and non-covalent inhibitors, referring to the ability of the compound to make an adduct with the hydroxyl group of the N-terminal catalytic threonine residue within the proteasome active site.
COVALENT INHIBITORS OF THE 20S PROTEASOME
Boronic Acids and Esters
Peptide boronic acids and esters thereof that are structural analogs of enzyme substrates have been extensively employed as serine protease inhibitors, most notably as thrombin inhibitors (Seufer-Wasserthal et al., 1994; Coutts et al., 1996; Martichonok and Jones, 1996; Tian et al., 1997). It is assumed that the activity of these compounds as reversible serine protease inhibitors is due to an empty p-orbital on the boron atom that is positioned to accept the oxygen lone pair of the active site serine residue to form a stable, nearly tetrahedral complex (Bone et al., 1987; Bone et al., 1989). The unique nature of the boron/oxygen interaction is also thought to provide selectivity for serine proteases over cysteine proteases (Martichonok and Jones, 1997), owing to the weak nature of bonding between the boron and the thiol group of the catalytic cysteine of cysteine proteases. By analogy, it was presumed that C-terminal boronic “war-heads” in peptide and derivative thereof could also form stable adducts with the N-terminal threonine residues of the proteasome active sites. A variety of peptide-like boronates have been reported in the literature or covered in several patents demonstrating the ability of the boronic acid pharmacophore to modulate proteasome enzymatic activity.
Bortezomib (PS-341, LDP-341, Velcade™; ProScript, now Millennium; compound 1, Figure 1) has been the first and probably the only 20S proteasome inhibitor to enter clinical trials thus far (Chauhan et al., 2005). This dipeptide boronic acid derivative is a slow, tight-binding inhibitor of the chymotryptic activity of the 20S proteasome (Ki = 0.6 nM) that shows at least 500-fold selectivity over a series of enzymes (e.g., Ki = 2.3 μM, human leukocyte elastase; Ki = 630 nM, human cathepsin G; Ki = 320 nM, human chymotrypsin; and Ki = 13 μM, thrombin) (Adams et al., 1998, 2000). Bortezomib showed antitumor activity and prevented carcinoma metastases alone and in combination with other standard anticancer agents (e.g., fluorouracil, gemcitabine, etoposide) or radiation in a variety of hematological malignancies and solid tumor in mice models (e.g., prostate, colon and lung human xenografts). All combination therapies were generally well tolerated. In the case of multiple myeloma – the indication in which the best results have so far been observed in the clinic – the compound abrogated TNF-α induced NF-κB activation and IL-6 secretion, and decreased the binding of multiple myeloma cells to bone marrow stromal cells. The compound passed all the preclinical requirements, and a broad Phase I trial to asses its maximum tolerated dose in several tumors was initiated in 1998. Based largely on the results obtained in Study 025 (response rate was 35%, including 10% positive complete responses), the compound, which was granted fast track statues for the treatment of multiple myeloma in 2002, was approved in May 2003 by the US FDA for the treatment of patients with relapsed or refractory multiple myeloma – patients that have previously been treated with conventional chemotherapy alone or followed by high dose chemotherapy and stem cell transplant (Chauhan et al., 2005). The most clinically significant toxicity was cumulative dose-related peripheral sensory neuropathy. At this moment, ongoing clinical trials are evaluating bortezomib in a variety of cancers, including refractory metastatic colorectal cancers, non-small cell lung cancers, lymphomas and leukemias. These trials are expected to provide critical data to confirm the activity of this agent in a variety of cancers and to clarify its potential adverse effect profile further.
Fig. 1.
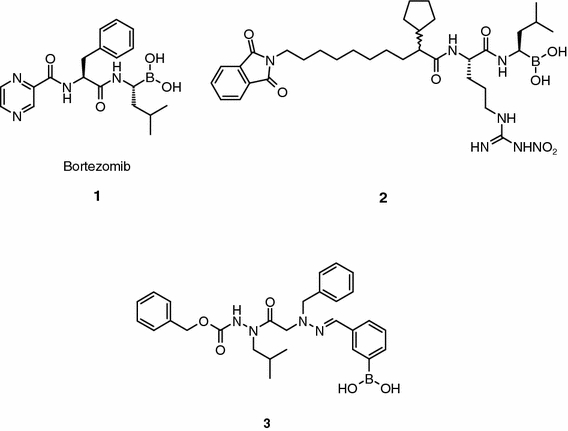
Peptide and peptoid boronic acids.
In addition to bortezomib, other proteasome inhibitors based on a boronic electrophile have been reported (Siman et al., 1999; Gardner et al., 2000). For example, significant inhibition in B16 tumor growth was observed in female C57BL mice after i.p. administration of compound 2 (Figure 1) at 10 mg Kg−1 day−1 (Siman et al., 1999). This compound is an analogue of CEP-1612 (Iqbal et al., 1995), a C-terminal aldehyde peptidomimetic (see next section).
To overcome some of the pharmaceutical limitations inherent in peptides, hydrazino-azapeptoids (Aubin et al., 2005) derivatives containing a reactive boronic acid in a phenyl group (Figure 1) were synthesized (e.g., compound 3, Figure 1; Ki = 53 μM, chymotrypsin-like activity). In spite of having the reactive “war-head” group, these peptidomimetics have in general weak activity in inhibiting the chymotrypsin-like activity of the 20S proteasome, and show modest antiproliferative activity against a panel of tumor cell lines (IC50s ranging from 1–80 μM). Although not supported with structural data, the low affinity of these boronic acid derivatives for the proteasome was explained due to steric hindrance effects, but it is at this point unclear which of the structural features of the molecule are involved in these negative interactions.
Peptide Aldehydes, α-Keto Aldehydes, and α-Keto Amides
The access to commercially available aldehydes (e.g., the calpain inhibitors Ac-Leu-Leu-norlecucinal and Ac-Leu-Leu-methional) (Wilk and Orlowski, 1983; Figueiredo-Pereira et al., 1994a) lead to the early identification and development of 20S proteasome inhibitor containing this functional group. The aldehyde forms a reversible covalent hemiacetal intermediate with the hydroxyl group of the amino terminal threonine, suggestive of the tetrahedral intermediate of a serine/cysteine protease reaction (Lowe et al., 1995).
One of the major medicinal chemistry challenges encountered with the early aldehyde derivatives was selectivity and considerable effort had to be expended in overcoming this hurdle (Stein et al., 1983; Figueiredo-Pereira et al., 1994a, 1994b; Rock et al., 1994; Iqbal et al., 1995; McCormack et al., 1998; Anma et al., 1999; Mundy, 2000). A representative example of the success achieved in this synthetic and drug design endeavor is CEP-1612 (compound 4, Figure 2). This aldehyde is a potent and reversible inhibitor of the chymotrypsin-like activity of the 20S proteasome (IC50 = 2 nM) that does not block the trypsin-like activity of this enzyme at concentrations up to 1 μM (Iqbal et al., 1995), and exhibits 50-fold selectivity against calpain and cathepsin B. CEP-1612 induces apoptosis in a panel of human tumor cell lines. Tumor cell death is p53-independent and is associated with accumulation of the cyclin-dependent kinase inhibitors p21 and p27 (An et al., 1998). Other peptide and peptoid aldehydes (compounds 5–10, Figure 2) have been described in several patent applications and publications to develop 20S proteasome subunit specific inhibitors for the treatment of cancer or other indications (e.g., inflammation, autoimmune disorders) (Stein et al., 1997; Anma et al., 1999; Loidl et al., 1999a, 1999b; Mundy, 2000; Delcros et al., 2003). Of special scientific interest, it is the 20S proteasome trypsin-like inhibitory selectivity achieved with bifunctional inhibitors (compound 8, Figure 2; IC50 = 0.5 μM, trypsin-like activity; IC50 > 100 μM, chymotrypsin) (Loidl et al., 1999a). These peptides contain a maleimido moiety to react with the side chain of cysteine-118 in the S3 binding pocket of the trypsin-like site, and a C-terminal aldehyde group for hemicacetal formation with the catalytic threonine residue. High potency against the 20S proteasome trypsin-like subunit was also obtained by linking the N-terminus of two peptide aldehydes with a polyoxyethylene spacer that is appropriate for simultaneous binding at two active sites (e.g., compound 9, Figure 2) (Loidl et al., 2000). The bivalent inhibitor is two orders of magnitude more potent than the parent peptide aldehyde (ICtrypsin-like50 = 0.071 μM for compound 9versus ICtrypsin-like50 = 6.4 μM for Ac-Arg-Val-Arg-CHO). Although compounds 8 and 9 are good examples of structure-based design, the reactivity of the maleimido group towards thiols and the high molecular weight of the bivalent inhibitors are important pharmaceutical limitations and can restrict the utility of these compounds only to cell-free systems.
