

Abstract
Two types of 32 arm star polymers incorporating amphiphilic block copolymer arms have been synthesized and characterized. The first type, stPCL-PEG32, is composed of a polyamidoamine (PAMAM) dendrimer as the core with radiating arms having poly(ε-caprolactone) (PCL) as an inner lipophilic block in the arm and poly(ethylene glycol) (PEG) as an outer hydrophilic block. The second type, stPLA-PEG32, is similar but with poly(l-lactide) (PLA) as the inner lipophilic block. Characterization with SEC, 1H NMR, FTIR, and DSC confirmed the structure of the polymers. Micelle formation by both star copolymers was studied by fluorescence spectroscopy. The stPCL-PEG32 polymer exhibited unimolecular micelle behavior. It was capable of solubilizing hydrophobic molecules, such as pyrene, in aqueous solution, while not displaying a critical micelle concentration. In contrast, the association behavior of stPLA-PEG32 in aqueous solution was characterized by an apparent critical micelle concentration of ca. 0.01 mg/mL. The hydrophobic anticancer drug etoposide can be encapsulated in the micelles formed from both polymers. Overall, the stPCL-PEG32 polymer exhibited a higher etoposide loading capacity (up to 7.8 w/w % versus 4.3 w/w % for stPLA-PEG32) as well as facile release kinetics and is more suitable as a potential drug delivery carrier.
INTRODUCTION
Micelles formed from amphiphilic block copolymers have recently attracted significant attention in diverse fields of medicine and biology. In particular, polymeric micelles have been developed as drug and gene delivery systems (1-3) as well as carriers for various contrast agents in diagnostic imaging applications (4). In an aqueous environment, the hydrophobic blocks of the copolymer are expected to segregate into the core of the micelle, while the hydrophilic blocks form the corona or outer shell. Such a core–shell architecture is essential for the utility of the polymeric micelles as functional materials for pharmaceutical applications. The hydrophobic micelle core serves as a microenvironment for the incorporation of various therapeutic compounds, while the corona, the outer shell, serves as a stabilizing interface between the hydrophobic core and the external medium. As a result, polymeric micelles can be used as effective “nanocontainers” for reagents with poor solubility and/or low stability in physiological environments (5, 6). Interest in polymeric micelles for drug delivery has increased rapidly since the late 1980s (7). Most of the work has focused on classical micelles formed by intermolecular aggregation of amphiphilic block copolymers as the drug delivery vehicle, and the advantages of using their micelle structures as drug delivery systems have been demonstrated (8, 9).
The major factors that influence the performance of polymeric micelles for drug delivery are loading capacity, release kinetics, circulation time, biodistribution, size, stability, and stability on dilution (2). One of the approaches to improve the micelles’ stability is to develop star-shaped molecules in which the lipophilic components are covalently bound together within the micelle core. Core polymerization is an effective method to prevent dissociation of the block copolymer micelle. Kataoka's group has successfully employed this idea (10). In their study, the micelles were prepared from an amphiphilic block copolymer in which the hydrophobic block contained a polymerizable end group. After micellation, the end groups on the hydrophobic block were polymerized to form a stable core for the star-shaped polymer structure. The resulting micelles showed fairly high stability and maintained their small size. As anticipated, the core-polymerized micelle showed excellent solubilization of rather large molecules such as taxol (11). Another approach was reported by Uhrich et al. (12) with a star-shaped polymer composed of mucic acid substituted with a fatty acid as the lipophilic inner block and PEG as the hydrophilic outer block. This new type of molecule was capable of encapsulating a hydrophobic model drug in aqueous media.
The increased interest in amphiphilic star polymers for drug delivery has been demonstrated by several more recent publications. One paper (13) described the synthesis of a 4 arm star block copolymer of PCL and PEG using chemistry similar to that reported in this manuscript. Another report (14) described the preparation of a 4 arm star PCL-b-PEG polymer using a diethyl zinc catalyst. However, the molecular weight distribution of the block copolymer was unacceptably wide. Allen et al. (15) described the synthesis of a 6 arm star poly(δ-valerolactone)-b-PEG copolymer via a cationic polymerization method using fumaric acid as a catalyst. The micelle formation and biocompatibility of the star copolymer were demonstrated.
