

Abstract
High Temperature Requirement A2 (HtrA2/Omi) is a mitochondrial protease that exhibits pro-apoptotic and cell protective properties and has been linked to Parkinson disease (PD). Impaired mitochondrial function is a common trait in PD patients, and is likely to play a significant role in pathogenesis of parkinsonism, but the molecular mechanisms remain poorly understood. Genetic studies in Drosophila have provided valuable insight into the function of other PD-linked genes, in particular PINK1 and parkin, and their role in maintaining mitochondrial integrity. Recently, HtrA2 was shown to be phosphorylated in a PINK1-dependent manner, suggesting it might act in the PINK1 pathway. Here, we describe the characterization of mutations in Drosophila HtrA2, and genetic analysis of its function with PINK1 and parkin. Interestingly, we find HtrA2 appears to be dispensable for developmental or stress-induced apoptosis. In addition, we found HtrA2 mutants share some phenotypic similarities with parkin and PINK1 mutants, suggesting that it may function in maintaining mitochondrial integrity. Our genetic interaction studies, including analysis of double-mutant combinations and epistasis experiments, suggest HtrA2 acts downstream of PINK1 but in a pathway parallel to Parkin.
Introduction
The mammalian HtrA (High Temperature Requirement) family of proteins show high homology to the bacterial chaperones DegS and DegP. 1,2 DegS, a serine protease, acts as a stress sensor by interacting with unfolded outer membrane porins via its PDZ domain. 3 This releases DegS proteolytic activity, triggering a cascade that results in activation of stress response genes via the transcription factor σE. Mammalian HtrA2/Omi, is a mitochondrial protein with both pro-apoptotic and cell protective roles. 4 Its pro-apoptotic function is exerted through binding and cleavage of IAPs (Inhibitor of Apoptosis Proteins) upon its release into the cytoplasm following a pro-apoptotic stimulus. 5-8 In Drosophila, HtrA2 has also been reported to be released from the mitochondria upon cellular insults such as UV irradiation, and to cleave DIAP1, the principal Drosophila IAP. 9-11
However, genetic data have suggested that mammalian HtrA2, like its bacterial counterpart, normally functions as a stress response gene, preserving mitochondrial integrity. mnd2 (motor neuron degeneration) mice, which have an inactivating mutation in the HtrA2 protease domain, show muscle wasting and neurodegeneration. 12 HtrA2 knockout mice have neuronal degeneration in a subset of striatal neurons and exhibit a parkinsonian phenotype, abnormal mitochondria and reduced lifespan. 13
In further support of a protective role, growing evidence suggests a link between HtrA2 and Parkinson disease (PD), a progressive neurodegenerative disorder of unknown aetiology. Two mutant alleles of HtrA2 (A141S and G399S) have been found in PD patients, leading to the classification of HtrA2 as PARK13 by OMIM. 14 Although one of these genetic variants was later found in non-PD controls, 15,16 Bogaerts et al. identified a new mutation (Arg404) in a large cohort of Belgian PD patients, confirming a role for HtrA2 in PD susceptibility. 17 Importantly, recent studies have shown that HtrA2 forms a complex with the PD-related factor PINK1, a mitochondrial-targeted kinase. 18 Moreover, HtrA2 is phosphorylated in a PINK1-dependent manner in response to p38 SAPK (Stress Activated Protein Kinase) pathway activation, suggesting that PINK1 can modulate HtrA2 activity as part of mitochondrial stress response. Overexpression studies in flies suggest PINK1 and HtrA2 may be functionally related 19,20, although their precise relationship remains unclear. We took advantage of Drosophila genetics to examine in vivo the function of HtrA2 in the PINK1 pathway and assess its putative role in apoptosis.
Results
The Drosophila HtrA2 homologue, encoded by CG8464, contains a predicted N-terminal mitochondrial targeting sequence (MTS), a trans-membrane domain (TM), a central protease domain, a C-terminal PDZ domain and an unconventional IAP-binding motif (Figure S1a and 10). HtrA2 encodes a full-length protein of ~46kDa. Upon mitochondrial import, HtrA2 is cleaved to yield two products of 37 and 35kDa. We have expressed and purified HtrA2 in bacteria and tested its activity towards an HtrA2 fluorescent peptide substrate (H2-Opt) as well as a control peptide as previously described. 8 These experiments revealed that HtrA2 efficiently cleaves the H2-Opt substrate but not a control peptide, suggesting that Drosophila HtrA2 has similar substrate specificity to its mammalian homologue (Figure S1b).
