

Abstract
To identify the structural elements of the prion protein (PrP) necessary for its protective function against Bax, we performed structure-function analyses of the anti-Bax function of cytosolic PrP (CyPrP) in MCF-7 cells. Deletions of 1, 2, or 3 N-terminal BH2-like octapeptide repeats (BORs), but not deletion of all 4 BORs, abolish CyPrP’s anti-Bax function. Deletion of α-helix 3 (PrP23-199) or further C-terminal deletions of α-helix 1 and 2, and β-strand 1 and 2 (PrP23-172, PrP23-160, PrP23-143, PrP23-127) eliminates CyPrP’s protection against Bax-mediated cell death. The substitution of helix 3 amino acid residues K204, V210, and E219 by proline inhibits the anti-Bax function of CyPrP. The substitution of K204, but not V210 and E219, by alanine residues also prevents CyPrP’s anti-Bax function. Expression of PrP’s helix 3 displays anti-Bax activity in MCF-7 cells and in human neurons. Together, these results indicate that although the BOR domain has an influence on PrP’s anti-Bax function, the helix 3 is necessary and sufficient for the anti-Bax function of CyPrP. Identification of helix 3 as the structural element for the anti-Bax function thus provides a molecular target to modulate PrP’s anti-Bax function in cancer and neurodegeneration.
Keywords: Prion protein, Bax, helix 3, neuron, MCF-7 cells, structure-function
INTRODUCTION
While the role of prion protein (PrP) in transmissible prion diseases is highly investigated, less is known on the physiological function of the normal cellular PrP. PrP is a highly expressed glycoprotein in brain and peripheral tissues (Prusiner et al. 1998). Most of the PrP is secreted and accumulates on the cell surface as a glycosylphosphatidylinositol (GPI)-anchored protein. However, it was discovered that some PrP is also retrotranslocated in the cytosol where it accumulates in a subset of neurons in the hippocampus, neocortex, and thalamus (Zanusso et al. 1999; Ma and Lindquist 2001; Yedidia et al. 2001; Mironov et al. 2003; Roucou et al. 2003). PrP promotes neuroprotection in vivo and in vitro. PrP null mice are more susceptible to ischemia, Doppel and N-terminally truncated PrP-induced cytotoxicity (Shmerling et al. 1998; Moore et al. 2001; Mitteregger et al. 2007). Furthermore, PrP null mice are more susceptible to axotomy-induced cell death and kainic acid-induced seizures (Walz et al. 1999; Coulpier et al. 2006). In vitro, PrP protects against oxidative stress (Brown et al. 1997a; Brown et al. 1999), serum deprivation (Kuwahara et al. 1999), and anisomycin-induced cell death (Chiarini et al. 2002; Zanata et al. 2002). PrP renders human breast carcinoma MCF-7 cells resistant to tumor necrosis factor α (Diarra-Mehrpour et al. 2004). Down-regulation of PrP increases sensitivity to adriamycin in adriamycin-resistant MCF-7 cells (Meslin et al. 2007a). Accordingly, breast tumors resistant to adjuvant chemotherapy express higher levels of PrP (Meslin et al. 2007b). Moreover, PrP is overexpressed and promotes cell survival in gastric cancer cells (Liang et al. 2006).
At the molecular level, PrP inhibits Bax-mediated cell death in human primary neurons, human differentiated neuronal NT2 teratocarcinoma cells, Saccharomyces Cerevisiae, and MCF-7 cells (Bounhar et al. 2001; Roucou et al. 2004; Li and Harris 2005; Roucou et al. 2005; Bounhar et al. 2006). The anti-Bax function of PrP is physiological since PrP antisense constructs promote susceptibility to Bax-mediated cell death in human primary neurons (Bounhar et al. 2001). In human neurons and MCF-7 cells, PrP prevents the initial conformational change of Bax resulting in Bax translocation to the mitochondrial membrane, cytochrome c release and cell death. PrP is thus considered a bona fide Bax inhibitor (Roucou et al. 2005). The anti-apoptotic effect of PrP is specific to Bax since PrP does not inhibit Bak and Bid (Roucou et al. 2005). Despite being Bax-specific, PrP does not interact directly with Bax (Lin et al. 2008). This suggests that PrP requires an intermediate to carry out its anti-Bax function. This intermediate is most likely not a Bcl-2 protein because PrP can protect against Bax-mediated cell death in yeast, which are genetically deficient for Bcl-2 gene family members (Li and Harris 2005; Bounhar et al. 2006). Yet the predominant anti-Bax form of PrP is the retrotranslocated cytosolic PrP (CyPrP) and not the more abundant cell surface GPI-anchored PrP (Lin et al. 2008).
Given the role of PrP in neuroprotection or survival of breast cancer cells, understanding the functional region or domain of PrP that is responsible for the anti-Bax function could provide a therapeutic target against neurodegenerative diseases and drug resistance in cancer cells. Structure-function analyses are often useful in determining underlying molecular mechanisms of protein action through the identification of regulatory or functional domains. The structural domains susceptible to be involved in the anti-Bax function of PrP include the octapeptide repeat (OR) region (residues 51-91) and the globular C-terminal region of PrP. The ORs are implicated in copper binding (Brown et al. 1997b; Viles et al. 1999), as well as protection against oxidative stress (Rachidi et al. 2003; Dupiereux et al. 2007) and Doppel neurotoxicity (Drisaldi et al. 2004). Interestingly, embedded in this motif are four other octarepeats (residues 56-87) that show similarity to the Bcl-2 homology domain 2 (BH2 domain) of the anti-apoptotic Bcl-2 protein (LeBlanc 1998). In Bcl-2, the BH2 domain is required for protection against Bax-mediated cell death (Yin et al. 1994) as are these BH2-like octapeptide repeats (BORs) since their deletion abolishes the anti-Bax function of PrP in human primary neurons (Bounhar et al. 2001). Moreover, both Bcl-2 and PrP are able to rescue PrP null hippocampal cell lines against serum deprivation (Kuwahara et al. 1999) and PrP null mice against N-truncated PrP-mediated toxicity (Nicolas et al. 2007). A second potential anti-Bax domain of PrP is its structured C-terminal region. This globular domain contains two β-strands and three α-helices (Riek et al. 1996; Donne et al. 1997; Zahn et al. 2000). Most of the familial human PrP mutations associated with prion diseases are localized in the globular domain. Some mutations can destabilize PrP structure (Swietnicki et al. 1998; Liemann and Glockshuber 1999) or cause misfolding into a form partially resistant to proteinase K digestion (Vanik and Surewicz 2002; Kiachopoulos et al. 2005), a property associated with the disease form of PrP (McKinley et al. 1983).
In this study, we investigated the role of the BORs and the globular domain of PrP in the anti-Bax function by generating a number of deletions and single point mutations of PrP to identify the region of PrP necessary for PrP’s anti-Bax function. While the BORs influence the ability of PrP to inhibit Bax-mediated cell death, the helix 3 region is sufficient to confer anti-Bax activity in MCF-7 cells and in human neurons.
EXPERIMENTAL PROCEDURES
Cell cultures
Human primary neurons were cultured from fetal brains, obtained with ethical approval from the McGill University Institutional Review Board, as described previously (LeBlanc et al. 1997). All cell lines were obtained from American Type Culture Collection (Manassas, VA). MCF-7 cells (Soule et al. 1973) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium containing 10% fetal bovine serum (FBS, HyClone, Logan, UT). Mouse neuroblastoma N2a cells were cultured in Minimal Essential Medium (MEM) and 10% FBS.
Site-directed mutagenesis, cloning and sequencing of PrP mutants
All human PrP mutants carried a valine residue at position 129 and were cloned into the bigenic pBudCE4.1 vector (Invitrogen, Carlsbad, CA). EGFP or EGFP-Bax cDNA were subcloned under the EF-1α promoter of pBudCE4.1 and PrP cDNA was subcloned under the CMV promoter as described previously (Jodoin et al. 2007). The details of the primers, templates and techniques used to create the mutations are found in Table 1. Briefly, the mutants were either created by PCR amplification of a given region of PrP, which was then subcloned into pBud-EGFP or pBud-EGFP-Bax or generated by the Quikchange Site-Directed Mutagenesis protocol (Stratagene, LaJolla, CA).