In a previous report (16), we described the synthesis and characterization of a 16 arm star PCL-PEG copolymer. The star structure of the polymers was confirmed by several physicochemical methods. The micellar properties of the star amphiphilic polymer in aqueous media were characterized by fluorescence techniques and dynamic light scattering (DLS). A series of entrapment experiments showed that various drugs could be incorporated in such micelles. These results indicate that the star-PCL-PEG micelle system is a promising drug carrier for the delivery of lipophilic drugs. The aggregation from unimolecular micelles into larger associated micelles was observed by DLS and pyrene fluorescence studies. It is likely that the 16 arm star polymer forms a relatively loose outer PEG shell, which is not sufficient to hinder the intermolecular association of the inner hydrophobic PCL blocks. It is expected that more stable unimolecular micelles may be obtainable by increasing the number of block copolymer arms. It has been demonstrated that when the number of arms reaches 64, star polymers exhibit characteristics of a hard sphere, and the repulsion between the star molecules becomes much stronger (17).
In this paper, we describe the synthesis and characterization of two types of 32 arm star polymers composed of amphiphilic block copolymer arms. The first type, stPCL-PEG32, is composed of a polyamidoamine (PAMAM) dendrimer as the core with radiating arms having poly(ε-caprolactone) (PCL) as an inner lipophilic block in the arm and poly(ethylene glycol) (PEG) as an outer hydrophilic block. The second type, stPLA-PEG32, is similar but with poly(l-lactide) (PLA) as the inner lipophilic block. The micelle formation of both star copolymers was studied by fluorescence spectroscopy. The affinity of the star polymer micelles for solubilizing small hydrophobic molecules was assessed by determining the aqueous partition coefficient of the lipophilic fluorescence probe pyrene. Finally, solubilization of the anticancer drug etoposide into the 32 arm star polymer micelles was studied to evaluate the relevance of such systems for drug delivery.
EXPERIMENTAL SECTION
Materials and Methods
PEG (Mn = 5000) with methoxy and carboxymethyl terminal groups was used as received from Nektar. ε-Caprolactone from Sigma-Aldrich was dried by stirring with CaH2 and distilled before use. l-Lactide was from Lancaster and was recrystallized twice from ethyl acetate before use. Dichloromethane was dried by stirring over and distilling from P2O5. All other reagents and solvents were used as received from Sigma-Aldrich.
1H NMR spectra were recorded on a Hitachi R-1500 FT-NMR spectrometer at 60 MHz. Infrared spectra were recorded on a Nicolet 750 FTIR spectrometer. UV–vis spectra were recorded on a HP 8452A UV–vis diode array spectrophotometer. Size exclusion chromatography (SEC) was used to determine the molecular weights, polydispersity, and degree of branching of the synthesized polymers. SEC measurements were conducted using a Hewlett-Packard series 1050 HPLC equipped with one Polymer Laboratories PL gel 5 μ mixed-D column and a Hewlett-Packard 1047A refractive index detector as well as Viscotek T60 light scattering (LS) and viscosity detectors. Tetrahydrofuran was used as the eluting solvent at a flow rate of 1 mL/min. Data analysis was performed using Viscotek Trisec GPC software version 3.0.
The thermal transitions of the polymers were determined using a Perkin-Elmer DSC7 differential scanning calorimeter under nitrogen purge. The samples were heated and cooled at a rate of 10 °C/min.
Synthesis of the 32 Arm Star-PCL Polymer (stPCL32)
A 100 mL three-neck flask equipped with a condenser and argon gas inlet was charged with PAMAM-OH Dendrimer Generation 3 with 32 terminal OH groups (20 wt % solution in methanol, 5.26 g, 0.151 mmol). Methanol was then removed under vacuum. A predetermined amount of ε-caprolactone (14.42 g, 126 mmol) followed by 15 mL toluene were added into the reaction flask. The mixture was heated to reflux using a Dean–Stark trap for 30 min, and the toluene was distilled off. The heating of the reaction mixture up to 145 °C resulted in the dissolution of the dendrimer in ε-caprolactone. After cooling the reaction mixture to 130 °C, 5 mg (0.012 mmol) of tin(II) 2-ethylhexanoate (Sn(Oct)2) was introduced, followed by stirring at 115−120 °C for 20 h. The reaction mixture was cooled to room temperature, dissolved in THF, and precipitated into cold methanol. After drying, 15.3 g (99% yield) of the white solid product was obtained. The polymer was further purified by fractionation. 3.6 g of polymer was dissolved in 70 mL of THF in a round-bottom flask, and methanol was added until the solution became cloudy. The solution was heated in a water bath at 60 °C and became clear. About 10 mL of methanol was added to reach a cloudy point, and the mixture was transferred into a hot separation funnel and allowed to stand at room temperature for three days. The polymer solution separated into two layers, and the top layer was removed without disturbing the lower layer. The lower layer was diluted with THF and precipitated into cold methanol. 2.76 g of polymer was obtained. 1H NMR (CDCl3): δ = 4.07 (t, 2H, OCH2), 2.31 (t, 2H, COCH2), 1.1−2 (m, 6H, (CH2)3). The characteristic peaks of dendrimer and polymer were overlapping, and peak integration was inconclusive.