To address the in vivo function of HtrA2, we generated a mutant allele by imprecise P-element excision of G4907 (Genexel Inc.), which is inserted between HtrA2 and mRpL11 (mitochondrial Ribosomal Protein-like 11; CG3351) (Figure 1a). The G4907 stock is homozygous lethal and could be mutant for either mRpL11, HtrA2 or both. We mobilized G4907 and generated a deletion removing 1037bp from the insertion site to exon 1 of HtrA2 leaving 8bp of exon 1 (Figure 1a). This allele (HtrA2Δ1) is homozygous lethal and lacks both the transcription and translation start sites of HtrA2, as well as the promoter region and transcription start site of mRpL11, and is therefore likely to be a null allele for both genes. We also recovered a precise excision as a control.
Figure 1. Generation of HtrA2 mutants.

(a) Schematic representation of the HtrA2 genomic region. The P-element G4907 (black triangle) was excised to produce a deletion of 1037bp (red line). Position of start (ATG) and stop (*) codons are indicated. Lengths of the genomic rescue constructs are indicated. (b) The HtrA2Δ1 deletion mutant was balanced over TM3, primers from the genomic rescue construct were used to amplify a 2.5kb (WT) product from TM3 and the resulting 1.8kb deletion product (deletion). The positive control (+) was the amplification of the genomic product from wiso flies, (-) Negative control (no DNA). (c) RT-PCR shows the absence of HtrA2 transcript in mutants (HtrA2Δ1). The HtrA2 transcript can be amplified from the genomic rescued line (HtrA2Δ1 rescue), negative control (-, no RNA). (d) Western blot shows the absence of HtrA2 protein in mutants (HtrA2Δ1) and its presence in control and rescue flies.
We generated genomic rescue constructs for both HtrA2 and mRpL11 (gHtrA2 and gmRpL11, Figure 1a and methods). The lethality of HtrA2Δ1 was rescued by gmRpL11 but not by gHtrA2, indicating that mRpL11 is required for viability, as expected for a ribosomal component. HtrA2Δ1 homozygotes bearing a gmRpL11 rescue transgene represent null alleles solely for HtrA2, indicating HtrA2 is non-essential.
Therefore, to further investigate the phenotypes of HtrA2 mutant flies, we compared flies mutant for HtrA2 and mRPL11 bearing two copies of the gmRpL11 construct (gmRpL11; HtrA2Δ1), which will be referred to as ‘HtrA2Δ1’, with precise excision control flies as a wild type. As an additional control, reversion of phenotypes was tested in flies carrying the HtrA2Δ1 deletion and bearing two copies each of rescue constructs for both mRpL11 and HtrA2 (gmRpL11,gHtrA2; HtrA2Δ1), which will be referred to as ‘HtrA2Δ1 rescued’.
The HtrA2 transcripts are expressed ubiquitously and uniformly in all tissues tested (Figure S2a-f). The precise excision, HtrA2Δ1 and HtrA2Δ1 rescued flies were tested by genomic PCR, RT-PCR, and western blot to confirm the deletions and the absence of both transcript and HtrA2 protein in mutants (Figure 1b-d).
Several reports have suggested that HtrA2 can promote apoptosis by cleaving DIAP1, thus releasing the apical caspase Dronc in response to pro-apoptotic stimuli. 9-11 We therefore monitored cell death using an antibody directed against cleaved-Caspase 3 in wing imaginal discs treated with γ-rays (4Gy), Staurosporine (STS - 4μM) and ultraviolet (UV) light (2.5 kJ/m2). Apoptosis induction was identical in control (precise excision) and HtrA2Δ1 mutant discs, both without treatment or upon treatment (Figure 2a-f). In addition, the development of HtrA2Δ1 animals is completely normal, in particular showing normal eye morphology, indicating that HtrA2 is dispensable for retinal apoptosis during development (data not shown). This is consistent with findings in HtrA2 knockout mice 13 and suggests that HtrA2 is dispensable for developmental and apoptosis induced by multiple stimuli in vivo.
Figure 2. Apoptosis is normal in HtrA2 mutants.

(a-d) Confocal micrographs of the pouch region of wing imaginal discs stained with anti-cleaved caspase 3 (red) and phalloidin (green) after mock treatment or exposure to 4Gy γ-rays. No difference was detected between precise excision controls (a, b) and HtrA2Δ1 mutants (c, d). Posterior is to the right. Scale bar = 50μm. (e) Quantification of the apoptotic index from wing discs after treatment with γ-rays (see materials and methods). n=10 for each genotype. (f) Quantification of the apoptotic index from wing discs after treatment with Staurosporine or UV light (see materials and methods). n=8 for each genotype. Error bars represent standard deviations.