Fig. 2.
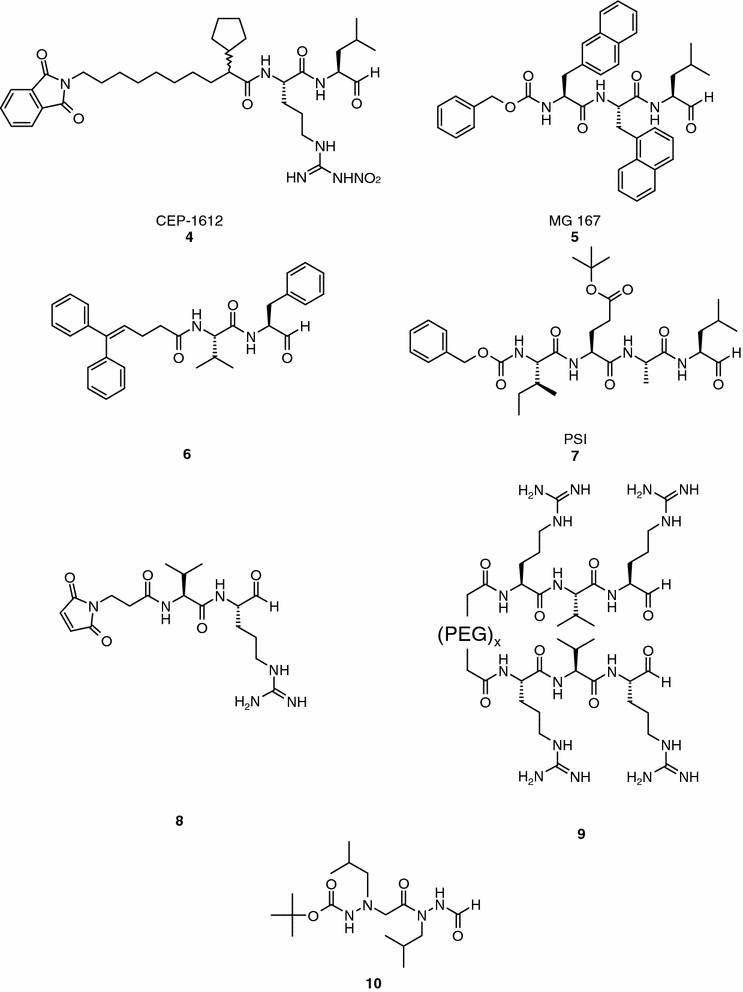
Peptide and peptoid aldehydes.
To overcome the poor stability of the aldehyde group, a number of carbonyl replacements have been developed. Thus, di- and tri-peptide α-keto aldehydes (e.g., compounds 11 and 12, Figure 3), which are slow-binding reversible inhibitors, were synthesized as putative modulators of the chymotrypsin-like activity of the 20S proteasome (Ki = 3.1 nM and 3.7 nM for 11 and 12, respectively). Along the same theme of using an α-keto group, compound 13 (Figure 3) emerged as the most potent chymotrypsin-like activity inhibitor (Ki = 1.1 nM) of a series of P’-extended α-keto amide derivatives. This compound shows good selectivity over the trypsin-like activity of the 20S proteasome and calpain 1 (>150-fold), and reduces the in vivo growth of B16-FO murine melanoma tumors (Chatterjee et al., 1999; Chatterjee and Mallamo, 1999). The α-keto amide moiety has also been introduced at the N-terminus of lipophilic peptides (compound 14, IC50 = 1 μg ml−1, bovine 20S proteasome; Figure 3), and exploited in hydrazino aza and N-aza peptoids containing trifluoromethyl, bromomethyl and cyanomethyl moieties (Delcros et al., 2003).
Fig. 3.
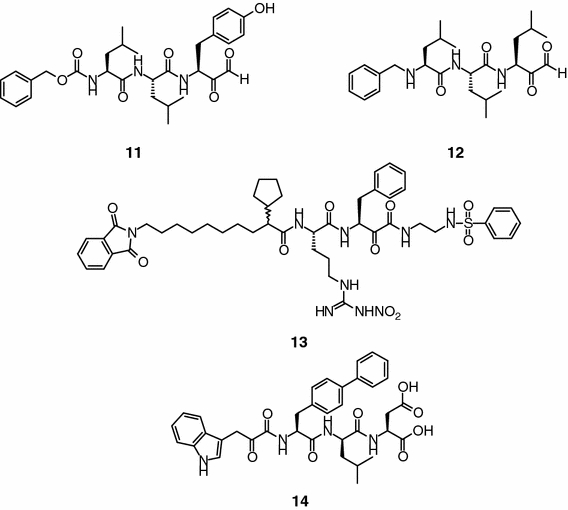
Peptide α-keto aldehydes and α-keto amides.
Peptide Vinyl Sulfones and Esters
Another class of peptide-based inhibitors exploits a vinyl sulfone moiety at the C-terminus. This group, which was originally designed to target cysteine proteases (Palmer, 1995), was expected to form an irreversible covalent adduct with the hydroxyl group of the catalytic threonine residue (Bogyo et al., 1997). Confirmation of this mechanism of action has been obtained by solving the X-ray structure of the 20S proteasome in complex with peptide vinyl sulfone inhibitors (Groll et al., 2002).
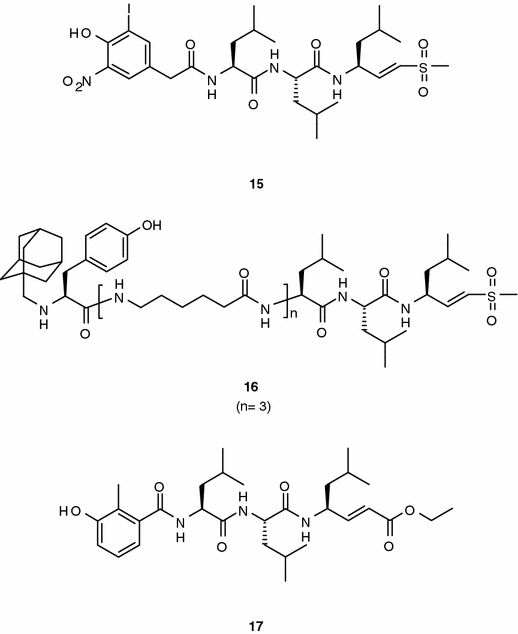
Initial attempts with tri- and tetrapeptides vinyl sulfones (compound 15; chymotrypsin-like activity IC50 = 0.1–0.5 μM; trypsin-like and PGPH activities IC50 = 50–100 μM; Figure 4) were directed to use these peptides as probes to study substrate binding and specificity of the proteasome (Bogyo et al., 1998). To this end, the peptides were labeled with 125I to trace binding to individual β-subunits by two-dimensional gels. This study revealed that the P2–P4 substrate positions are critical for directing the peptide substrates to individual catalytic sites of the proteasome. Incorporation of a number of aminohexanoic acid spacers into the (leucinyl)3-vinyl-(methyl)-sulfone core and N-terminal capping with bulky groups resulted in cell-permeable derivatives with proteasome inhibitory activity (compound 16, Figure 4) (Kessler et al., 2001).