Table 1.
Mutagenesis and cloning methods used to generate the PrP mutants.
| Mutatgenesis | |||
|---|---|---|---|
| Mutant | Primers | Template | Restrictrion sites |
| pBSK-PrPΔBOR4/BH2 | 5’-TGGGATGCCTTTGTGGAACTGTACACCCACAGTCAGTGG-3’ 5’-GCCTCCGTTATCCTGGATCCAGGTACCACCGCCCTGAGG-3’ |
pBSK-PrPΔBOR4 | Non applicable (N/A) |
| pBSK-CyPrPΔBOR1 | 5’-GGCTGGGGGCAGCCTCATGGTGGTG-3’ 5’-ACCACCGCCCTGAGGTGGGTAGCGG-3’ |
pBSK-CyPrP | N/A |
| pBSK-CyPrPΔBOR2 | 5’-GGCTGGGGGCAGCCTCATGGTGGTG-3’ 5’-ACCACCGCCCTGAGGTGGGTAGCGG-3’ |
pBSK-CyPrP | N/A |
| pBSK-CyPrPΔBOR3 | 5’-GGATGGGGACAGCCTC-3’ 5’-ACCACCGCCCTGAGGTGGGTAGCGG-3’ |
pBSK-CyPrPΔBOR2 | N/A |
| Kozak sequence | 5’-GCGGTGGCGGCACCATGAAGAAGC-3’ 5’-GCTTCTTCATGGTGCCGCCACCGC-3’ |
CyPrP mutants in the pBSK vector | N/A |
| pBud-EGFP/ pBud-EGFP-Bax/CyPrP K204P | 5’-CCGAGACCGACGTTCCGATGATGGAGCGC-3’ 5’-GCGCTCCATCATCGGAACGTCGGTCTCGG-3’ |
pBud-EGFP/CyPrP | pBud-EGFP: N/A pBud-EGFP-Bax: SalI/EcoRI |
| pBud-EGFP/ pBud-EGFP-Bax/CyPrP V210P | 5’-GATGATGGAGCGCGTGCCTGAGCAGATGTGTATC-3’ 5’-GATACACATCTGCTCAGGCACGCGCTCCATCATC-3’ |
pBud-EGFP/CyPrP | pBud-EGFP: N/A pBud-EGFP-Bax: SalI/EcoRI |
| pBud-EGFP/ pBud-EGFP-Bax/CyPrP E219P | 5’-GTGTATCACCCAGTACCCGAGGGAATCTCAGGC-3’ 5’-GCCTGAGATTCCCTCGGGTACTGGGTGATACAC-3’ |
pBud-EGFP/CyPrP | pBud-EGFP: N/A pBud-EGFP-Bax: SalI/EcoRI |
| pBud-EGFP/ pBud-EGFP-Bax/CyPrP K204A | 5’-CCGAGACCGACGTTGCGATGATGGAGCGC-3’ 5’-GCGCTCCATCATCGCAACGTCGGTCTCGG-3’ |
pBud-EGFP/CyPrP | pBud-EGFP: N/A pBud-EGFP-Bax: SalI/EcoRI |
| pBud-EGFP/ pBud-EGFP-Bax/CyPrP V210A | 5’-GATGGAGCGCGTGGCTGAGCAGATGTG-3’ 5’-CACATCTGCTCAGCCACGCGCTCCATC-3’ |
pBud-EGFP/CyPrP | pBud-EGFP: N/A pBud-EGFP-Bax: SalI/EcoRI |
| pBud-EGFP/ pBud-EGFP-Bax/CyPrP E219A | 5’-CACCCAGTACGCGAGGGAATCTCAGGC-3’ 5’-GCCTGAGATTCCCTCGCGTACTGGGTG-3’ |
pBud-EGFP/CyPrP | pBud-EGFP: N/A pBud-EGFP-Bax: SalI/EcoRI |
| Cloning | |||
| pBSK-CyPrP 23-127 | 5’-ATAAGAATGCGGCCGCATGAAGAAGCGCCCGAAGCCTGGAG-3’ 5’-TTTATCGATTCAGCCAAGGCCCCCCACCACTG-3’ |
pCep4β-CyPrP | NotI/SmaI (destroyed) |
| pBSK-CyPrP 23-143 | 5’-ATAAGAATGCGGCCGCATGAAGAAGCGCCCGAAGCCTGGAG-3’ 5’-TTTATCGATTCAGCCGAAATGTATGATGGGC-3’ |
pCep4β-CyPrP | NotI/SmaI (destroyed) |
| pBSK-CyPrP 23-160 | 5’-ATAAGAATGCGGCCGCATGAAGAAGCGCCCGAAGCCTGGAG-3’ 5’-TTTATCGATTCAGTTGGGGTAACGGTGCATG-3’ |
pCep4β-CyPrP | NotI/SmaI (destroyed) |
| pBSK-CyPrP 23-172 | 5’-ATAAGAATGCGGCCGCATGAAGAAGCGCCCGAAGCCTGGAG-3’ 5’-TTTATCGATTCAGTTGCTGTACTCATCCATG-3’ |
pCep4β-CyPrP | NotI/SmaI (destroyed) |
| pBSK-CyPrP 23-199 | 5’-ATAAGAATGCGGCCGCATGAAGAAGCGCCCGAAGCCTGGAG-3’ 5’-TTTATCGATTCAGGTGAAGTTCTCCCCCTTG-3’. |
pCep4β-CyPrP | NotI/SmaI (destroyed) |
| pBud-EGFP/ pBud-EGFP-Bax/CyPrP 23-227 | 5’-GCAAGTCGACATGAAGAAGCGCCCGAAGCCTG-3’ 5’-GCCAGTACTCACTGGTAATAGGCCTGAGATTCCCTC-3’ |
pBud-EGFP/PrP | SalI/ScaI |
| pBSK-CyPrP | 5’-ATAAGAATGCGGCCGCATGAAGAAGCGCCCGAAGCCTGGAG-3’ 5’-CGCGGATCCTCACATGCTCGATCCTCTCTGGTAATAG-3’ |
pCep4β-CyPrP | NotI/SmaI (destroyed) |
| pBud-EGFP/ or pBud-EGFP-Bax/CyPrPΔBOR4 | 5’-GCAAGTCGACATGAAGAAGCGCCCGAAGCCTG-3’ 5’-GCCAGTACTCAGCTCGATCCTCTCTGGTAATAGGC-3’ |
pBud-EGFP/PrPΔBOR4 | SalI/ScaI |
| pBud-EGFP/ pBud-EGFP-Bax/PrP helix 3 tag | 5’-GCACTGCAGAATGTTCACCGAGACCGACGTTAAG-3’ 5’-GCGAGTACTGCTCGATCCTCTCTGGTAATAGGC-3’ |
pBud-EGFP/CyPrP | pBud-EGFP: PstI/ScaI pBud-EGFP-Bax: HindIII/EcoRI |
| pBud-EGFP/ pBud-EGFP-Bax/PrP helix 3 | 5’-GCACTGCAGAATGTTCACCGAGACCGACGTTAAG-3’ 5’-GCCAGTACTCAGCTCGATCCTCTCTGGTAATAGGC-3’ |
pBud-EGFP/CyPrP | pBud-EGFP: PstI/ScaI pBud-EGFP-Bax: HindIII/EcoRI |
| Direct subcloning | |||
| CyPrP mutants in pBud-EGFP or pBud EGFP-Bax vector | 5’-CGAGCGGTCGACTATAGGGCGAATTGGAGC-3’ 5’-GCCGGCAGTACTCGAGGTCGACGGTATCG-3’ |
CyPrP mutants in the pBSK vector | SalI/ScaI |
| PrP mutants in p Bud EGFP/ or pBud-EGFP-Bax | N/A | N/A | SalI/XbaI |
Full-length PrP lacking all four BORs (PrPΔBOR4) has been described previously under the name PrPΔOR (Bounhar et al. 2001). The BH2 domain of the Bcl-2 protein was inserted in PrP in place of the BOR domain to create the PrPΔBOR4/BH2 mutant. CyPrPΔBOR1, CyPrPΔBOR2, CyPrPΔBOR3, and CyPrPΔBOR4 are CyPrP lacking one, two, three, or all four BORs. α-helices and β-strands from the C-terminal globular domain of PrP were sequentially removed from CyPrP to create the deletion mutants CyPrP23-227, CyPrP23-199, CyPrP23-172, CyPrP23-160, CyPrP23-143, and CyPrP23-127. K204, V210 or E219 were replaced by proline or alanine residues in CyPrP to generate K204P, V210P, E219P, K204A, V210A, and E219A. The PrP α-helix 3 DNA encoding amino acid residues 198 to 231 was amplified by PCR with a primer introducing a Kozak sequence and an N-terminal methionine start codon. The amplified DNA was introduced in frame with the myc epitope and His tag of pBud-EGFP or pBud EGFP-Bax. A version without tags was also produced. The PrP mutants were verified by sequencing at the Genome Quebec Innovation Centre sequencing platform (Montréal, QC).