To determine whether HtrA2 plays a role in maintenance of cellular homoeostasis by the PINK1 pathway, we assessed whether HtrA2Δ1 flies share phenotypic similarities with PINK1 mutants. Drosophila PINK1 mutants exhibit locomotor defects in flight and climbing behaviours, widespread degeneration of indirect flight muscles (IFMs) with accompanying mitochondrial morphology defects, male sterility and partial loss of dopaminergic neurons. 21-23 Strikingly, all these phenotypes are phenocopied in Drosophila mutants of another PD-linked gene, parkin 24-27, which was shown to act genetically downstream of PINK1. 21-23 Thus, we investigated such phenotypes in HtrA2 mutants.
Myopathology was investigated using Toluidine Blue staining of IFMs in longitudinal sections of adult thoraces (Figure 3a-c). While marked myopathology akin to that described in parkin and PINK1 mutants was not observed (in 5 or 30 day old animals), more detailed investigation will be needed to determine whether Htr2A dysfunction leads to overt myodegeneration. However, examination of IFM ultrastructure from aged HtrA2Δ1 mutants revealed increased numbers of defective mitochondria, displaying reduced electron density and open cristae, compared with wild type and HtrA2Δ1 rescued controls (Figures 3c-f and S3). It should be noted that the mitochondrial abnormalities seen in HtrA2Δ1 mutants are not widespread, suggesting this does not reflect a general mitochondrial defect. To confirm this, we measured mitochondrial activity by high-resolution respirometry in whole flies using 30 day-old males. Coupled mitochondria from WT and HtrA2Δ1 showed no difference in oxygen consumption with substrates of complex I and complex II in the presence of ADP (Figure S4a, b). This was confirmed by measuring respiratory activities of complex II and complex IV under uncoupled conditions (FigureS4c, d). Thus, there is no systemic mitochondrial deficiency in HtrA2Δ1 mutants compared with controls.
Figure 3. HtrA2 mutants show mild mitochondrial disruption and progressive locomotor deficits.

(a-c) Toludine blue stained, whole thorax sections of 30 day old flies revealed overall IFM structure is maintained in HtrA2Δ1 mutant flies. (d-f) IFM mitochondria in TEM micrographs exhibit morphological abnormalities, typically reduced electron density and disrupted cristae in HtrA2Δ1 mutant (arrow in e), compared to wild type (d) and HtrA2Δ1 rescue flies (f). (g) Quantification of the mitochondrial phenotype from multiple individuals. Bars show mean and standard error (see methods). HtrA2Δ1 mutant flies have an increased proportion of mitochondria with reduced electron density, as determined by a Fisher exact test (*p<0.01, # = 0.058). HtrA2Δ1 mutants show progressive deficits in flight (h) and climbing (i). 5 day old mutant flies have wild type flight and climbing ability. Aged (30 day old) mutants show reduced flight and climbing, which is restored in HtrA2Δ1 rescue flies. Bars show mean and standard error, inset numbers show n. ***, p<0.0001; **, p<0.001; *p<0.01, by Kruskal-Wallis non-parametric analysis and Dunn’s pair-wise comparison.
To determine if, like PINK1 and parkin mutants, HtrA2Δ1 mutant flies present locomotor deficits, HtrA2Δ1 mutants were assayed for climbing and flight ability. HtrA2Δ1 mutants showed wild-type flight and climbing ability at 5-days of age (Figure 3h, i). However, 30-day-old HtrA2Δ1 flies show significantly reduced flight and climbing ability compared with wild type flies. This defect was completely restored in HtrA2Δ1 rescued flies (Figure 3h, i). Thus, the progressive mitochondrial morphological abnormalities seen in HtrA2Δ1 mutants correlates with progressive motor deficits.
In further similarity to parkin/PINK1 mutants, we also find HtrA2Δ1 males are sterile, while female fertility is unaffected. HtrA2Δ1 males are almost completely sterile (4% fertility compared to 100% fertility in control flies - Figure 4a). Importantly, male fertility is restored to wild-type levels in HtrA2Δ1 rescued lines (Figure 4a). However, while PINK1/parkin mutants exhibit distinct morphological defects during spermatogenesis, 21,22,27,28 the ultrastructural analysis of HtrA2Δ1 testes revealed no observable defects (Figure 4b). Indeed, mature individualized sperm are formed properly, however, we noted that HtrA2Δ1 sperm were completely immotile, as was the case for PINK1 mutants (see movies S1-3 in supplementary materials).
Figure 4. HtrA2Δ1 have normal spermatogenesis but are male sterile.