Fig. 4.
Peptide vinyl sulfones and esters.
In addition to sulfones, the vinyl group has been used in conjunction with esters (Marastoni et al., 2005). The incorporation of the ethyl acrylate group at the C-terminus of tripeptide-based compounds has resulted in potent and selective inhibitors of the trypsin-like activity (e.g., compound 17; IC50 = 0.041 μM, trypsin-like; IC50 = 4.21 μM, chymotrypsin-like; and IC50 > 10 μM, PGPH; Figure 4). Enzymatic dilution experiments indicate irreversible inhibition of the enzyme suggesting that the ethyl acrylate group functions as a substrate of the catalytic threonine. As expected from the in vitro activity profile, the compounds showed negligible tumor cell antiproliferative activity (IC50 > 10 μM), but were able to modulate the generation and presentation of the subdomain CLG epitope in cellular settings (HLA-A2-positive RG lymphobastoid cells).
5-Methoxy-1-Indanone Dipeptide Benzamides
An alternative to the traditional electrophilic head groups used to target serine proteases is the 5-methoxy-1-indanone moiety. This group, which can be considered less reactive but hydratable, was incorporated at the N-terminus of a series of dipeptide derivatives (e.g., compound 18–20, Figure 5) (Lum et al., 1998b) to target the chymotrypsin-like activity of the 20S proteasome (IC50 = 0.20 and 0.10 μM for 18 and 19, respectively), but they show modest antiproliferative activity in cellular assays (IC50 = 8 μM, RAW cells, murine macrophage cell line) (Lum et al., 1998a, b). Inhibition of activation of NF-κB in vitro and lipopolysaccharide-induced tumor necrosis factor synthesis in RAW cells have also been described for this compounds class (Lum et al., 1998a).
Fig. 5.
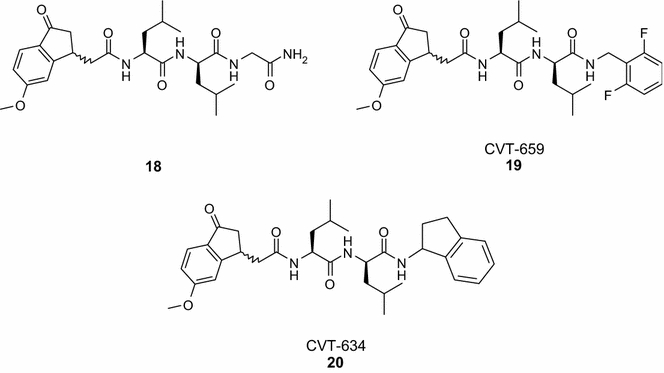
5-Methoxy-1-indanone dipeptide benzamides.
Natural Products and Derivatives Thereof
Proteasome inhibitors from natural sources have been intensively pursued both as molecular probes of the proteasome biology and as potential therapeutic agents. Representative examples of natural products that have been described as proteasome inhibitors in the past few years are briefly reported in this section.
Natural products containing an α′,β′-epoxyketone pharmacophore group and a liner peptide backbone have exhibited potent proteasome inhibitory activity in biochemical and cellular settings. Dihydroeponemycin (compound 21, Figure 6) (Sugawara et al., 1990; Oikawa et al., 1998) inhibits competitively and irreversibly the enzymatic activities of the β1, β2, and β5 subunits of the 20S proteasome, but at different rates (Meng et al., 1999); thus, inhibition of the chymotrypsin-like (kassociation = 66.4 ± 8.9 M−1 s−1) and PGPH activities (kassociation = 60.5 ± 8.8 M−1 s−1) proceeded more than 10-fold faster than inhibition of the trypsin-like activity (kassociation = 4.4 ± 0.4 M−1 s−1). Moreover, this natural product does not inhibit calpain and trypsin, although minor inhibition is observed against cathepsin B and chymotrypsin at higher concentration. Another example of a proteasome inhibitor containing an α′,β′-epoxyketone group is epoxomicin (compound 22, Figure 6) (Hanada et al., 1992), which is a natural peptide isolated from the actinomycete strain No. Q996-17. This compound potently and irreversibly inhibits the chymotrypsin-like (kobs/[I] = 37,200 M−1 s−1) activity of the 20S proteasome, and to a lesser extent the trypsin-like (kobs/[I] = 287 M−1 s−1) and PGPH activities (kobs/[I] = 34 M−1 s−1) of this protease complex. Interestingly, and unlike most other proteasome inhibitors, epoxomicin did not inhibit papain, chymotrypsin, trypsin, cathepsin B or calpain at concentrations up to 50 μM. The first insights into its mechanism of action and high selectivity for the proteasome were obtained by solving the X-ray structure of epoxomicin in complex with the yeast S. cerevisiae 20S proteasome (Groll et al., 2000). The structure revealed the presence of an unexpected morpholino ring derived from the interaction of the α′,β′-epoxyketone pharmacophore of epoxomicin with the N-terminal Thr Oγ and N atoms. The occurrence of the morpholine derived adduct is also supported by mass spectrometric analysis (Groll et al., 2000). Additional natural products containing an α′,β′-epoxyketone moiety have been obtained by culturing Streptomyces (e.g., compound 23, Figure 6), and this reactive group has also been exploited in the design and synthesis of epoxomicin-like proteasome inhibitors (compound 24–25, Figure 6) (Elofsson et al., 1999; Kim et al., 1999).
Fig. 6.
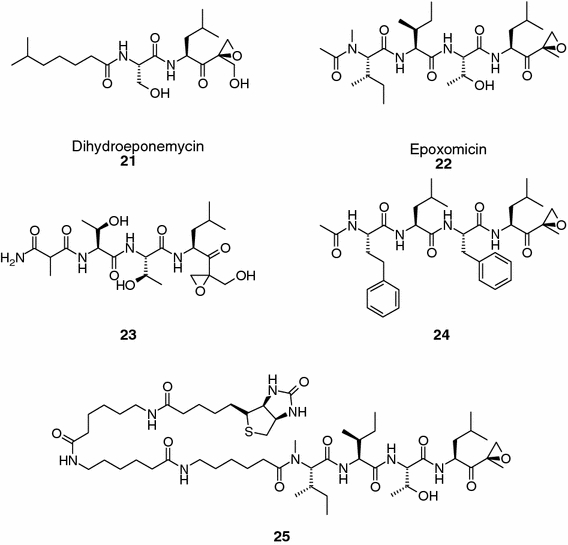
Natural and synthetic α′,β′-epoxyketone containing compounds.