Synthesis of the 32 Arm Star-PCL-PEG Polymer (stPCL-PEG32)
The stPCL32 (1.3 g, 0.0152 mmol), PEG (2.64 g, 0.527 mmol), and 4-(dimethylamino)pyridinium 4-toluenesulfonate (DPTS) (0.155 g, 0.527 mmol) were dissolved in 80 mL of dry CH2Cl2 by stirring at room temperature. Then diisopropylcarbodiimide (DIPC) (0.2 g, 1.58 mmol) was added into the reaction mixture via syringe. The reaction mixture was stirred overnight at room temperature. An additional amount of DIPC (0.15 g) was added to the reaction mixture followed by stirring for a total of 7 days. The reaction was stopped, and fractionation was used to purify the Star-PCL-PEG. Briefly, 20 mL CH2Cl2 was added to dilute the product. Hexane was slowly added to the resulting solution at a water bath of 45 °C until the appearance of cloudiness. Then the mixture was transferred into a hot separation funnel and was allowed to stand at room temperature. The top layer was removed, and the lower layer refractionated by the same procedure. Finally, the lower layer was diluted with CH2Cl2 and precipitated into hexane. After drying, 3.1 g (83% yield) of white solid product was obtained. 1H NMR (CDCl3): δ = 4.07 (t, 2H, COOCH2), 3.64 (s, 18.8H, OCH2CH2O), 2.31 (t, 2H, COCH2), 1.1−1.9 (m, 6H, (CH2)3).
Synthesis of the 32 Arm Star-PLA polymer (stPLA32)
A 100 mL three-neck flask equipped with a condenser and argon gas inlet was charged with PAMAM-OH Dendrimer Generation 3 (20 wt % solution in methanol, 5.26 g, 0.151 mmol). Methanol was then removed under vacuum. A predetermined amount of l-lactide (14.71 g, 102 mmol) followed by 15 mL toluene was added into the reaction flask. The mixture was heated to reflux using a Dean–Stark trap for 30 min and toluene was distilled off. Heating of the reaction mixture up to 135 °C resulted in the dissolution of the dendrimer in l-lactide. After cooling the reaction mixture to 125 °C, 5 mg (0.012 mmol) of Sn(Oct)2 was introduced, followed by stirring at 105 °C for 20 h. The reaction mixture was cooled to room temperature, dissolved in THF, and precipitated into cold methanol. After drying, 15.4 g (97% yield) of the white solid product was obtained. SEC analysis indicated that there were some low molecular weight impurities mixed with the star polymer. The polymer was further purified by fractionation using THF as the solvent and methanol as the nonsolvent. However, after repeated trials, including the use of chloroform as the solvent, the fractionation was found to be unsuccessful. Low molecular weight materials remained in the star polymer. 1H NMR (CDCl3): δ = 5.17 (q, 1H, -CH(CH3)-), 1.57 (d, 3H, CH3). The characteristic peaks of the dendrimer were too broad and integration was inconclusive.
Synthesis of the 32 Arm Star-PLA-PEG Polymer (stPLA-PEG32)
The stPLA32 (1.2 g, 0.0141 mmol), PEG (2.6 g, 0.52 mmol), and DPTS (0.155 g, 0.527 mmol) were dissolved in 80 mL of dry CH2Cl2 by stirring at room temperature. Then DIPC (0.2 g, 1.58 mmol) was added into the reaction mixture via syringe. The reaction mixture was stirred overnight at room temperature. An additional amount of DIPC (0.15 g) was added to the reaction mixture followed by stirring for total of 7 days. The product was purified by fractionation twice with CH2Cl2 as the solvent and hexane as the nonsolvent. After workup and drying, 2.53 g (72% yield) of white solid product was obtained. 1H NMR (CDCl3): δ = 5.18 (q, 1H, -CH(CH3)-), 3.64 (s, 12.6H, OCH2CH2O), 1.58 (d, 3H, -CH3).