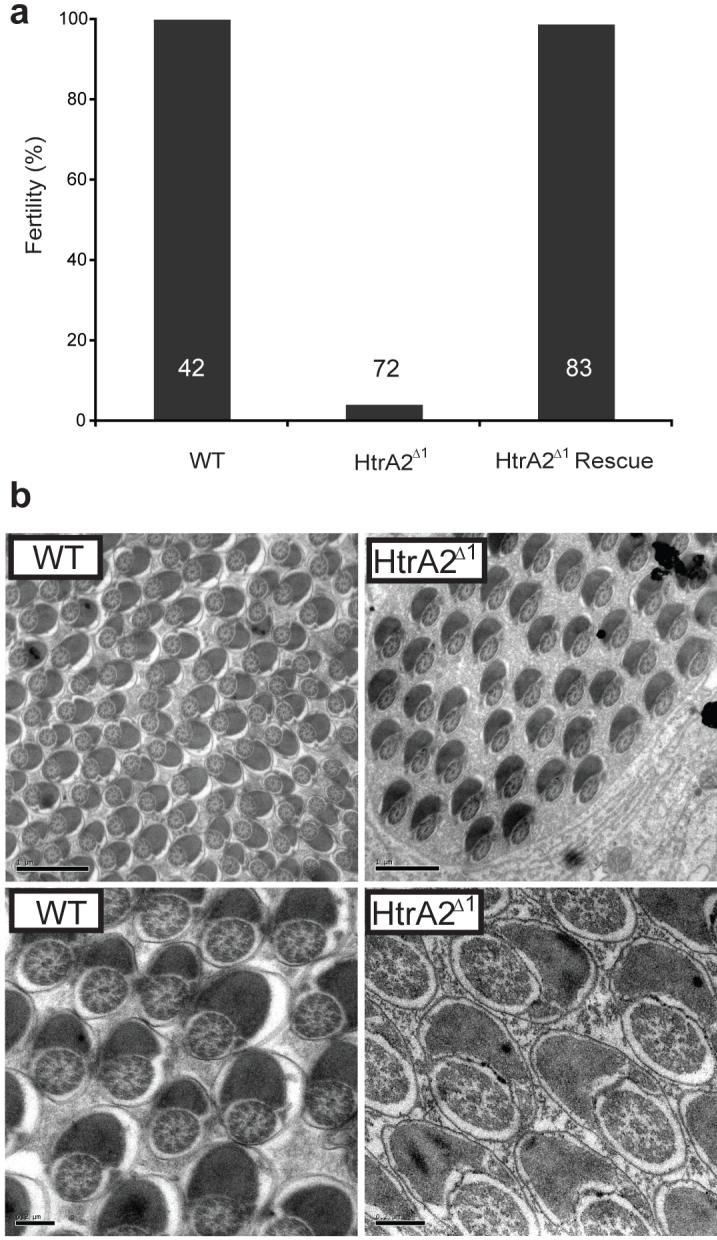
(a) HtrA2Δ1 males have greatly reduced fertility compared to controls. Normal fertility is regained in HtrA2Δ1 rescued flies. Inset numbers represent n value. (b) TEM micrographs of mature individualized spermatids show no structural defects in the axoneme or Nebenkern. Scale bar shows 1 μm and 0.2 μm in upper and lower panels respectively.
Recent evidence indicates HtrA2 likely plays a role in cellular stress response mechanisms. 18 To test this we determined the effect of loss of HtrA2Δ1 function on longevity and stress resistance. HtrA2Δ1 mutants have a reduced lifespan compared to rescued or precise excision controls (Figure 5a). HtrA2Δ1 flies were also sensitive to paraquat-induced oxidative stress and mitochondrial respiration deficit induced by complex I inhibitor rotenone (Figure 5b, c).
Figure 5. HtrA2Δ1 have reduced survival under normal and stressed conditions.

(a) Male HtrA2Δ1 flies (red) have reduced lifespan compared with wild type control (blue), which is restored with a genomic rescue construct (black) (n=75 for each genotype). (b) HtrA2Δ1mutant flies (red) are less resistant to oxidative stress resulting from paraquat ingestion compared to wild type controls (blue) or rescued flies (black). n= 200 for each genotype. (c) HtrA2Δ1mutant flies are more sensitive to rotenone-induced oxidative stress (red) than wild type controls (blue) or the HtrA2Δ1 rescued line (black). n= 200 for each class. Data is presented as Kaplan-Meier survival distributions, Log-rank tests were performed to determine statistical significance. Individual p values are shown with the figure.
Given the evidence linking HtrA2 to PD, we determined whether the dopaminergic neurons are affected in HtrA2Δ1 flies. While parkin and PINK1 mutants show age-dependent dopaminergic neuron loss, 21,23,25 we found 30 day old HtrA2Δ1 flies showed no difference in number of dopaminergic neurons (Figure 6). However, this is also consistent with the weaker phenotypes in other tissues.