Molecular Modeling
The 3D model of the globular domain of PrP showing the position of the K204, V210 and E219 amino acid residues was made using Pymol molecular visualization software (DeLano 2002) (DeLano Scientific, Palo Alto, CA) and the Protein Databank file 1qlx (Zahn et al. 2000).
Transfections
MCF-7 cells were either plated on glass coverslips in 24 well plates (2.5×105 cells/well) for cell death assays or directly in 6 well plates (1.5×106 cells/well) for expression assays. Human primary neurons (1.5×105 cells) were plated on poly-L-lysine (Sigma-Aldrich, St-Louis, MI) coated plastic Aclar coverslips for cell death assays. N2a cells were plated directly in 6 well plates at a density of 0.8×106 cells/well for expression assays. The N2a and MCF-7 cells were transfected with 0.8μg of DNA for the 24 well plates or 4 μg of DNA for the 6 well plates with the Lipofectamine™ 2000 transfection reagent (Invitrogen, Carlsbad, CA). Lipofectamine™ 2000 is slightly toxic in MCF-7 cells and we realized that despite being simpler as a technique and yielding statistically significant results, that results obtained with lipofectamine (Figures 2A, 3A&B, 4A) had high cell death values in EGFP-transfected cells. We therefore switched to the Helios Gene Gun system (BioRad, Mississauga, ON), for MCF-7 and human neuron transfections, as described previously (Jodoin et al. 2007). The DNA used for the transfections was purified with the UltraClean™ Endotoxin Removal Kit (MoBio, Carlsbad, CA) according to the manufacturer’s protocol.
Figure 2. Expression and anti-Bax function of CyPrP with deletion of one to four BORs.

A. Schematic diagram of the sequential CyPrP BOR deletion mutants showing the main domains and structural elements. Right panel: Percentage (mean and SEM) of condensed chromatin positive cells in transfected MCF-7 cells. Top line represents EGFP-or EGFP-Bax-transfected cells in absence of PrP constructs. Data represents results from six independent experiments. At least 200 cells were counted in each experiment. Asterisk indicates a statistically significant difference from pBud-EGFP or pBud-EGFP-Bax (p≤0.05). B. Western blot of total proteins extracted from CyPrP and CyPrPΔBOR deletion mutants with 3F4 anti-PrP, anti-GFP and anti-β-actin antibodies.
Figure 3. Deletions in the globular domain of CyPrP eliminate PrP’s anti-Bax function.
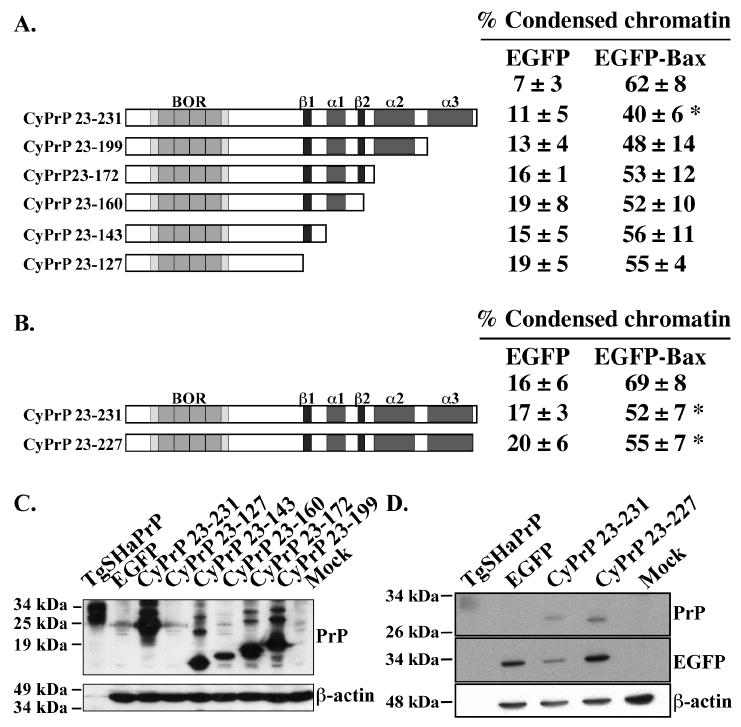
A & B. Schematic diagram of the sequential C-terminal CyPrP deletion mutants showing the main domains and structural elements. Right panel: Percentage (mean and SEM) of condensed chromatin positive cells in transfected MCF-7 cells. Data represents results from 4 to 5 independent experiments. At least 100 cells were counted for each experiment. Asterisk indicates a statistically significant difference from pBud-EGFP or pBud-EGFP-Bax (p≤0.05). C. Western blot analyses with 3F4 and β-actin antibodies of proteins extracted from cells transfected with CyPrP or CyPrP C-terminal globular domain deletion mutants. D. Western blot of total protein extracts from CyPrP23-231- or CyPrP23-227-transfected cells with 3F4, anti-GFP and anti-β-actin antibodies.
Figure 4. Effect of CyPrP helix 3 mis-sense mutations on the anti-Bax function of PrP.
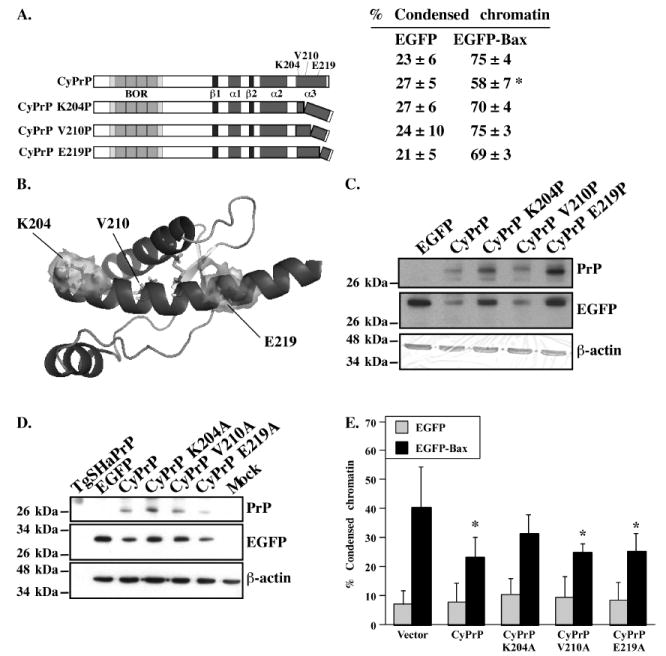
A. Schematic diagram of the CyPrP mutants with proline substitutions in helix 3.Right panel: Percentage (mean and SEM) of condensed chromatin positive cells in transfected MCF-7 cells. Data represents the results from 6 independent experiments. At least 100 cells were counted in each experiment. Asterisk indicates a statistically significant difference from pBud-EGFP or pBud-EGFP-Bax (p≤0.05). B. Location of the residues K204, V210 and E219 on the 3-dimensional structure of the CyPrP globular domain. C and D. Western blot analyses in protein extracts from cells transfected with CyPrP, the CyPrP proline substitution mutants (C) or the CyPrP alanine substitution mutants (D) with 3F4, anti-GFP or anti-β-actin antibodies. E. Percentage of cell death assessed by condensed chromatin in MCF-7 cells transfected with pBud-EGFP, pBud-EGFP/CyPrP, pBud-EGFP/CyPrP K204A, pBud-EGFP/CyPrP V210A, pBud-EGFP/CyPrP E219A, pBud-EGFP-Bax, pBud-EGFP-Bax/CyPrP, pBud-EGFP-Bax/CyPrP K204A, pBud-EGFP-Bax/CyPrP V210A, or pBud-EGFP-Bax/CyPrP E219A. Data represents the mean ± SEM of 3 independent experiments. At least 150 cells were counted in each experiment. Asterisk indicates a statistical difference from pBud-EGFP or pBud-EGFP-Bax-transfected cells (p≤0.05).