Synthesis of Linear-PLA (l-PLA)
A linear PLA was synthesized in a similar procedure using l-lactide (5.1 g, 35.4 mmol), di(ethylene glycol) benzyl ether (0.05 g, 0.25 mmol) as an initiator, and two drops of Sn(Oct)2. Isolation was conducted with the same procedure as described previously. After drying, 4.8 g (95% yield) of the white solid product was obtained.
Preparation of Polymer Micelles
Aqueous dispersions of stPCL-PEG32 and stPLA-PEG32 were prepared using the dialysis technique. Initially, the copolymer was dissolved in N,N-dimethylformamide (DMF) at polymer concentrations ranging from ∼100−200 mg/mL. Next, distilled water was added to the mixture dropwise in a very slow fashion (25 μL of water per 2−3 min) until the desired water content was achieved (67−87.5 vol %). Finally, the water–organic mixture was dialyzed against 3 L distilled water using Membra-Cel MD-25−03.5 membrane tubing. The water was changed every hour for the first 4 h and then every 4−6 h for the next two days. The concentration of the star copolymers after dialysis was determined gravimetrically.
Micelle Characterization
Dynamic laser light scattering (DLS) measurements on polymer micelles were carried out using a “ZetaPlus” Zeta Potential Analyzer (Brookhaven Instrument Co.) equipped with the Multi Angle Option and with a 30 mW solid state laser operated at a wavelength of 635 nm. The sizing measurements were performed at 25 °C at an angle of 90°. All solutions were filtered repeatedly through Millipore membranes with pore sizes of 0.45 and 0.22 μm prior to measurements. Software provided by the manufacturer was used to calculate the effective hydrodynamic diameters of the micelles. DLS measurements were also used to monitor the aggregation stability of micelle solutions. Consequently, size measurements were carried out every 2−3 days over a one month period. Steady-state fluorescence spectra were recorded using a Shimadzu RF5000U spectrofluorophotometer with a bandwidth of 2.5 nm for both excitation and emission. For fluorescence emission spectra, λex was 336 nm, and for excitation spectra, λem was 390 nm. Negative staining was used for the transmission electron microscopy (TEM) studies. A drop of the filtered (0.22 μm filter) sample solution was allowed to settle on a Formvar precoated copper grid for one minute. Excess solution was wicked away with filter paper, and a drop of 1% uranyl acetate solution was allowed to contact the sample for one minute. The samples were studied using a Phillips 410LS electron microscope with a digital imaging camera. The diameters of the particles were determined using 127 measurements randomly collected from TEM images using the Image Pro program (Media Sybernetics).
Drug Encapsulation and Release
The two star copolymers with different lipophilic components were evaluated to determine which structure was more suitable for the delivery of etoposide. A micelle extraction method (2) was used to encapsulate etoposide into the 32 arm star polymer micelles. 100 μL of etoposide stock solution (20 mg/mL) in acetonitrile was added to the empty vial, and the solvent was evaporated to dryness. Then 1 mL of micelle solution was added to the vial and agitated for 2 days. The concentration of micelle solution was varied from 2 mg/mL to 15 mg/mL. The solution was centrifuged (5 min at 10 000 × g) to remove any insoluble drug. It should be noted that centrifugation of the polymer micelle solutions under these conditions did not change the polymer content in the supernatant. The etoposide concentration in the supernatant was determined by HPLC using a Shimadzu LC-10AT system equipped with a reverse-phase C18 column (4.6 mm × 15 cm), mobile phase: 25% (acetonitrile/1% acetic acid)/75% (water/1% acetic acid), flow rate at 1 mL/min, T = 32 °C. The detection of etoposide was performed by UV absorption at 230 nm.