Figure 6. Loss of HtrA2 does not cause dopaminergic neurons loss.

Number of neurons staining positive with anti-tyrosine hydroxylase antiserum from aged (30-day) precise excision control (black) and HtrA2 mutants (grey). No difference is seen in any of the clusters. Bars show mean and standard error (n≥10).
Thus, HtrA2Δ1 mutant flies share a number of phenotypic similarities with parkin/PINK1 mutants, though generally the HtrA2Δ1 phenotypes are weaker. To address whether HtrA2 functions in a common or divergent pathways to PINK1/Parkin we generated double mutant combinations and determined the effect on climbing ability. Previously, PINK1 and Parkin have been reported to act in a common pathway, consistent with the observation that PINK1:parkin double mutants do not have a more severe phenotype than either single mutant. 21-23 Corroborating these findings we find PINK1:parkin double mutants do not show a difference in climbing ability compared to single mutants of either PINK1 or parkin (Figure 7a). Similarly, we find PINK1:HtrA2 double mutants show no difference in climbing ability compared with PINK1 mutants (Figure 7b), consistent with them acting in a common pathway. In contrast, we find that HtrA2:parkin double mutants show a dramatically enhanced climbing defect in comparison to parkin mutants (Figure 7c), suggesting that HtrA2 might act in a different pathway from Parkin. The PINK1 mutant defects are rescued by overexpression of parkin, leading to the suggestion that Parkin acts downstream of PINK1. 21-23 We therefore generated transgenic animals expressing HtrA2 under GAL4/UAS control and tested its ability to rescue PINK1 phenotypes. Interestingly, ubiquitous expression of the HtrA2 transgene significantly rescued the PINK1 climbing defects, (Figure 8). Together these results suggest HtrA2 likely plays a role in the PINK1 pathway.
Figure 7. HtrA2 mutants enhance the locomotor deficit exhibited by parkin mutants but not PINK1 mutants.

(a) Climbing ability of PINK1B9, park25 and PINK1B9:park25 double mutants. (b) Climbing ability of HtrA2Δ1, PINK1B9 and PINK1B9:HtrA2Δ1 double mutants. (c) Climbing ability of HtrA2Δ1, park25 and park25:HtrA2Δ1 double mutants. Bars show mean and standard error. n ≥ 50.
Figure 8. Overexpression of HtrA2 can rescue PINK1 climbing defect.

Compared to heterozygous controls, PINK1 mutants have reduced climbing ability, which is suppressed by overexpression of HtrA2 under the control of the actin5c promoter. Bars show mean and standard error, n>50. Significance was established using a Kruskal Wallis test and Dunn’s comparison (***, p<0.0001; *, p<0.01).
Discussion
We have characterized the Drosophila homologue of the HtrA2 protease and found that HtrA2 mutants share some phenotypic characteristics with previously described Drosophila models of PD. HtrA2 null mutants are viable but exhibit mild mitochondrial defects, loss of flight and climbing ability, male infertility and sensitivity to oxidative stress and mitochondrial toxins (Figures 3-6). In contrast, we find that HtrA2 is not required for developmental or stress-induced apoptosis (Figure 2). These data are consistent with findings in mice and humans, which suggest that, HtrA2 functions primarily to maintain mitochondrial integrity and protect cells against oxidative stress12,13, and contradicts previous reports that HtrA2 is a pro-apoptotic factor. 9-11
Recently, Yun et al have reported a characterization of ethanemethylsulfonate-induced alleles of Drosophila HtrA2/Omi. 20 In accordance with our results, they show that HtrA2 is required for male fertility, normal lifespan and stress resistance. They do not report a mitochondrial defect, though the mild nature of the defect observed in our hands and allelic or environmental differences might explain this discrepancy. Both of our reports suggest that HtrA2 mutants have a markedly weaker phenotype than PINK1, which is consistent with the idea that HtrA2 does not play a critical role in the PINK1-Parkin interaction. However, our genetic interaction studies presented here, in conjunction with our previous evidence19, support the view that HtrA2 acts downstream of PINK1 in a divergent pathway parallel to Parkin.
We have previously shown that a PINK1 over-expression phenotype is partially suppressed by loss of HtrA2, while conversely an HtrA2 overexpression phenotype is not suppressed by PINK1 mutations19, suggesting that HtrA2 functions downstream of PINK1. In a complementary approach using a loss of function phenotype, we show here that overexpression of HtrA2 can partially substitute for loss of PINK1 (Figure 8). These findings are consistent with our previous report and suggest that HtrA2 acts downstream of PINK1. While the study of Yun et al. did not uncover evidence for HtrA2 acting downstream of PINK1, technical differences, particularly reflecting transgenic expression levels, may account for this discrepancy.