Cell death measurements
Condensed chromatin: Twenty-four hours after transfection, cells were fixed in a solution of 4% paraformaldehyde (Sigma-Aldrich) and 4% sucrose (Bioshop, Burlington, ON) in phosphate-buffered saline (PBS: 150 mM NaCl, 2.7 mM KCl, 1.3 mM KH2PO4, 8.1 mM Na2HPO4 pH 7.4) and the chromatin was stained for 20 minutes with 0.5 μg/ml Hoechst 33342 (Sigma-Aldrich) in PBS. Cell death was identified as Hoechst-stained chromatin condensation in EGFP-or EGFP-Bax-positive cells by fluorescence microscopy (Nikon eclipse TE2000-U microscope, Mississauga, ON). The percentage of cell death was calculated as the number of EGFP-positive cells displaying condensed chromatin over the total number of EGFP-positive cells. TUNEL: Twenty-four hours after transfection, cells were fixed and TUNEL labeling was performed with the In Situ Cell Death Detection Kit, TM red, according to the manufacturer’s protocol (Roche Applied Science, Laval, QC). TUNEL positive EGFP-or EGFP-Bax-transfected cells were detected by fluorescence microscopy. The percentage of TUNEL positive cells was calculated as the number of EGFP-positive cells with red fluorescence over the total number of EGFP-positive cells. Caspase activity: Twenty-four hours after transfection, cells were labeled for active caspases with the SR FLICA Poly Caspases kit (AbD Serotec, Raleigh, NC) according to the manufacturer’s instructions. EGFP- or EGFP-Bax-transfected cells displaying active caspases were identified by fluorescence microscopy. The percentage of FLICA positive cells was calculated as the number of EGFP-positive cells with red fluorescence over the total number of EGFP-positive cells.
Subcellular fractionation
Subcellular fractionation of PrPΔBOR4 and wild-type PrP was performed as described previously (Jodoin et al. 2007).
Western blot analyses
Twenty-four hours after transfection, cells were either directly harvested or treated for 24h with 0.25 μM epoxomicin (BioMol, Plymouth Meeting, PA). Cellular proteins were extracted in non-ionic detergent Nonidet P-40 (NP-40) lysis buffer (50 mM Tris-Cl pH 8.0, 150 mM NaCl, 1% NP-40, 5 mM EDTA pH 8.0) for 10 min on ice. Detergent-insoluble proteins were separated by centrifugation at 16 245 × g for 10 min at 4°C and insoluble proteins were re-solubilized in 2% sodium dodecyl sulfate (SDS). Fifty or 100 μg of proteins, as quantified by bicinchoninic acid (BCA) assay (Pierce, Rockford, IL), were precipitated in four volumes of ice-cold 100% methanol overnight at -20°C. The proteins were centrifuged at 16,245 × g for 15 minutes at 4°C and resuspended in 20 μl Laemmli sample buffer (0.5% SDS (w/v), 1.25% β-mercaptoethanol, (v/v), 2.5% glycerol (v/v), 0.01% bromophenol blue (w/v), 15.6 mM Tris-HCl, pH 6.8). Proteins were submitted to 15% SDS-PAGE and transferred to PVDF membranes. The membranes were probed with the anti-PrP 3F4 (Kascsak et al. 1987) (1:1000), anti-β-actin (1:1000, Clone AC-15, Sigma, Oakville, ON), anti-mtHsp70 (1/1000, Clone JG1: Affinity Bioreagents, Golden, CO), anti-His Tag (1:200, EMD Bioscience, Gibbstown, NJ), and anti-GFP (1:1000, B-2, Santa Cruz Biotechnology, Santa Cruz, CA) antibodies. Immunoreactivity was revealed with horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (Jackson Immnunoresearch Laboratories, West Grove, PA) and chemiluminescence reagents (Millipore, Billerica, MA).
Statistical analysis
Statistical analyses were done with StatView 5.0 software (SAS Institute Inc., Cary, NC) by an analysis of variance (ANOVA) followed by a Post-hoc Scheffé test. A p value under 0.05 was considered a significant difference.
RESULTS
Substitution of the BOR domain of full-length PrP by the BH2 domain of the Bcl-2 protein does not rescue the anti-Bax function of PrPΔBOR4
It was previously shown that the deletion of the BOR domain from full-length PrP (PrPΔBOR4) abolishes its anti-Bax function in human neurons (Bounhar et al. 2001). To investigate the role of the BOR domain in MCF-7 cells, the PrPΔBOR4 was subcloned under the CMV promoter in the bigenic vector pBudCE4.1 containing EGFP or EGFP-Bax cDNA under the EF-1α promoter (Fig. 1A). The expression of N-terminally EGFP-fused Bax is sufficient to activate Bax-mediated cell death and assures specific Bax activation in comparison to using an apoptotic insult that would activate several cell death pathways concomitantly. In transfected cells, PrPΔBOR4 migrates as several protein bands ranging from 23 to 36 kDa whereas full-length wild type PrP migrates mostly as a mature 36 kDa protein, similar to PrP from Syrian Hamster PrP transgenic mouse brain protein extracts (Fig. 1B). This difference in glycosylation suggests that PrPΔBOR4 accumulates as an immature glycosylated protein. Neither PrP nor PrPΔBOR4 are toxic to MCF-7 cells (Fig. 1C). Wild-type PrP significantly protects against Bax-mediated cell death in MCF-7 cells but PrPΔBOR4 does not (Fig. 1C), consistent with our previous observations in human neurons (Bounhar et al. 2001). The loss of anti-Bax function in PrPΔBOR4 is not due to decreased retrotranslocation since cytosolic PrPΔBOR4 is easily detected in subcellular cytosolic and membrane fractions of transfected cells (Fig. 1D). Thus, it is likely that the loss of function is attributable to the altered structure of the protein.
Figure 1. Expression and anti-Bax function of full-length PrPΔBOR4 and PrPΔBOR4/BH2 mutants.
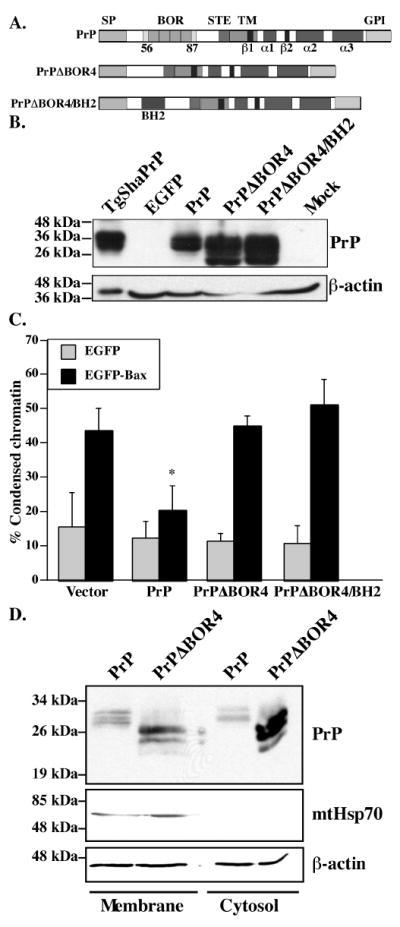
A. Schematic diagram showing the main domains and structural elements of PrP, PrPΔBOR4 and PrPΔBOR4/BH2. Abbreviations: SP: N-terminal ER-targeting signal peptide (residues 1-23), BOR: BH2-like octapeptide repeats (residues 56-87), as opposed to standard octapeptide repeats (residues 51-91), STE: Stop-transfer effector sequence (residues 104-111), TM: transmembrane domain (residues 112-135), β1 and β2: beta-strands 1 (residues 128-131) and 2 (residues 161-164), α1, α2 and α3: alpha-helices 1 (residues 144-154), 2 (residues 173-194) and 3 (residues 200-227), GPI: glycosyl phosphatidylinositol anchor signal (residues 232-253). B. Western blot of 100 μg SDS-soluble proteins extracted from EGFP, PrP, PrPΔBOR4, PrPΔBOR4/BH2, or Mock-transfected cells with 3F4 anti-PrP and anti-β-actin antibodies. Proteins (1.05 μg) extracted from Syrian Hamster PrP transgenic mice brains (TgSHaPrP) were used as a positive control for PrP immunodetection. C. Percentage of cell death assessed by chromatin condensation in MCF-7 cells transfected with pBud-EGFP, pBud-EGFP/PrP, /PrPΔBOR4, or /PrPΔBOR4/BH2, and pBud-EGFP-Bax, pBud-EGFP-Bax/PrP, /PrPΔBOR4 or /PrPΔBOR4/BH2. Data represents the mean ± standard error of the mean (SEM) of 3 independent experiments. At least 100 cells were counted in each experiment. Asterisk sign indicates a statistically significant difference from pBud-EGFP or pBud-EGFP-Bax (p≤0.05). D. Western blot analysis of proteins from membrane and cytosolic subcellular fractions with 3F4, mtHsp70 and β-actin antibodies.