Lactacystin (compound 26, Figure 7) is an irreversible, covalent inhibitor of the chymotrypsin-like and trypsin-like activities and a weak, reversible inhibitor of the PGPH activity of the 20S proteasome (Fenteany et al., 1996; Omura and Takeshima, 1996; Omura and Tomoda, 1996; Ostrowska et al., 1997). This natural product, which is a Streptomyces metabolite (Omura et al., 1991a, 1991b) undergoes in solution a spontaneous conversion – lactonization – to the active proteasome inhibitor, clasto-lactacystin β-lactone also known as omuralide (compound 27, Figure 7), which is the active compound that deactivates the 20S proteasome (Dick et al., 1996; Fenteany et al., 1996). The acylation of the N-terminal threonine β5 subunit of the 20S proteasome by the reactive clasto-lactacystin β-lactone has been confirmed by X-ray crystallography studies (Groll et al., 1997). Furthermore, in vitro experiments with radiolabelled lactacystin in whole cells have shown that almost all the radioactivity becomes associated with the proteasome, demonstrating a remarkable specificity for that entity over a very large number of other proteins. In spite of these experimental results, lactacystin is not absolutely specific for the proteasome, as it has been reported to inhibit other enzymes (Geier et al., 1999).
Fig. 7.
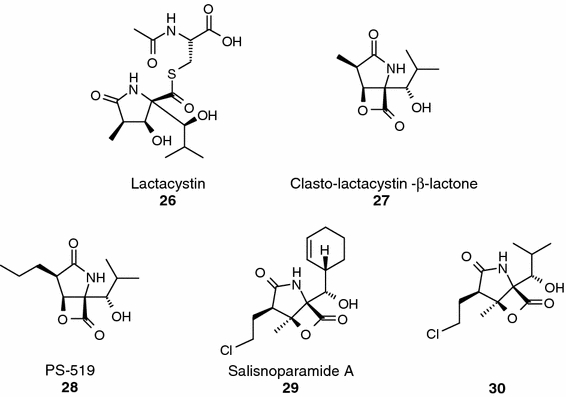
Lactacystin and β-lactones.
A substantial effort was devoted to the total synthesis of lactacystin, and the successful identification of different synthetic routes (Corey and Reichard, 1992; Corey et al., 1993; Sunazuka et al., 1993; Uno et al., 1994; Chida et al., 1995; Nagamitsu et al., 1996; Chida et al., 1997; Corey et al., 1998;) allowed the preparation of lactacystin analogs (e.g., compound 28, Figure 7) (Soucy et al., 1999) and initial studies on the molecular basis of its selectivity and potency (Corey et al., 1999). The initial SAR data revealed that the original groups at C5 and C9 seem to be optimal for proteasome inhibition and only the replacement of the 7-methyl substituent by ethyl, n-butyl or isopropyl improved 2- to 2.8-fold the chymotrypsin-like peptidase inhibitory activity of the parent compound.
A structurally related analogue of clasto-lactacystin β-lactone is salinosporamide A (NPI-0052; compound 29, Figure 7), a compound isolated from the Marine Actinomycete Salinispora tropica. This natural product is a potent and selective inhibitor of the proteasome (Feling et al., 2003) that exhibits low nM IC50 values against a broad panel of tumor cell lines (e.g., MDA-MB435 and SK-MEL28, IC50 < 11 nM) (Feling et al., 2003; Williams et al., 2005). Interestingly, salinosporamide A did show potent activity when tested in antimicrobial and antiviral assays. Cellular SAR data obtained with seven related salinosporamide analogs indicate that the β-lactone and the cloroethyl group of salinosporamide A are key pharmacophores for this compound class. Further confirmation of these initial observations was obtained by testing a series of analogues generated through fermentation or synthetic derivatization in biochemical and cellular assays (Macherla et al., 2005). Thus, marked reductions in potency, particularly in cellular settings (3 to 2-logs), accompanied the replacement of the cloroethyl group with unhalogenated alkyl substituents. The in vitro SAR data did not provide a clear understanding of the potential mechanistic role of the halogen and, probably, additional studies and the resolution of the X-ray structure of salinosporamide A complexed with the 20S proteasome will be required to clarify this interesting point.
Recently, the synthesis and biological characterization of an omuralide–salinosporin hybrid has been reported (Reddy et al., 2005). The synthetic hybrid (compound 30, Figure 7) is about 2.5 times more potent than oromulide in inhibition of the chymotrypsin-like activity of the 20S proteasome, and it is slightly less potent than salisnosporamide A.
Tyropeptin A and B (compounds 31 and 32, Figure 8) are proteasome inhibitors isolated from Kitasatospora sp. MK993-dF2 (Momose et al., 2001a, b). They inhibit the chymotrypsin-like activity and trypsin-like activities of the 20S proteasome with IC50 values of 0.1/1.5 and 0.2/4.0 μg ml−1, respectively. No inhibition of the PGPH activity is observed at concentration of 100 μg ml−1. Other enzymes (e.g., chymotrypin, cathepsin L, and m-calpain) are also inhibited by these natural peptides. To increase its potency, a structural model of tyropeptin A bound to the chymotrypsin-like activity site of the mammalian 20S proteasome was constructed. Analyses of the interactions of the inhibitor with the protein identified an open space in the vicinity of the N-terminus of tyropeptin A. The possibility to increase inhibitory activity by increasing the number of van der Waals interactions with the protein fueled the synthesis of a series of analogues containing bulky N-terminal moieties (Momose et al., 2005). This synthetic effort resulted in the identification of TP-110 (compound 33, Figure 8), which inhibits the chymotrypsin-like activity of the 20S proteasome with an IC50 value of 0.027 μM and shows an excellent selectivity over the other two catalytic sites (IC50 > 100 μM). No data have been provided so far on the anticancer activity of TP-110 or derivatives thereof.
Fig. 8.
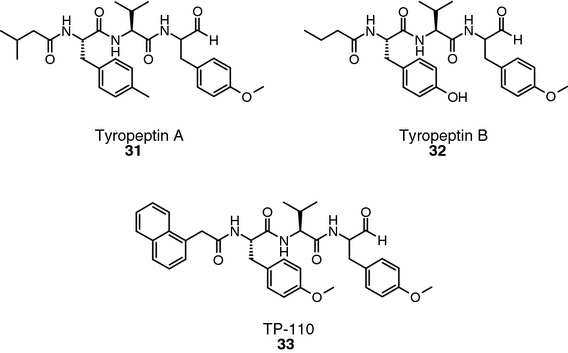
Tyropeptin A and B, and a synthetic analogue.
Belactosin A (compound 34, Figure 9) is a Streptomyces metabolite (KY11780) identified in a yeast based assay (Asai et al., 2000) that has been reported to block 20S proteasome activity in biochemical assays (IC50 = 0.4 μM, chymotrypsin-like). Structural features of belactosin A – in particular, the β-lactone and the 3-(2-aminocyclopropyl)-alanine moiety – were exploited in the design of a new class of proteasome inhibitors (e.g., KF33955; compound 35, Figure 9; IC50 = 0.04 μM, chymotrypsin) that showed in vivo activity in tumor bearing animals (T/C of 49% in nude mice implanted with WiDr tumor xenografts) (Yamaguchi et al., 2000). Recently, the enantioselective total synthesis of belactosin A, C and its homo-analogue has been reported (Larionov and de Meijere, 2004).
Fig. 9.
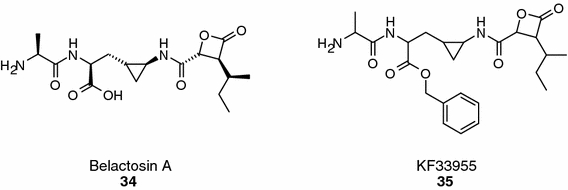
Belactosin A and a synthetic analogue.