In agreement with Yun et al. we have shown that, similar to PINK1:parkin double mutants, PINK1:HtrA2 double mutants display an identical phenotype to PINK1 mutants alone, suggesting they act in a common pathway (Figure 7). However, we also showed that parkin:HtrA2 double mutants display a stronger phenotype than either mutant alone (Figure 7), suggesting HtrA2 acts in parallel pathway to Parkin. These results are also consistent with our previous findings that loss of either HtrA2 or parkin can partially suppress a PINK1 overexpression phenotype, while attenuating both genes together is sufficient to completely suppress the overexpression of PINK1. 19 Together these findings support the notion that HtrA2 and Parkin are acting as downstream effectors of PINK1 but in parallel pathways. The relative weakness of the HtrA2 mutant phenotype suggests that it is not the predominant PINK1 effector.
The most pressing questions remain how this pathway is triggered and how its activation leads to protection from mitochondrial stress. By analogy to bacterial DegS, it is possible that the PDZ domain of HtrA2 acts as a sensor for unfolded proteins. In addition, we and others have shown that the mitochondrial protease Rhomboid-7/PARL can process both PINK1 and HtrA2, 19,29 which may represent a regulatory process. Lastly, the p38 SAPK pathway is known to be triggered by reactive oxygen species, which are produced when mitochondrial electron transport is perturbed. 30 Mitochondrial dysfunction may therefore modulate HtrA2 activity at several levels. Once activated, HtrA2 may cleave unfolded protein and/or elicit a transcriptional response to clear the damaged proteins, like DegS 4. Further work will be required to fully elucidate the functional relationship between PINK1 and HtrA2, and the involvement of this putative pathway in PD pathogenesis.
EXPERIMENTAL PROCEDURES
Drosophila genetics
HtrA2 mutants were created by imprecise excision of P-element G4907 (Genexel Inc.). Breakpoints were mapped by genomic PCR and sequencing. PINK1B9 mutants were a kind gift from J. Chung. 21 park25 mutants have been described previously. 27 All fly experiments were carried out at 25°C and on the same food batch for each experiment.
For crosses to combine PINK1 mutants with GAL4/UAS transgenes, paternal males with marked X chromosomes (y- or FM6) were used to ensure correct identification of PINK1 mutant progeny.
Fertility assays
Single 1-day-old males of each genotype were individually placed in a vial with 3 virgin females. The number of vials with larvae was recorded after 5 days.
Flight/climbing assay
Flight/climbing assays were performed as previously described. 27 n = ≥30 per genotype, unless otherwise stated within figures. Statistical significance calculated by Kruskal-Wallis non-parametric analysis and Dunn’s pair-wise comparison.
Longevity assay
50 0-4-hours-old males and females of each genotype were placed into separate vials in groups of 25 for each gender. The flies were transferred to fresh food every 2 days and the number of live flies recorded. Data is presented as Kaplan-Meier survival distributions and significance determined by Log-rank tests.
Oxidative stress assay
the procedure used for longevity assay was repeated for oxidative stress assays using food containing 20mM paraquat or 20mM rotenone. Data is presented as Kaplan-Meier survival distributions and significance determined by Log-rank tests.
Molecular Biology
RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA). RT-PCR was performed using the kit ‘SuperScript™ III One-step RT-PCR with Platinum Taq (Invitrogen). The 5′ and 3′ sequences used to amplify the dHtrA ORF are ATGGCTTTGCGCGGTTCC and CTAGGGATCTTCTGGCGTA respectively. Primers used to amplify 5′ and 3′ dPINK1 ORF are ATGTCTGTGAGACTGCTGAC and CTACGCCACCACATTCTGGA respectively. 5′ primer GTTGTGCCGCTGTCGTCTAT and 3′ primer CGTCGTGCGAAACGTACG were used to amplify a product of 1kb between exon 2 and exon 3 respectively to detect transcripts by RT-PCR in mutants.
Genomic Rescue Constructs
The predicted full-length HtrA2 gene, 700bp of upstream promoter region (including the complete 3′ UTR) was amplified by PCR using 5′ primer CAACTCGAGGAAGTACATTGGGCGGGTC and 3′ primer GGGACTAGTGGGTTTGTCAGCGATTTC, sequenced and subcloned into pCaSpeR-HS using XhoI and SpeI restriction sites. mRPL11 was constructed by including 463bp of upstream promoter region, the entire predicted gene and 108bp of downstream genomic region using 5′ primer GGGTCTAGAGCAGCTGATTTCAATTTGGC with XbaI restriction site and 3′ primer GGGGAATTCCACTGGAAAACTCAACAAGC with EcoRI restriction site, sequenced and subcloned into pCaSpeR-HS.
pUASp construct
the dHtrA2 ORF was amplified using 5′ primer TCACGCGGCCGCATGGCTTTGCGCGGTTCC containing NotI restriction site, and 3′ primer ACAGCTCGAGCTAGGGATCTTCTGGCGTA containing XhoI restriction site. The sequenced, digested product was subcloned into pUASp. Western blotting was performed by standard procedures. The anti-HtrA2 antibody (used at 1:1000) was a kind gift from Dr E. Alnemri (Kimmel Cancer Institute, USA).