Next, we assessed if the loss of function in PrPΔBOR4 could be rescued by replacing the BORs by the BH2 domain of Bcl-2 (Fig. 1A). The results show that PrPΔBOR4/BH2 does not rescue the loss of anti-Bax function in PrPΔBOR4 (Fig. 1C) although it is expressed at higher levels than PrP (Fig. 1B) and is not cytotoxic (Fig. 1C). This result indicates that despite a similarity in their sequences, the BOR domain of PrP is not functionally similar to the BH2 domain of Bcl-2.
Partial but not complete deletion of BOR abolishes CyPrP anti-Bax function
Because CyPrP is the form of PrP responsible for the anti-Bax function (Lin et al. 2008), we re-examined the involvement of the BOR domain in CyPrP’s anti-Bax function. We further tested if these repeats act in cooperation by sequentially deleting each repeat from CyPrP (Fig. 2A). In transfected cells, each mutant migrates as a progressively shorter protein with more extensive deletions, as expected (Fig. 2B). Deletion of one, two, or three repeats eliminates the anti-Bax function (Fig. 2A). In the case of CyPrPΔBOR1, statistically significant cytotoxicity is also observed, thus it is difficult to determine the anti-Bax function for this mutant. Unexpectedly, the deletion of all four BORs retains the anti-Bax function (Fig. 2A). This contrasts with the loss of anti-Bax function of PrPΔBOR4 in MCF-7 cells (Fig. 1C) and in human neurons (Bounhar et al. 2001). Nevertheless, the loss of anti-Bax function in the partial BOR deletions indicates that this domain is involved in PrP’s anti-Bax function.
Deletion of the C-terminal helix 3 abolishes CyPrP anti-Bax function
Mutants carrying sequential deletions of the cDNA regions encoding the 3 α-helices and 2 β-strands of PrP were tested (Fig. 3A). None of the deletions causes toxicity in the cells (Fig. 3A). Deletion of amino acid residues 200 to 231 from CyPrP (CyPrP23-199) abolishes the anti-Bax function in MCF-7 cells (Fig. 3A). Further deletions of α-helices 2 and 1 and β-strands 1 and 2 also eliminate PrP’s role against Bax-mediated cell death (Fig. 3A). All the deletion mutants are highly expressed except for CyPrP23-127 (Fig. 3C). Therefore, the loss of function is not due to faulty expression. These results indicate that amino acid residues 200 to 231 of CyPrP, which contain helix 3 and four non-helical C-terminal amino acid residues of CyPrP, are required for CyPrP’s anti-Bax function.
To verify if those four C-terminal amino acid residues are required for PrP’s anti-Bax function, we constructed a CyPrP23-227 mutant lacking these amino acid residues (Fig. 3B). This deletion mutant is expressed in cells (Fig. 3D) and retains the anti-Bax function (Fig. 3B). Together, these results indicate that the helix 3 region is required for the anti-Bax function of PrP.
Substitution of helix 3 K204, V210 and E219 amino acid residues by prolines causes a loss of CyPrP’s anti-Bax function
To determine if the helical structure of helix 3 is necessary for the protection against EGFP-Bax-mediated cell death, amino acid residues at positions 204, 210 and 219 in CyPrP were substituted by a proline to disrupt its helical conformation (Fig. 4A). These mutants represent a substitution in each turn of the helix, omitting the C214 residue involved in the disulfide bond and amino acid residues after E219 since the helical structure is already unstable beyond this point (Zahn et al. 2000). K204 and E219 are on the surface of the folded structure while V210 is part of the hydrophobic core of PrP (Fig. 4B). Each of these mutants is expressed at equivalent levels in transfected cells considering their transfection efficiency determined by anti-GFP immunoblotting (Fig. 4C). None of the mutants are toxic. The introduction of proline residues in K204, V210 and E219 results in a loss of anti-Bax function (Fig. 4A). These results indicate that helix 3 stability is a requirement for CyPrP anti-Bax function.
Substitution of helix 3 amino acid residues by alanines shows that the N-terminal portion of helix 3 is required for CyPrP anti-Bax function
Because disruption of the helix 3 region with prolines in CyPrP can affect the structure of the entire globular domain of CyPrP, we replaced helix 3 amino acid residues 204, 210 and 219 by alanines to further study the involvement of this helix 3 in the anti-Bax function. These mutations are predicted to have little effect on the structure of CyPrP (Blaber et al. 1993). However, replacing K204 by an alanine residue does break an ionic bond with E200, which destabilizes the structure of a helix 3 peptide (Gallo et al. 2005). These alanine mutants are all expressed (Fig. 4D). The replacement of K204 by an alanine abolishes the anti-Bax function while the replacement of V210 and E219 by alanine residues has no effect (Fig. 4E). These results show that the K204 amino acid residue of helix 3 is most important for the anti-Bax function.
Helix 3 of PrP encompasses the anti-Bax function in MCF-7 cells and in human primary neurons
To assess if the helix 3 region is directly responsible for PrP’s anti-Bax function, we cloned amino acid residues 198 to 231 of PrP in frame with the myc and His tags (helix 3 tag) in pBud vectors and tested these constructs’ ability to prevent Bax-mediated cell death (Fig. 5A). The expression of helix-3 from this construct is observed with an anti-His Tag western blot (Fig. 5B). Similar to CyPrP, helix 3 tag significantly prevents Bax-mediated cell death in MCF-7 cells as observed with either Hoechst staining of condensed chromatin (Fig. 5C) or FLICA detection of active caspases (Fig. 5D).
Figure 5. Effect of PrP helix 3 on the anti-Bax function of PrP in MCF-7 cells.

A. Schematic diagram of the helix 3 construct relative to the full length CyPrP. B. Western blot with anti-His Tag and anti-β-actin antibodies. Lower panel shows Coomassie blue stain of the area depicted in the western blots. C. Percentage cell death assessed by condensed chromatin in MCF-7 cells transfected with pBud-EGFP or pBud-EGFP/CyPrP, or /PrP helix 3 tag, and pBud-EGFP-Bax, pBud-EGFP-Bax/CyPrP or /PrP helix 3 tag. Data represents the mean ± SEM of 7 independent experiments. At least 100 cells were counted in each experiment. Asterisk indicates a statistically significant difference from pBud-EGFP or pBud-EGFP-Bax-transfected cells (p≤0.05). D. Percentage of cells with caspase activity assessed by FLICA in MCF-7 cells transfected with pBud-EGFP or pBudEGFP/CyPrP or /PrP helix 3 tag and pBud-EGFP-Bax or pBud-EGFP-Bax/CyPrP or /PrP helix 3 tag. Data represents the mean ± SEM of 3 independent experiments. At least 100 cells were counted in each experiment. Asterisk indicates a statistically significant difference from pBud-EGFP or pBud-EGFP-Bax (p≤0.05).