The release of etoposide from the polymer micelles was evaluated by the dialysis method. The polymer micelles loaded with etoposide in phosphate buffered saline (PBS, pH 7.4, 0.14 M NaCl, 1 mL) were transferred to a dialysis bag (molecular weight cutoff 3500). The dialysis bag was immediately placed in 20 mL of PBS. At regular time intervals, 1 mL of release medium was sampled and replaced with an equal volume of PBS. The release experiments were performed at room temperature. The concentration of etoposide present in the dialysate was monitored using HPLC as described above.
RESULTS AND DISCUSSION
Synthesis of Star Copolymers
The synthesis of stPCL-PEG32 was carried out following the synthetic scheme shown in Scheme 1. The expected PEG formula weight is 5000 D and the PCL formula weight is 3000 D, corresponding to m = 26. PAMAM-OH Dendrimer Generation 3 was used as the initiator core. Starting from this core, the star-PCL polymer with 32 arms was synthesized by the ring-opening polymerization of ε-caprolactone with Sn(Oct)2 as the catalyst. The stPCL32 product was obtained in almost quantitative yield. After two fractionations, the pure star polymer was obtained. Next, PEG polymers were attached to the stPCL32 polymers using a commercial product from Nektar. The structure of this product is a PEG polymer (5000 D), with one end capped by a methoxy and the other end by a carboxylic acid group. The carboxylic acid end-group on the PEG was esterified by the terminal hydroxyl groups in the stPCL32 polymer to produce the desired final star polymer stPCL-PEG32. Excess PEG polymer was used, and the reaction was allowed to continue for seven days. After the reaction was complete, the product was purified twice by fractionation to remove the excess PEG and other impurities. The final stPCL-PEG32 polymer was obtained in 83% yield.
Scheme 1.

Synthesis of Star Polymers
Using a similar approach, a 32-arm stPLA-PEG32 was synthesized as shown in Scheme 1. Here, we started with a PAMAM-OH Dendrimer Generation 3 as the initiator to polymerize l-lactide, using Sn(Oct)2 as a catalyst. Similar to the polymerization of ε-caprolactone, the reaction was well-controlled, and the final star-PLA polymer had an arm length determined by the feed ratio of monomer/initiator. The reaction yield was 97%. SEC analysis indicated that the star-PLA polymer contained some low molecular weight side products. Purification by fractionation was attempted but unsuccessful, possibly because of interfering crystallization (see DSC study below). The impure stPLA32 polymer was therefore used in the next step, with the expectation that the low molecular weight side product will be removed when purifying the final product. The stPLA-PEG32 polymer was prepared by the same esterification reaction described above. The stPLA-PEG32 polymer was purified by two fractionations, and the yield was 72%. The low molecular weight impurity was completely removed, and pure stPLA-PEG32 product was obtained.
To verify that l-lactide polymerization under Sn(Oct)2 catalysis conditions can produce PLA with a narrow molecular weight distribution, a linear PLA polymer was synthesized using di(ethylene glycol) benzyl ether as the initiator. The yield was 94%. SEC analysis confirmed that this linear-PLA polymer indeed has a very narrow molecular weight distribution (shown in Table 1).
Table 1.
SEC Results for the Polymers
| theoretical mol. wt | number-average mol. wt | weight-average mol. wt | polydispersity Mw/Mn | |
|---|---|---|---|---|
| StPCL32 | 102 900 | 89 200 | 92 600 | 1.04 |
| stPCL-PEG32 | 262 900 | 215 400 | 231 400 | 1.07 |
| StPLA32 | 102 900 | 80,500a | 87,100a | 1.08 |
| stPLA-PEG32 | 262 900 | 221 400 | 230 000 | 1.04 |
| Linear-PLA | 20 000 | 18 100 | 19 300 | 1.07 |
When calculating the molecular weights, the integration was limited only to the major star polymer peak.
Characterization of Star Polymers
Spectroscopic measurements confirmed the structures of the star polymer products. In the 1H NMR spectrum of stPCL32 (shown in Figure 1), four major peaks were observed: the triplet peak at 3.98 ppm is assigned as (-CH2-O-) protons; the triplet at 2.23 ppm is assigned as (-CH2-CO-) protons; the multiple peak at 1.57 ppm are the protons of (-O-CH2-CH2-CH2-CH2-CH2-CO-); the multiple peak at 1.30 ppm are the protons in the middle. The peaks from the dendrimer core have a low intensity due to the low content. The small triplet peak at 3.58 ppm is assigned as the chain end terminal protons (-CH2-OH). The integration ratio of this peak vs peaks at 3.98 ppm is used to estimate the degree of polymerization of the PCL polymer arms and was found to be about 1/26. This corresponds to a PCL block formula weight of about 3000 D. However, this estimation is based on the ratio of a very small terminal proton peak to a large polymer peak.