NON-COVALENT 20S PROTEASOME INHIBITORS
Non-covalent inhibitors of the proteasome seem to have been investigated less extensively judging from the paucity of reports on this type of compounds. In principle, such inhibitors should be devoid of the inherent drawbacks often associated with the reactive groups – lack of specificity, excessive reactivity and instability – used for the establishment of the covalent interaction. This section highlights key achievement in the identification and development of 20S proteasome inhibitors that do not covalently modify the protein.
2-Aminobenzylstatine Derivatives and Synthetic Analogs
A series of 2-aminobenzylstatine derivatives, which were originally designed to target the HIV-1 protease, have shown proteasome-inhibitory activity in biochemical assays (Furet et al., 2001; García-Echeverría et al., 2001). Compound 36 (Figure 10) was identified by HTS and represented one of the first non-covalent proteasome inhibitors reported in the literature. This compound inhibits the chymotrypsin-like activity of the 20S proteasome with an IC50 value of 0.9 μM and shows good selectivity over the trypsin-like and PGPH activities (IC50 > 20 μM). The active site responsible for the chymotrypsin-like proteolytic activity of the human 20S proteasome is formed by the association of β-subunits X and HC5. Exploiting the X-ray crystal structure of the yeast proteasome, a structural model of the preceding β-subunits was constructed (Furet et al., 2001). The model served to understand the structural basis of its inhibitory activity and to guide the medicinal chemistry optimization of this class of non-covalent inhibitors. According to this model, which was supported by extensive structure-activity relationships, a large number of complementary hydrophobic and hydrogen bond interactions between the inhibitor and the S1 and S3 pockets of the enzyme may account for the ability of this class of inhibitors to inhibit selectivity of the chymotrypsin-like activity site of the human 20S proteasome without forming an adduct with the catalytic threonine residue. The homology model in conjunction with a modular chemistry strategy was used to improve the inhibitory activity of the original hit (García-Echeverría et al., 2001; Furet et al., 2002). A major boost in activity was obtained by improving the interactions of the benzylamino group of the inhibitor with the side chains of Ser 157 and Tyr 135 in the S3 pocket of the enzyme. Compound 37 (Figure 10) that can form the targeted hydrogen bond interactions with the preceding residues inhibits the chymotrypsin-like activity of the human 20S proteasome with an IC50 value of 0.05 μM. Parallel to the optimization of the central motif, the possibility of enhancing potency by creating additional van der Waals interactions with a small cavity accessory pocket near the S3 binding site was explored by modifying the N-terminus of compound 37. The replacement of the original benzyloxycarbonyl group by naphthalene-1-yl-acetic acid improved substantially the inhibitory potency (IC50 = 0.007 μM) of the new peptidomimetic (compound 38, Figure 10). The chemical modifications introduced in the original hit did not affect the specificity profile of this class of inhibitors. Thus, compound 38 has a modest activity against the trypsin-like and PGPH activities of the 20S proteasome (IC50 > 20 μM). However, and despite of the potency and selectivity observed in biochemical assays, compound 38 did not show good activity in cellular settings (e.g., IC50 = 1.5 μM, inhibition of proliferation of MDA-MB-435 cells). Assuming that poor cellular permeability was the main reason for the modest cellular activity of these 2-aminobenzylstatine derivates, the SAR data generated during the optimization of this compound class and the binding model were exploited to design an alternative inhibitor scaffold with reduced size and attenuated peptidic character (Furet et al., 2004). The starting hypothesis was that the two amide bonds flanking the tert-leucine amino acid and the statine 4-benzyl group have no significant interactions with the enzyme. A representative example of this design strategy is compound 39 (Figure 10). In this new scaffold, the non-productive structural features have been removed and the critical interactions of the 2-aminobenzylstatine inhibitors have been incorporated or mimicked. Thus, the two amide bonds flanking the valine residue and the C-terminal benzylamide group filling the S1 pocket are conserved whereas the statine moiety is replaced by a non-proteinogenic residue -3,4,5-trimethoxy-l-phenylalinine-, which is able to mimic the interactions with the S3 pocket. The tert-leucine side chain and the hydrophobic N-terminal group are mimicked by a single phenoxy substituted benzylic synthon. Overall, the high potency and selectivity of compound 38 were matched by compound 39 in biochemical assays: IC50 = 15 nM, chymotrypsin-like activity and IC50 > 20 μM, trypsin-like and PGPH proteolytic activities. However, and in contrast to compound 38, compound 39 also showed good activity in cellular settings. Thus, it blocks proteasome activity in cultured cells (IC50 = 20 nM) and inhibits in a dose-dependent manner the proliferation of different cell lines (e.g., IC50 = 60 nM, MDA-MB-435 cells).
Fig. 10.
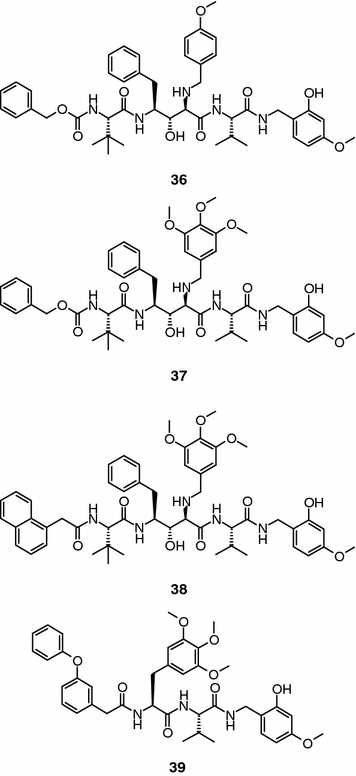
2-Aminobenzylstatine derivatives and a de novo design inhibitor.
HIV-1 Inhibitors as Proteasome Modulators
Ritonavir (compound 40, Figure 11) is a potent inhibitor of the human immunodeficiency virus (HIV)-1 protease and is clinically applied to suppress HIV-1 replication in AIDS patients. In addition to its biological activity against its original target, it inhibits in vitro the chymotrypsin-like activity of the 20S proteasome while the trypsin-like activity is markedly enhanced (Andre et al., 1998; Schmidtke et al., 1999). Kinetic studies revealed that the effect on the trypsin-like enzymatic activity is due to binding of ritonavir to a non-catalytic modifier site (Schmidtke et al., 1999). Apart from ritonavir, saquinavir also inhibits the chymotrypsin-like activity of the 20S proteasome although much less efficiently. Other HIV-1 inhibitors like indinavir or nelfinavir do not affect 20S proteasome activity (Andre et al., 1998).
Fig. 11.
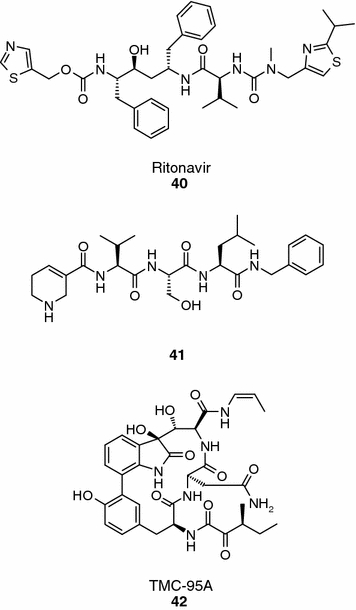
Ritonavir, arecoline tripeptide inhibitor and TMC-95A.