Cell Biology
Tissue sectioning
Thoraces and testes were prepared from 30-day old and 2-day old, adult flies, respectively and fixed overnight in 2% paraformaldehyde, 2.5% gluteraldehyde in sodium cacodylate (0.1M). Thoraces were rinsed in sodium cacodylate (0.1M) and 1% tannic acid and post-fixed in 1:1 2% OsO4 and sodium cacodylate (0.2M) for 1h. After rinsing, thoraces were dehydrated in an ethanol series and embedded using Spurr’s resin. Semi-thin sections were then taken and stained with Toludine blue, while ultra-thin sections were examined using TEM. Quantification of the mitochondrial phenotype was carried out under blinded conditions for both imaging and analysis. 10 randomly selected fields of view were captured per individual and the number of mitochondria exhibiting a disrupted morphology phenotype and those with normal mitochondria were counted. Approximately 1500 mitochondria were examined per genotype. Pair-wise differences in proportion of mitochondria exhibiting the phenotype were made utilizing the conservative Fisher exact test.
Dopaminergic neuron counts
Brains were dissected from 30-day old flies and fixed in 4% para-formaldehyde, rinsed in PBS and incubated with anti-TH antibody (Immunostar Inc., Hudson, WI, USA) in PBS-T BSA (0.5%) overnight (4°C). After washing with PBS-T, brains were incubated (2h) with Alexafluor 488 secondary antibody (Invitrogen) in PBS-T-BSA (0.5%). After washing brains were mounted and TH-positive neurons imaged by confocal microscopy. In all cases, TH-positive neurons were counted with the investigator blinded to the genotypes.
Apoptosis assay
third instar wing imaginal discs were exposed to 4Gy γ-rays, fixed and stained with anti-caspase 3 after 2h (Cell Signaling Technologies, Danvers, MA, USA) and FTIC-phalloidin (Sigma, Highland, IL, USA) as previously described 31. For STS (4μM) and UV treatment (2.5 kJ/m2), larvae were dissected and discs were incubated in Schneider’s medium (Invitrogen, Carlsbad, CA, USA), then fixed and stained after 4h. Images were acquired on a Zeiss LSM 510 confocal microscope. The apoptotic index corresponds to the percentage of caspase 3-positive pixels relative to the total area, and was determined as described using Image J. 31 For consistency, the analysis was performed on the pouch region of the wing discs.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank J. Chung and the Bloomington Drosophila Stock Center for fly stocks. T. Gilbank, S. Murray and F. Earl for fly transgenics, G. Kelly for help with statistical analysis, C. Gray for help with movies and K. Klupsch for comments on the manuscript. We are grateful to M. Guo for sharing data prior to publication. AW and LT acknowledge the Department of Biomedical Science, Centre for Electron Microscopy for assistance, and the Light Microscopy Facility is supported by the Wellcome Trust (Grant No. GR077544AIA). AW is funded by the Parkinson’s Disease Society (G-4063, G-0713) and the Wellcome Trust (081987). The MRC Centre for Developmental and Biomedical Genetics is supported by Grant G070091. LMM is supported by the MRC (UK). Work in the Tapon and Downward laboratories is supported by Cancer Research UK. HPF is supported by the MRC Grant G0700183.