The MCF-7 cells constitute a simpler system to perform structure-function analyses than primary human neurons. After we found the important anti-Bax region in helix 3, we verified if helix 3 could also prevent Bax-mediated cell death in primary human neurons (Fig. 6). Similar to our observations in MCF-7 cells, the helix 3 tag prevents Bax-mediated cell death in human neurons as shown with Hoechst staining (Fig. 6B) or TUNEL labeling (Fig. 6C). To determine if helix 3 is functional in absence of the tags, we made a second construct without the myc and His tags (helix 3) (Fig. 6A). Again, helix 3 inhibits Bax-mediated cell death assayed with Hoechst (Fig. 6B) or TUNEL staining (Fig. 6C). Compared to MCF-7 cells, CyPrP and PrP helix 3 have a stronger protection against EGFP-Bax-mediated cell death in human neurons. The incomplete protection of CyPrP or PrP helix 3 in MCF-7 cells is likely due to Bax-mediated activation of PrP insensitive pro-apoptotic pathways such as Bak or Bid (Roucou et al. 2005). Together, these results show that helix 3 is sufficient for the anti-Bax function of PrP.
Figure 6. Effect of PrP helix 3 on the anti-Bax function of PrP in human primary neurons.
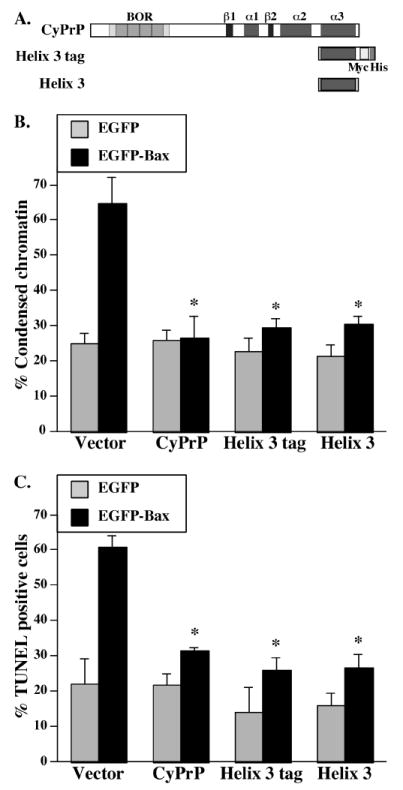
A. Schematic diagram of the helix 3 constructs with and without tags and the full length CyPrP. B. Percentage cell death assessed by condensed chromatin in human neurons transfected with pBud-EGFP, pBud-EGFP/CyPrP, /PrP helix 3 tag, /PrP helix 3, and pBud-EGFP-Bax, pBud-EGFP-Bax/CyPrP, /PrP helix 3 tag, or /PrP helix 3. Data represents the mean ± SEM of 7 independent experiments. At least 100 to 200 cells were counted. Asterisk indicates a statistically significant difference from pBud-EGFP or pBud-EGFP-Bax (p≤0.05). C. Percentage of apoptotic cells displaying DNA fragmentation assayed by TUNEL in human neurons transfected with pBud-EGFP, pBud-EGFP/CyPrP, /PrP helix 3 tag, or /PrP helix 3, and pBud-EGFP-Bax, pBud-EGFP-Bax/CyPrP, /PrP helix 3 tag, or /PrP helix 3. Data represents the mean ± SEM of 3 independent experiments. At least 50 to 100 cells were counted. Asterisk indicates a statistically significant difference from pBud-EGFP or pBud-EGFP-Bax (p≤0.05).
DISCUSSION
In this manuscript, we conclude that the helix 3 region is necessary and sufficient for the anti-Bax function of PrP. This conclusion is based on the fact that [1] deletion of the C-terminal 200-227 amino acids of PrP that encompass helix 3 results in a loss of anti-Bax function, [2] substitution of the K204, V210 and E219 amino acid residues with the helix disrupting proline amino acid residue destroys the anti-Bax function of PrP, [3] substitution of K204 with alanine also disrupts PrP anti-Bax function, and [4] expressing the helix 3 alone is sufficient to inhibit Bax-mediated cell death in MCF-7 cells and in primary human neurons. A search for similarities between the amino acid sequence of helix 3 and other proteins revealed only prion proteins from various species and no similarities between this sequence and other anti-Bax proteins. Therefore, the anti-Bax function of helix 3 is quite unique and differs from that found in a number of cytosolic anti-Bax proteins such as humanin, clusterin or Ku70 (reviewed by (Walensky 2006)). However, involvement of helix 3 in PrP’s anti-Bax function is reminiscent of the survival or cell death role of α-helices in Bcl-2 family members (Walensky 2006). Interestingly, deletion of amino acids 201-217 in mouse PrP containing a deletion of amino acid 23-88 results in a neurodegenerative disease showing some nerve cell loss, cytoplasmic inclusions in enlarged neurons and excess endoplasmic reticulum characteristic of a neuronal storage disease (Muramoto et al. 1997). However, simply deleting the amino acids 23-88 does not induce neurodegeneration. Therefore, deletion of the anti-Bax function in helix 3 could be responsible for the observed pathologies in these mice, but only if PrP plays an important physiological role in preventing Bax activation in neurons. Alternatively, the deletion mutant protein causes toxicity. Our findings warrant revisiting this model to elucidate the underlying molecular mechanism of this neurodegeneration.
The structural element that is necessary for helix 3 anti-Bax function is likely the helical structure itself. Disruption of the helical structure by the substitution of K204, V210 and E219 with the helix breaking proline amino acid eliminates the function of PrP against Bax-mediated cell death. With milder alanine substitutions, only the K204A mutant loses the anti-Bax function. This substitution disrupts the helical structure of the helix 3 peptide which in vitro displays structural autonomy (Gallo et al. 2005). In Gallo et al. (Gallo et al. 2005), it is believed that the K204 forms an ionic bond with amino acid residue E200, hence disruption of this bond could alter the structural features of the N-terminus of helix 3. However, the E200K familial PrP mutant with a Val at codon 129 (Val 129) retains the anti-Bax function of PrP entirely and the Met129 E200K PrP mutant retains partial function (Jodoin et al. 2007). This indicates that the disruption of the E200-K204 ionic bond may not be sufficient to explain the loss of helical structure and anti-Bax function. Alternatively, the K204 could be part of an amino acid motif that is responsible for PrP’s anti-Bax function. However, while K204 is entirely conserved in the PrP of all mammalian species and in chicken, amino acid variations are observed at V203, M205, M206 and E207 (Kim et al. 2008). Furthermore, the helix 3 V203I, R208H and E211Q familial PrP mutants retain the anti-Bax function of PrP (Jodoin et al. 2007), and the V210A and E219A PrP mutants studied in this paper also retain anti-Bax function in MCF-7 cells. Therefore, the results indicate that the K204 performs an essential function in PrP’s anti-Bax function. Exactly how the K204 amino acid residue functions will only be elucidated by identifying the molecular mechanism involved in PrP’s anti-Bax function. We have previously excluded a direct interaction of PrP with Bax and also other Bcl-2 family members (Bounhar et al. 2006; Lin et al. 2008). We can now use this sequence to search for interacting proteins that would mediate PrP’s anti-Bax function.
While the helix 3 region is most important for the anti-Bax function of CyPrP, the N-terminal BOR domain also is implicated. Deletion of the four BORs does not affect the anti-Bax function of CyPrP, but deletion of the same BORs in the full length PrP loses the anti-Bax function in both human primary neurons and MCF-7 cells (Bounhar et al. 2001). A number of factors may explain the difference between retrotranslocated and cytosolically expressed CyPrPΔBOR4. We exclude the possibility that the loss of PrP anti-Bax function in PrPΔBOR4 is due to a defect of PrP retrotranslocation, as previously shown with a number of Creutzfeldt-Jakob disease-associated PrP mutants (Jodoin et al. 2007). However, the retrotranslocated, but not cytosolically expressed, CyPrPΔBOR4, is glycosylated. The glycosylation is expected since the epoxomicin proteasomal inhibitor added to increase the detection of CyPrPΔBOR4 blocks both the deglycosylation and degradation of retrotranslocated cytosolic proteins (Karaivanova and Spiro 2000). Glycosylation may alter the structure of the protein resulting in a loss of the anti-Bax function of PrP. Alternatively, different chaperones could be involved in the folding of secreted and cytosolically synthesized proteins (Ellgaard and Helenius 2001). Furthermore, our data showing that deletion of only one, two, or three of the four BORs in the CyPrP results in a loss of anti-Bax function whereas deletion of the four BORs retains function could be explained by the possibility that these deletions differentially affect the structure of CyPrP. Therefore, it is possible that retro-translocated PrPΔBOR4 is folded differently than CyPrPΔBOR4.