Arecoline Tripeptides
Although originally designed as potential covalent inhibitors, arecoline tripeptides seem to inhibit the human proteasome in biochemical assays (e.g., compound 41, Figure 11; IC50 = 0.52 μM, chymotrypsin-like site) without forming an adduct with the protein (Marastoni et al., 2004). The arecoline moiety, which could act as a Michael substrate for the catalytic threonine, did not improve the inhibitory activity of reference compounds when introduced at the C-terminus, but proved to be beneficial when incorporated at the N-terminus of selected sequences. The active arecoline peptides inhibit 20S proteasome in cellular settings with activities similar to the ones obtained in the biochemical assays, and have good stability in culture media (t1/2 > 360 min).
Macrocycle Peptides
A series of 17-membered macrocycle peptides (TMC-95A, -B, -C, and -D) isolated from the fermentation broth of Apiospora montagnei Sacc. TC1093 have been shown to potently inhibit the proteasome in biochemical assays (e.g., TMC-95A, compound 43, IC50 = 5.4, 200 and 60 nM for the chymotrypsin-like, trypsin-like and PGPH activities, respectively; Figure 11) (Koguchi et al., 2000). The biological profile of TMC-95A elicited a great interest in the scientific community and several groups have reported different synthetic approaches to prepare this natural product and synthetic analogues thereof (Lin and Danishefsky, 2002; Berthelot et al., 2003; Inoue et al., 2003). The X-ray structure of the yeast proteasome in complex with TMC-95A revealed multiple hydrogen-bond interactions between the inhibitor and the protein that, in the absence of a covalent bond, may contribute to the high-affinity binding with the protein. In the structure, the n-propyline group protudes into the S1 pocket and the side chain of asparagine is inserted deeply into the S3 pocket. This last interaction is believed to play a major role in the selectivity of TMC-95A over other enzymes (e.g., IC50 > 30 μM for m-calpain, cathepsin L and trypsin). Furthermore, synthetic activities around the TMC-95A skeleton have shown that not all of the elements of the natural product are required for proteasome inhibition (Kaiser et al., 2002), and that simpler compounds with improved affinity and selectivity could be identified and developed.
Allosteric Inhibitors
Atomic force microscopy and in vitro 26S proteasome assembly experiments have shown that PR39, a natural 39-mer antibacte9rial peptide (RRRPRPPYLPRPRPPPFFPPRLPPRIPPGFPPRFPPRFP), interferes with the conformation of both 20S and 26S proteasomes leading to inhibition of its peptidase activities (e.g., Ki = 25 nM, chymotrypsin-like activity) (Gaczynska et al., 2003). Although the possibility of contacts with sites on the 19S particles are not ruled out, it seems that the N-terminal arginine residues of this non-competitive and reversible inhibitor interacts with the acidic tail of the non-catalytic subunit α7 perturbing the “normal” conformational dynamics of the enzyme. Biological evaluation of truncated peptides of PR39 and alanine scans identified RRRPRPPYLPR as the shortest functional sequence.
CONCLUSIONS AND OUTLOOK
As shown in the preceding sections, a variety of synthetic and natural inhibitors are capable of modulating the subunit-specific enzymatic activities of the 20S proteasome complex in ways not previously possible. Retrospectively, it seems that the transition from the original peptide-like molecules to potential drug candidates has proved rather difficult and has been meet with limited success. Even if some of the new 20S proteasome modulators have both in vitro and in vivo activity against a variety of cancer cell lines and tumor xenografts in preclinical studies (Adams, 2004), only one 20S proteasome inhibitor has so far entered clinical trials and received approval by the health authorities for the treatment of patients with relapsed and refractory multiple myeloma. Overall, we can conclude from the current preclinical and clinical information that the introduction of proteasome inhibitors as potential anticancer drugs is still in an early stage. Definitely, the clinical efficacy of 20S proteasome modulators in other malignancies besides multiple myeloma is eagerly awaited.
Notes
The 26S proteasome is formed by the 20S proteasome capped by another multimeric component called the 19S complex or PA700.
This catalytic site is also termed post-acidic or caspase-like activity.
It has been shown that malignant cells are more susceptible to the pro-apoptotic effects of proteasome inhibition than normal cells.
Abbreviations
Amino acid and peptide nomenclature conforms to IUPAC-IUB guidelines [Eur. J. Biochem. 138 (1984) 9].
- FDA
Food and Drug Administration
- IL-6
interleukin-6
- NF-κB
nuclear transcription factor-κB
- PGPH-activity
peptidylglutamyl-peptide hydrolytic-activity
- Ub
ubiquitin
- TNF
tumor necrosis factor
References
- Adams J. 2001, Semin. Oncol. 28: 613–619 [DOI] [PubMed]
- Adams J. 2002, Trends Mol. Med. 8: S49–S54 [DOI] [PubMed]
- Adams, J.: 2004, in Proteasome Inhibitors in Cancer Therapy, Humana Press
- Adams J., Behnke M., Chen S., Cruickshank A. A., Dick L. R., Grenier L., Klunder J. M., Ma Y. T., Plamondon L., Stein R. L. 1998, Bioorg. Med. Chem. Lett. 8: 333–338 [DOI] [PubMed]
- Adams, J., Ma, Y. T., Stein, R., Baevsky, M., Grenier, L. and Plamondon, L.: 2000, US 6,083,903
- An B., Goldfarb R. H., Siman R., Dou Q. P. 1998 Cell Death Diff. 5: 1062–1075 [DOI] [PubMed]
- Andre P., Groettrup M., Klenerman P., De Giuli R., Booth B. L., Jr., Cerundolo V., Bonneville M., Jotereau F., Zinkernagel R. M., Lotteau V. 1998, Proc. Natl. Acad. Sci. USA 95: 13120–13124 [DOI] [PMC free article] [PubMed]