REFERENCES
- 1.Clausen T, Southan C, Ehrmann M. The HtrA family of proteases: implications for protein composition and cell fate. Mol Cell. 2002;10:443–55. doi: 10.1016/s1097-2765(02)00658-5. [DOI] [PubMed] [Google Scholar]
- 2.Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun. 2003;304:499–504. doi: 10.1016/s0006-291x(03)00622-3. [DOI] [PubMed] [Google Scholar]
- 3.Sohn J, Grant RA, Sauer RT. Allosteric Activation of DegS, a Stress Sensor PDZ Protease. Cell. 2007;131:572–83. doi: 10.1016/j.cell.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 4.Vaux DL, Silke J. HtrA2/Omi, a sheep in wolf’s clothing. Cell. 2003;115:251–3. doi: 10.1016/s0092-8674(03)00851-1. [DOI] [PubMed] [Google Scholar]
- 5.Srinivasula SM, Gupta S, Datta P, Zhang Z, Hegde R, Cheong N, et al. Inhibitor of apoptosis proteins are substrates for the mitochondrial serine protease Omi/HtrA2. J Biol Chem. 2003;278:31469–72. doi: 10.1074/jbc.C300240200. [DOI] [PubMed] [Google Scholar]
- 6.Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17:1487–96. doi: 10.1101/gad.1097903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Loo G, van Gurp M, Depuydt B, Srinivasula SM, Rodriguez I, Alnemri ES, et al. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002;9:20–6. doi: 10.1038/sj.cdd.4400970. [DOI] [PubMed] [Google Scholar]
- 8.Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–44. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 9.Igaki T, Suzuki Y, Tokushige N, Aonuma H, Takahashi R, Miura M. Evolution of mitochondrial cell death pathway: Proapoptotic role of HtrA2/Omi in Drosophila. Biochem Biophys Res Commun. 2007;356:993–7. doi: 10.1016/j.bbrc.2007.03.079. [DOI] [PubMed] [Google Scholar]
- 10.Challa M, Malladi S, Pellock BJ, Dresnek D, Varadarajan S, Yin YW, et al. Drosophila Omi, a mitochondrial-localized IAP antagonist and proapoptotic serine protease. EMBO J. 2007;26:3144–56. doi: 10.1038/sj.emboj.7601745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan FS, Fujioka M, Datta P, Fernandes-Alnemri T, Jaynes JB, Alnemri ES. The interaction of DIAP1 with dOmi/HtrA2 regulates cell death in Drosophila. Cell Death Differ. 2008 doi: 10.1038/cdd.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–7. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- 13.Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–62. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, et al. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson’s disease. Hum Mol Genet. 2005;14:2099–111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- 15.Ross OA, Soto AI, Vilarino-Guell C, Heckman MG, Diehl NN, Hulihan MM, et al. Genetic variation of Omi/HtrA2 and Parkinson’s disease. Parkinsonism Relat Disord. 2008;14:539–43. doi: 10.1016/j.parkreldis.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simon-Sanchez J, Singleton AB. Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum Mol Genet. 2008;17:1988–93. doi: 10.1093/hmg/ddn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bogaerts V, Nuytemans K, Reumers J, Pals P, Engelborghs S, Pickut B, et al. Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson disease. Hum Mutat. 2008;29:832–40. doi: 10.1002/humu.20713. [DOI] [PubMed] [Google Scholar]
- 18.Plun-Favreau H, Klupsch K, Moisoi N, Gandhi S, Kjaer S, Frith D, et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat Cell Biol. 2007;9:1243–52. doi: 10.1038/ncb1644. [DOI] [PubMed] [Google Scholar]
- 19.Whitworth AJ, Lee JR, Ho VM, Flick R, Chowdhury R, McQuibban GA. Rhomboid-7 and HtrA2/Omi act in a common pathway with the Parkinson’s disease factors Pink1 and Parkin. Dis Model Mech. 2008;1:168–74. doi: 10.1242/dmm.000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yun J, Cao JH, Dodson MW, Clark IE, Kapahi P, Chowdhury RB, et al. Loss-of-function analysis suggests that Omi/HtrA2 is not an essential component of the PINK1/PARKIN pathway in vivo. J Neurosci. 2008;28:14500–10. doi: 10.1523/JNEUROSCI.5141-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–61. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 22.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–6. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. 2006;103:10793–8. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, et al. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–94. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- 25.Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2005;102:8024–9. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum Mol Genet. 2005;14:799–811. doi: 10.1093/hmg/ddi074. [DOI] [PubMed] [Google Scholar]
- 27.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–83. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riparbelli MG, Callaini G. The Drosophila parkin homologue is required for normal mitochondrial dynamics during spermiogenesis. Dev Biol. 2007;303:108–20. doi: 10.1016/j.ydbio.2006.10.038. [DOI] [PubMed] [Google Scholar]
- 29.Chao JR, Parganas E, Boyd K, Hong CY, Opferman JT, Ihle JN. Hax1-mediated processing of HtrA2 by Parl allows survival of lymphocytes and neurons. Nature. 2008;452:98–102. doi: 10.1038/nature06604. [DOI] [PubMed] [Google Scholar]
- 30.Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle. 2006;5:824–8. doi: 10.4161/cc.5.8.2685. [DOI] [PubMed] [Google Scholar]
- 31.Colombani J, Polesello C, Josue F, Tapon N. Dmp53 activates the Hippo pathway to promote cell death in response to DNA damage. Curr Biol. 2006;16:1453–8. doi: 10.1016/j.cub.2006.05.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.