It appears that even if the N-terminal region influences the anti-Bax function of PrP, it is not sufficient because the C-terminal deletion constructs do not have any anti-Bax activity despite retaining intact BOR domains. It may be that the N-terminal BOR region regulates the C-terminal helix 3 anti-Bax function. Indeed, it has been shown that the N-terminus of PrP regulates the C-terminal structure of PrP (Zahn et al. 2000). Together, the influence of complete or partial BOR deletions in either full length or CyPrP on the anti-Bax function of PrP is consistent with the role of the OR against Doppel-and ischemia-mediated cell death in vivo (Atarashi et al. 2003; Mitteregger et al. 2007).
This study is the first to implicate the helix 3 region of PrP in a protective function. Elucidating the anti-Bax region of PrP provides a molecular handle upon which to develop therapeutic strategies against Bax in neurodegenerative diseases and an essential tool to further characterize the underlying molecular mechanism of PrP inhibition of Bax activation. Furthermore, helix 3 of PrP can be used to develop therapeutic strategies to prevent PrP’s anti-Bax function in cancer cells. Since host PrP is essential to transmissible spongiform encephalopathies (TSEs), deletion of the PRNP gene is considered as a mean of disease prevention. An alternative approach to prevention against TSEs could involve selective deletion of the PrP regions implicated in disease transmission while leaving the protective helix 3 intact.
Acknowledgments
This work was supported by the National Institutes for Health (1RO1 NS-40431), the Canadian Institutes of Health Research (MOP49594), and the Fonds de Recherche en Santé du Québec. The authors gratefully acknowledge Dr. Cynthia G. Goodyer (Department of Pediatrics, McGill University, Montréal, QC) and the Birth Defects Research Laboratory (University of Washington, Seattle, WA) for providing conceptal brain tissues (NIH HD 000836). The authors would like to thank Jennifer Hammond for the preparation of the human neurons, Michaël Baril and Younes Bounhar for their initial cloning of some of the PrP mutant constructs, and Dalia Halawani for help with the Pymol software.
Abbreviations used
- Bax
Bcl-2 associated protein X
- Bcl-2
B-cell lymphoma 2
- BH2
Bcl-2 homology domain 2
- BOR
BH2-like octapeptide repeat
- CyPrP
cytosolic prion protein
- EGFP
enhanced green fluorescent protein
- GPI
glycosylphosphatidylinositol
- mtHsp70
mitochondrial heat shock protein 70
- OR
octapeptide repeat
- PrP
prion protein
- SP
signal peptide
- STE
stop transfer effector domain
- TgSHaPrP
transgenic mice expressing the Syrian Hamster PrP gene
- TM
transmembrane domain
- TSEs
transmissible spongiform encephalopathies
References
- Atarashi R, Nishida N, Shigematsu K, Goto S, Kondo T, Sakaguchi S, Katamine S. Deletion of N-terminal residues 23-88 from prion protein (PrP) abrogates the potential to rescue PrP-deficient mice from PrP-like protein/doppel-induced Neurodegeneration. J Biol Chem. 2003;278:28944–28949. doi: 10.1074/jbc.M303655200. [DOI] [PubMed] [Google Scholar]
- Blaber M, Zhang XJ, Matthews BW. Structural basis of amino acid alpha helix propensity. Science. 1993;260:1637–1640. doi: 10.1126/science.8503008. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion Protein Protects Human Neurons against Bax-mediated Apoptosis. J Biol Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Mann KK, Roucou X, LeBlanc AC. Prion protein prevents Bax-mediated cell death in the absence of other Bcl-2 family members in Saccharomyces cerevisiae. FEMS Yeast Res. 2006;6:1204–1212. doi: 10.1111/j.1567-1364.2006.00122.x. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol. 1997a;146:104–112. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- Brown DR, Wong BS, Hafiz F, Clive C, Haswell SJ, Jones IM. Normal prion protein has an activity like that of superoxide dismutase. Biochem J. 1999;344(Pt 1):1–5. [PMC free article] [PubMed] [Google Scholar]
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H. The cellular prion protein binds copper in vivo. Nature. 1997b;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21:3317–3326. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulpier M, Messiaen S, Boucreaux D, Eloit M. Axotomy-induced motoneuron death is delayed in mice overexpressing PrPc. Neurosci. 2006;141:1827–1834. doi: 10.1016/j.neuroscience.2006.05.037. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System DeLano Scientific. Palo Alto, CA: 2002. [Google Scholar]
- Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Pietu G, Pitaval A, Ripoche H, Eloit M, Dormont D, Chouaib S. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004;64:719–727. doi: 10.1158/0008-5472.can-03-1735. [DOI] [PubMed] [Google Scholar]
- Donne DG, Viles JH, Groth D, Mehlhorn I, James TL, Cohen FE, Prusiner SB, Wright PE, Dyson HJ. Structure of the recombinant full-length hamster prion protein PrP(29-231): the N terminus is highly flexible. Proc Natl Acad Sci U S A. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drisaldi B, Coomaraswamy J, Mastrangelo P, Strome B, Yang J, Watts JC, Chishti MA, Marvi M, Windl O, Ahrens R, Major F, Sy M-S, Kretzschmar H, Fraser PE, Mount HTJ, Westaway D. Genetic Mapping of Activity Determinants within Cellular Prion Proteins: N-Terminal Modules in PrPC Offset Pro-apoptotic Activity of the Doppel Helix B/B’ Region. J Biol Chem. 2004;279:55443–55454. doi: 10.1074/jbc.M404794200. [DOI] [PubMed] [Google Scholar]
- Dupiereux I, Falisse-Poirrier N, Zorzi W, Watt NT, Thellin O, Zorzi D, Pierard O, Hooper NM, Heinen E, Elmoualij B. Protective effect of prion protein via the N-terminal region in mediating a protective effect on paraquat-induced oxidative injury in neuronal cells. J Neurosci, Res. 2007;86:653–659. doi: 10.1002/jnr.21506. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. ER quality control: towards an understanding at the molecular level. Curr Op Cell Biol. 2001;13:431–437. doi: 10.1016/s0955-0674(00)00233-7. [DOI] [PubMed] [Google Scholar]
- Gallo M, Paludi D, Cicero DO, Chiovitti K, Millo E, Salis A, Damonte G, Corsaro A, Thellung S, Schettini G, Melino S, Florio T, Paci M, Aceto A. Identification of a conserved N-capping box important for the structural autonomy of the prion alpha 3-helix: the disease associated D202N mutation destabilizes the helical conformation. Int J Immunopathol Pharmacol. 2005;18:95–112. doi: 10.1177/039463200501800111. [DOI] [PubMed] [Google Scholar]
- Jodoin J, Laroche-Pierre S, Goodyer CG, LeBlanc AC. Defective Retrotranslocation Causes Loss of Anti-Bax Function in Human Familial Prion Protein Mutants. J Neurosci. 2007;27:5081–5091. doi: 10.1523/JNEUROSCI.0957-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaivanova VK, Spiro RG. Effect of proteasome inhibitors on the release into the cytosol of free polymannose oligosaccharides from glycoproteins. Glycobiol. 2000;10:727–735. doi: 10.1093/glycob/10.7.727. [DOI] [PubMed] [Google Scholar]
- Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987;61:3688–3693. doi: 10.1128/jvi.61.12.3688-3693.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiachopoulos S, Bracher A, Winklhofer KF, Tatzelt J. Pathogenic mutations located in the hydrophobic core of the prion protein interfere with folding and attachment of the glycosylphosphatidylinositol anchor. J Biol Chem. 2005;280:9320–9329. doi: 10.1074/jbc.M412525200. [DOI] [PubMed] [Google Scholar]