- Anma, T., Yamada, T., Miyashita, H. and Sauta, T.: 1999, JP 11292833
- Asai A., Hasegawa A., Ochiai K., Yamashita Y., Mizukami T. 2000, J. Antibiot. 53: 81–83 [DOI] [PubMed]
- Aubin S., Martin B., Delcros J.-G., Arlot-Bonnermains Y., Baudy-Floch M. 2005, J. Med. Chem. 48: 330–334 [DOI] [PubMed]
- Baumeister W., Walz J., Zühl F., Seemüller E. 1998 Cell 92: 367–380 [DOI] [PubMed]
- Baumeister W., Lupas A. 1997, Curr. Opin. Struct. Biol. 7: 273–278 [DOI] [PubMed]
- Berthelot A., Piguel S., Le Dour G., Vidal J. 2003 J. Org. Chem. 68: 9835–9838 [DOI] [PubMed]
- Bogyo M., McMaster J. S., Gaczynska M., Tortorella D., Goldberg A. L., Ploegh H. 1997, Proc. Natl. Acad. Sci. USA 94: 6629–6634 [DOI] [PMC free article] [PubMed]
- Bogyo M., Shin S., McMaster J. S., Ploegh H. L. 1998 Chem. Biol. 5: 307–320 [DOI] [PubMed]
- Bone R., Frank D., Kettner C. A., Agard D. A. 1989 Biochemistry 28: 7600–7609 [DOI] [PubMed]
- Bone R., Shenvi A. B., Kettner C. A., Agard D. A. 1987 Biochemistry 26: 7609–7614 [DOI] [PubMed]
- Chatterjee S., Dunn D., Mallya S., Ator M. A. 1999 Bioorg. Med. Chem. Lett. 9: 2603–2606 [DOI] [PubMed]
- Chatterjee, S. and Mallamo, J. P.: 1999, WO 9917778
- Chauhan D., Hideshima T., Mitsiades C., Richardson P., Anderson K. C. 2005 Mol. Cancer Ther. 4: 686–692 [DOI] [PubMed]
- Checler F., da Costa C. A., Ancolio K., Chevallier N., Lopez-Perez E., Marambaud P. 2000 Biochim. Biophys. Acta 1502: 133–138 [DOI] [PubMed]
- Chida N., Takeoka J., Ando K., Tsutsumi N., Ogawa S. 1997, Tetrahedron 53: 16287–16298
- Chida N., Takeoka J., Tsutsumi N., Ogawa S. 1995 J. Chem. Soc. Chem. Commun. 7: 793–794
- Corey E. J., Li W. D., Nagamitsu T., Fenteany G. 1999 Tetrahedron 55: 3305–3316
- Corey E. J., Li W., Reichard G. A. 1998 J. Am. Chem. Soc. 120: 2330–2336
- Corey E. J., Reichard G. A. 1992 J. Am. Chem. Soc. 114: 10677–10678
- Corey E. J., Reichard G. A., Kania R. 1993 Tetrahedron Lett. 34: 6977–6980
- Coutts S. J., Kelly T. A., Snow R. J., Kennedy C. A., Barton R. W., Adams J., Krolikowski D. A., Freeman D. M., Campbell S. J. 1996 J. Med. Chem. 39: 2087–2094 [DOI] [PubMed]
- Coux O., Tanaka K., Goldberg A. L. 1996 Ann. Rev. Biochem. 65: 801–847 [DOI] [PubMed]
- Delcros J. G., Floc M. B., Prigent C., Arlot-Bonnermains Y., 2003 Curr. Med. Chem. 10: 479–503 [DOI] [PubMed]
- Dick L. R., Cruikshank A. A., Grenier L., Melandri F. D., Nunes S. L., Stein R. L. 1996 J. Biol. Chem. 271: 7273–7276 [DOI] [PubMed]
- Dou Q. P., Nam S. 2000 Expert. Opin. Ther. Pat. 10: 1263–1272
- Elliott P. J., Pien C. S., McCormack T. A., Chapman I. D., Adams J. 1999, J. Allergy Clin. Immunol. 104: 294–300 [DOI] [PubMed]
- Elofsson M., Splittgerber U., Myung J., Mohan R., Crews C. M., 1999 Chem. Biol. 6: 811–822 [DOI] [PubMed]
- Feling R. H., Buchanan G. O., Mincer T. J., Kauffman C. A., Jensen P. R., Fenical W., 2003 Angew. Chem., Int. Ed. 42: 355–357 [DOI] [PubMed]
- Fenteany G., Standaert R. F., Lane W. S., Choi S., Corey E. J., Schreiber S. L. 1996, Science 268: 726–731 [DOI] [PubMed]
- Figueiredo-Pereira M. E., Banik N., Wilk S. 1994a J. Neurochem. 62: 1989–1994 [DOI] [PubMed]
- Figueiredo-Pereira M. E., Berg K. A., Wilk S. 1994b J. Neurochem. 63: 1578–1581 [DOI] [PubMed]
- Furet P., Imbach P., Fürst P., Lang M., Noorani M., Zimmermann J., García-Echeverría C., 2001 Bioorg. Med. Chem. Lett. 11: 1321–1324 [DOI] [PubMed]
- Furet P., Imbach P., Fürst P., Lang M., Noorani M., Zimmermann J., García-Echeverría C. 2002 Bioorg. Med. Chem. Lett. 12: 1331–1334 [DOI] [PubMed]
- Furet P., Imbach P., Noorani M., Koeppler J., Laumen K., Lang M., Guagnano V., Fürst P., Roesel J., Zimmermann J., García-Echeverría C. 2004 J. Med. Chem. 47: 4810–4813 [DOI] [PubMed]
- Gaczynska M., Osmulski P. A., Gao Y., Post M. J., Simons M. 2003, Biochemistry 42: 8663–8670 [DOI] [PubMed]
- García-Echeverría C., Imbach P., France D., Fürst P., Lang M., Noorani M., Scholz D., Zimmermann J., Furet P. 2001 Bioorg. Med. Chem. Lett. 11: 1317–1319 [DOI] [PubMed]
- García-Echeverría C. 2002 Mini-Rev. Med. Chem. 2: 247–259 [DOI] [PubMed]
- Gardner R. C., Assinder S. J., Christie G., Mason G. G. F., Markwell R., Wadsworth H., McLaughlin M., King R., Chabot-Fletcher M. C., Breton J. J., Allsop D., Rivett A. J. 2000 Biochem. J. 346: 447–454 [PMC free article] [PubMed]
- Geier E., Pfeifer G., Wilm M., Lucchiari-Hartz M., Baumeister W., Eichmann K., Niedermann G. 1999 Science 283: 978–981 [DOI] [PubMed]
- Goldberg A. L., Stein R., Adams J. 1995 Chem. Biol. 2: 503–508 [DOI] [PubMed]
- Groll M., Nazif T., Huber R., Bogyo M 2002 Chem. Biol. 9: 655–662 [DOI] [PubMed]
- Groll M., Ditzel L., Loewe J., Stock D., Bochtler M., Bartunik H. D., Huber R. 1997 Nature 386: 463–471 [DOI] [PubMed]
- Groll M., Huber R., 2004 Biochem. Biophys. Acta. 1695: 33–44 [DOI] [PubMed]
- Groll M., Kim K. B., Kairies N., Huber R., Crews C. M. 2000, J. Am. Chem. Soc. 122: 1237–1238
- Hanada M., Sugawara K., Kaneta K., Toda S., Nishiyama Y., Tomita K., Yamamoto H., Konishi M., Oki T. 1992 J. Antibiot. 45: 1746–1752 [DOI] [PubMed]
- Hochstrasser M., 1995, Curr. Opin. Cell Biol. 7: 215–223 [DOI] [PubMed]
- Inoue M., Sakzaki H., Furuyama H., Hirama M. 2003 Angew. Chem. Int. Ed. 42: 2654–2657 [DOI] [PubMed]
- Iqbal M., Chatterjee S., Kauer J. C., Das M., Messina P., Freed B., Biazzo W., Siman R., 1995 J. Med. Chem. 38: 2276–2277 [DOI] [PubMed]
- Kaiser M., Groll M., Renner C., Huber R., Moroder L. 2002 Angew. Chem. Int. Ed. 41: 780–782 [DOI] [PubMed]
- Kessler, B. M., Tortorella, D., Altun, M., Kisselev, A. F., Fiebiger, E., Hekking, B. G., Ploegh, H. L. and Overkleeft, H. S.: 2001, Chem. Biol. 913–929 [DOI] [PubMed]