- Kim Y, Lee J, Lee C. In silico comparative analysis of DNA and amino acid sequences for prion protein gene. Transbound Emerg Dis. 2008;55:105–114. doi: 10.1111/j.1865-1682.2007.00997.x. [DOI] [PubMed] [Google Scholar]
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Matsumoto Y, Yokoyama T, Itohara S, Onodera T. Prions prevent neuronal cell-line death. Nature. 1999;400:225–226. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- LeBlanc A. Unravelling the controversy of prion diseases. In: W E, S S, editors. Handbook of the Aging Brain. Academic Press; New York: 1998. pp. 202–214. [Google Scholar]
- LeBlanc AC, Papadopoulos M, Belair C, Chu W, Crosato M, Powell J, Goodyer CG. Processing of amyloid precursor protein in human primary neuron and astrocyte cultures. J Neurochem. 1997;68:1183–1190. doi: 10.1046/j.1471-4159.1997.68031183.x. [DOI] [PubMed] [Google Scholar]
- Li A, Harris DA. Mammalian prion protein suppresses Bax-induced cell death in yeast. J Biol Chem. 2005;280:17430–17434. doi: 10.1074/jbc.C500058200. [DOI] [PubMed] [Google Scholar]
- Liang J, Pan YL, Ning XX, Sun LJ, Lan M, Hong L, Du JP, Liu N, Liu CJ, Qiao TD, Fan DM. Overexpression of PrPC and its antiapoptosis function in gastric cancer. Tumour Biol. 2006;27:84–91. doi: 10.1159/000092488. [DOI] [PubMed] [Google Scholar]
- Liemann S, Glockshuber R. Influence of Amino Acid Substitutions Related to Inherited Human Prion Diseases on the Thermodynamic Stability of the Cellular Prion Protein. Biochem. 1999;38:3258–3267. doi: 10.1021/bi982714g. [DOI] [PubMed] [Google Scholar]
- Lin DT, Jodoin J, Baril M, Goodyer CG, LeBlanc AC. Cytosolic prion protein is the predominant anti-Bax prion protein form: exclusion of transmembrane and secreted prion protein forms in the anti-Bax function. Biochim Biophys Acta-Mol Cell Res. 2008;1783:2001–2012. doi: 10.1016/j.bbamcr.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc Natl Acad Sci U S A. 2001;98:14955–14960. doi: 10.1073/pnas.011578098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35:57–62. doi: 10.1016/0092-8674(83)90207-6. [DOI] [PubMed] [Google Scholar]
- Meslin F, Hamai A, Gao P, Jalil A, Cahuzac N, Chouaib S, Mehrpour M. Silencing of Prion Protein Sensitizes Breast Adriamycin-Resistant Carcinoma Cells to TRAIL-Mediated Cell Death. Cancer Res. 2007a;67:10910–10919. doi: 10.1158/0008-5472.CAN-07-0512. [DOI] [PubMed] [Google Scholar]
- Meslin F, Conforti R, Mazouni C, Morel N, Tomasic G, Drusch F, Yacoub M, Sabourin JC, Grassi J, Delaloge S, Mathieu MC, Chouaib S, Andre F, Mehrpour M. Efficacy of adjuvant chemotherapy according to Prion protein expression in patients with estrogen receptor-negative breast cancer. Ann Oncol. 2007b;18:1793–1798. doi: 10.1093/annonc/mdm406. [DOI] [PubMed] [Google Scholar]
- Mironov A, Jr, Latawiec D, Wille H, Bouzamondo-Bernstein E, Legname G, Williamson RA, Burton D, DeArmond SJ, Prusiner SB, Peters PJ. Cytosolic Prion Protein in Neurons. J Neurosci. 2003;23:7183–7193. doi: 10.1523/JNEUROSCI.23-18-07183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitteregger G, Vosko M, Krebs B, Xiang W, Kohlmannsperger V, Nolting S, Hamann GF, Kretzschmar HA. The Role of the Octarepeat Region in Neuroprotective Function of the Cellular Prion Protein. Brain Pathol. 2007;17:174–183. doi: 10.1111/j.1750-3639.2007.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D, DeArmond SJ, Tremblay P. Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci U S A. 2001;98:15288–15293. doi: 10.1073/pnas.251550798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramoto T, DeArmond SJ, Scott M, Telling GC, Cohen FE, Prusiner SB. Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an alpha-helix. Nat Med. 1997;3:750–755. doi: 10.1038/nm0797-750. [DOI] [PubMed] [Google Scholar]
- Nicolas O, Gavin R, Braun N, Urena JM, Fontana X, Soriano E, Aguzzi A, Antonio del Rio J. Bcl-2 overexpression delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice that overexpress amino-terminally truncated prion. FASEB J. 2007;21:3107–3117. doi: 10.1096/fj.06-7827com. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Scott MR, DeArmond SJ, Cohen FE. Prion protein biology. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- Rachidi W, Vilette D, Guiraud P, Arlotto M, Riondel J, Laude H, Lehmann S, Favier A. Expression of Prion Protein Increases Cellular Copper Binding and Antioxidant Enzyme Activities but Not Copper Delivery. J Biol Chem. 2003;278:9064–9072. doi: 10.1074/jbc.M211830200. [DOI] [PubMed] [Google Scholar]
- Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121-321) Nature. 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- Roucou X, Gains M, LeBlanc AC. Neuroprotective functions of prion protein. J Neurosci Res. 2004;75:153–161. doi: 10.1002/jnr.10864. [DOI] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005;12:783–795. doi: 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, Rulicke T, Flechsig E, Cozzio A, von Mering C. Expression of Amino-Terminally Truncated PrP in the Mouse Leading to Ataxia and Specific Cerebellar Lesions. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- Soule HD, Vazguez J, Long A, Albert S, Brennan M. A human cell line from a pleural effusion derived from a breast carcinoma. J Natl Cancer Inst. 1973;51:1409–1416. doi: 10.1093/jnci/51.5.1409. [DOI] [PubMed] [Google Scholar]
- Swietnicki W, Petersen RB, Gambetti P, Surewicz WK. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J Biol Chem. 1998;273:31048–31052. doi: 10.1074/jbc.273.47.31048. [DOI] [PubMed] [Google Scholar]
- Vanik DL, Surewicz WK. Disease-associated F198S mutation increases the propensity of the recombinant prion protein for conformational conversion to scrapie-like form. J Biol Chem. 2002;277:49065–49070. doi: 10.1074/jbc.M207511200. [DOI] [PubMed] [Google Scholar]
- Viles JH, Cohen FE, Prusiner SB, Goodin DB, Wright PE, Dyson HJ. Copper binding to the prion protein: Structural implications of four identical cooperative binding sites. Proc Natl Acad Sci U S A. 1999;96:2042–2047. doi: 10.1073/pnas.96.5.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD. BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ. 2006;13:1339–1350. doi: 10.1038/sj.cdd.4401992. [DOI] [PubMed] [Google Scholar]
- Walz R, Amaral OB, Rockenbach IC, Roesler R, Izquierdo I, Cavalheiro EA, Martins VR, Brentani RR. Increased Sensitivity to Seizures in Mice Lacking Cellular Prion Protein. Epilepsia. 1999;40:1679–1682. doi: 10.1111/j.1528-1157.1999.tb01583.x. [DOI] [PubMed] [Google Scholar]
- Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 2001;20:5383–5391. doi: 10.1093/emboj/20.19.5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X-M, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- Zahn R, Liu A, Luhrs T, Riek R, von Schroetter C, Lopez Garcia F, Billeter M, Calzolai L, Wider G, Wuthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanata SM, Lopes MH, Mercadante AF, Hajj GNM, Chiarini LB, Nomizo R, Freitas ARO, Cabral ALB, Lee KS, Juliano MA, De Oliveira E, Jachieri SG, Burlingame A, Huang L, Linden R, Brentani RR, Martins VR. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21:3307–3316. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanusso G, Petersen RB, Jin T, Jing Y, Kanoush R, Ferrari S, Gambetti P, Singh N. Proteasomal Degradation and N-terminal Protease Resistance of the Codon 145 Mutant Prion Protein. J Biol Chem. 1999;274:23396–23404. doi: 10.1074/jbc.274.33.23396. [DOI] [PubMed] [Google Scholar]