- Kim K. B., Myung J., Sin N., Crews C. M. 1999, Bioorg. Med. Chem. Lett. 9: 3335–3340 [DOI] [PubMed]
- Koguchi Y., Kohno J., Nishio M., Takahashi K., Okuda T., Ohnuki T., Komatsubara S. 2000, J. Antibiot. 53: 105–109 [DOI] [PubMed]
- Larionov O. V., de Meijere A. 2004 Org. Lett. 6: 2153–2156 [DOI] [PubMed]
- Lin S., Danishefsky S. 2002 Angew. Chem. Int. Ed. 41: 512–515 [DOI] [PubMed]
- Loewe J., Stock D., Jap B., Zwickl P., Baumeister W., Huber R. 1995 Science 268: 533–539 [DOI] [PubMed]
- Loidl G., Groll M., Musiol H. J., Ditzel L., Huber R., Moroder L. 1999a Chem. Biol. 6: 197–204 [DOI] [PubMed]
- Loidl G., Groll M., Musiol H. J., Huber R., Moroder L. 1999b Proc. Natl. Acad. Sci. USA 96: 5418–5422 [DOI] [PMC free article] [PubMed]
- Loidl G., Musiol H. J., Groll M., Huber R., Moroder L. 2000 J. Pept. Sci. 6: 36–46 [DOI] [PubMed]
- Lowe J., Stock D., Jap B., Zwickl P., Baumeister W., Huber R. 1995 Science 268: 533–539 [DOI] [PubMed]
- Lum R. T., Kerwar S. S., Meyer S. M., Nelson M. G., Schow S. R., Shiffman D., Wick M. M., Joly A. 1998a Biochem. Pharmacol. 55: 1391–1397 [DOI] [PubMed]
- Lum R. T., Nelson M. G., Joly A., Horsma A. G., Lee G., Meyer S. M., Wick M. M., Schow S. R. 1998b Bioorg. Med. Chem. Lett. 8: 209–214 [DOI] [PubMed]
- Macherla V. R., Mitchell S. S., Manam R. R., Reed K. A., Chao T. H., Nicholson B., Deyanat-Yazdi G., Mai B., Jensen P. R., Fenical W. F., Neuteboom S. T. C., Lam K. S., Palladino M. A., Potts B. C. M. 2005 J. Med. Chem. 48: 3684–3687 [DOI] [PubMed]
- Marastoni M., Baldisserotto A., Canella A., Gavioli R., De Risi C., Pollini G. P., Tomatis R. 2004 J. Med. Chem. 47: 1587–1590 [DOI] [PubMed]
- Marastoni M., Baldisserotto A., Cellini S., Gavioli R., Tomatis R. 2005 J. Med. Chem. 48: 5038–5042 [DOI] [PubMed]
- Martichonok V., Jones J. B. 1996 J. Am. Chem. Soc. 118: 950–958
- Martichonok V., Jones J. B. 1997 Bioorg. Med. Chem. 5: 679–684 [DOI] [PubMed]
- McCormack T. A., Cruikshank A. A., Grenier L., Melandri F. D., Nunes S. L., Plamondon L., Stein R. L., Dick L. R. 1998 Biochemistry 37: 7792–7800 [DOI] [PubMed]
- Mendoza F. J., Espino P. S., Cann K. L., Bristow N., McCrea K., Los M. 2005 Archivum Immunologiae et Therapiae Experimentalis 53: 47–60 [PubMed]
- Meng L., Kwok B. H. B., Sin N., Crews C. M. 1999 Cancer Res. 59: 2798–2801 [PubMed]
- Mitsiades C., Mitsiades N., Hideshima T., Richardson P. G., Anderson K. C. 2005 Exp. Rev. Anticancer Ther. 5: 465–476 [DOI] [PubMed]
- Momose I., Sekizawa R., Hashizume H., Kinoshita N., Homma Y., Hamada M., Iinuma H., Takeuchi T. 2001a J. Antibiot. 54: 997–1003 [DOI] [PubMed]
- Momose I., Sekizawa R., Hirosawa S., Ikeda D., Naganawa H., Iinuma H., Takeuchi T. 2001b J. Antibiot. 54: 1004–1012 [DOI] [PubMed]
- Momose I., Umezawa Y., Hirosawa S., Iinuma H., Ikeda D. 2005 Bioorg. Med. Chem. 15: 1867–1871 [DOI] [PubMed]
- Mundy, G. R.: 2000, WO 0061167
- Myung J., Kim K. B., Crews C. M. 2001 Med. Res. Rev. 21: 245–273 [DOI] [PMC free article] [PubMed]
- Nagamitsu T., Sunazuka T., Tanaka H., Omura S., Sprengeler P. A., Smith A. B. III, 1996 J. Am. Chem. Soc. 118: 3584–3590
- Oikawa T., Sasaki T., Nakamura M., Shimamura M., Tanahashi N., Omura S., Tanaka K. 1998, Biochem. Biophys. Res. Commun. 246: 243–248 [DOI] [PubMed]
- Omura S., Fujimoto T., Otoguro K., Matsuzaki K., Moriguchi R., Tanaka H., Sasaki Y. 1991a J. Antibiot. 44: 113–116 [DOI] [PubMed]
- Omura S., Matsuzaki K., Fujimoto T., Kosuge K., Furuya T., Fujita S., Nakagawa A. 1991b J. Antibiot. 44: 117–118 [DOI] [PubMed]
- Omura S., Takeshima H. 1996 Tanpakushitsu Kakusan Koso 41: 327–336 [PubMed]
- Omura S., Tomoda H. 1996 Saibo Kogaku 15: 929–940
- Orlowski M., Cardozo C., Michaud C. 1993 Biochemistry 32: 1563–1572 [DOI] [PubMed]
- Ostrowska H., Wojcik C., Omura S., Worowski K. 1997 Biochem. Biophys. Res. Commun. 234: 729–732 [DOI] [PubMed]
- Palmer J. T. 1995 J. Med. Chem. 38: 3193–3196 [DOI] [PubMed]
- Reddy L. R., Fournier J.-F., Subba R. B. V., Corey E. J. 2005 J. Am. Chem. Soc. 127: 8974–8976 [DOI] [PubMed]
- Rock K. L., Gramm C., Rothstein L., Clark K., Stein R., Dick L., Hwang D., Goldberg A. L. 1994 Cell 78: 761–771 [DOI] [PubMed]
- Schmidtke G., Holzhutter H. G., Bogyo M., Kairies N., Groll M., De Giuli R., Emch S., Groettrup M. 1999 J. Biol. Chem. 274: 35734–35740 [DOI] [PubMed]
- Seufer-Wasserthal P., Martichonok V., Keller T. H., Chin B., Martin R., Jones J. B. 1994 Bioorg. Med. Chem. 2: 35–48 [DOI] [PubMed]
- Siman, R., Jani, J., Goldfarb, R. and Dou, Q.: 1999, WO 9930707
- Soucy F., Grenier L., Behnke M. L., Destree A. T., McCormack T. A., Adams J., Plamondon L. 1999, J. Am. Chem. Soc. 121: 9967–9976
- Stein, R. L., Ma, Y. T. and Brand, S.: 1997, US 5,693,617
- Sugawara K., Hatori M., Nishiyama Y., Tomita K., Kamei H., Konishi M., Oki T. 1990 J. Antibiot. 43: 8–18 [DOI] [PubMed]
- Sunazuka T., Nagamitsu T., Matsuzaki K., Tanaka H., Omura S., Smith A. B. III, 1993 J. Am. Chem. Soc. 115: 5302
- Tanaka K. 1998 J. Biochem. 123: 195–204 [DOI] [PubMed]
- Tian Z. Q., Brown B. B., Mack D. P., Hutton C. A., Bartlett P. 1997 J. Org. Chem. 62: 514–522 [DOI] [PubMed]
- Uno H., Baldwin J. E., Russell A. T. 1994 J. Am. Chem. Soc. 116: 2139–2140
- Voges D., Zwickl P., Baumeister W. 1999 Annu. Rev. Biochem. 68: 1015–1068 [DOI] [PubMed]
- Wilk S., Orlowski M. 1983 J. Neurochem. 40: 842–849 [DOI] [PubMed]
- Williams, P. G., Buchanan, G. O., Feling, R. H., Kauffman, C. A., Jensen, P. R. and Fenical, W.: 2005, J. Org. Chem. 6193–6203 [DOI] [PubMed]
- Yamaguchi, H., Asai, A., Mizukami, T., Yamashita, Y., Akinaga, S., Ikeda, S. and Kanda, Y.: 2000, WO 0043000
- Zavrski I., Jakob C., Possinger K., Sezer O. 2005 Anti-Cancer Drugs 16: 475–481 [DOI] [PubMed]