Figure 1.

1H NMR spectrum of stPCL32.
In the 1H NMR spectrum of stPCL-PEG32, in addition to the peaks from PCL, the single peak at 3.56 ppm is assigned as the (-O-CH2CH2-O-) of the PEG unit. The integration ratio between PEG and PCL reflects the component ratio in the block copolymers and is consistent with the SEC results. The 1H NMR spectrum of stPLA-PEG32 is consistent with the structure, with a quartet peak observed at 5.08 ppm due to the (-CH(CH3)-) proton; a doublet at 1.49 ppm due to the (-CH(CH3)-) protons; and single peak at 3.56 ppm due to the (-O-CH2CH2-O-) in the PEG unit.
The molecular weights of the synthesized polymers were determined by SEC and are summarized in Table 1. The system is equipped with refractive index, viscosity, and light scattering detectors. The combination of these three detectors can provide the absolute molecular weights of the star polymers. The polydispersity of the star polymers was narrow (Mw/Mn < 1.08). All the star polymer samples had relatively low intrinsic viscosities and small radii of gyration, confirming their highly branched, compact structure. As shown in Table 1, all of the polymers had lower molecular weights than the theoretical values. This may reflect some uncertainty in the SEC-LS method. There are two possible explanations for this: (1) the polymer arms are shorter than expected; (2) the number of star polymer arms is less than desired. Data from the 1H NMR study indicate that the stPCL32 product had an arm length close to the expected value of about 3K. However, as discussed before, such an estimation is not precise. Some OH-containing impurities exist in the system that initiate the polymerization of monomer, as is evident from the low molecular weight polymer impurities in the PCL and PLA stars. These low molecular weight impurities consume some of the monomers, which can result in star polymers with shorter arms than the theoretical value. The 1H NMR analysis of stPCL-PEG32 indicates that the PEG/PCL ratio is 1.81, higher than the theoretical 1.67. This excludes a deficiency in the esterification reaction. It is also possible that our batch of PAMAM-OH dendrimer had some defects, so that the number of surface -OH groups available for polymerization was less than 32. Therefore, both shorter arms and a reduced number of arms may contribute to the overall lower molecular weight of the final star polymer. The number of arms equal to 27.8 is derived from [Mw(stPCL-PEG32) – Mw(stPCL32)]/Mw(PEG). Similarly, for stPLA-PEG32 the calculated number of arms is 28.6. Taking into account the various uncertainties, the estimated number of arms in these star polymers is in the range 26−30.
Thermal Behavior
The thermal properties of the polymers were investigated using differential scanning calorimetry (DSC) to determine the glass transition temperature (Tg) and melt temperature (Tm). For the stPCL32 and stPCL-PEG32 polymers, the thermal behavior was simple: Tm = 50 °C was observed for stPCL32 and Tm = 57 °C for stPCL-PEG32. For the PLA polymers, interesting thermal behavior was observed. The DSCs of stPLA32 and the linear PLA are shown in Figures 2 and 3, respectively. The heating trace for stPLA32 shows a Tg at about 56 °C, a crystallization transition temperature at 103 °C, and two melting transition peaks at 139 and 148 °C. The cooling trace shows no crystallization transition, and the Tg is at about 50 °C. In contrast, the heating trace for the linear-PLA shows no Tg, and a single melting transition is observed at 171 °C. The cooling trace shows a crystallization transition at 98 °C and no Tg. The linear-PLA polymer can reorganize more freely and has a higher molecular weight of 19K. This indicates a high proportion of crystalline phase in the sample and a small amorphous phase. For the stPLA32, the polymer chain is shorter (3K), and one end is bound to the dendrimer core. This means that it has less freedom to reorganize and therefore forms less perfect crystals and a larger amorphous fraction, leading to the observed Tg. In the cooling process, the nucleation of the crystallization (which dominates) is slow at high temperatures (above Tm), so the sample stays amorphous until it passes the Tg. In the heating process, the polymer chains become mobile (kinetics dominate) above the Tg. Nucleation is then very fast at low temperature (below Tm), and crystallization occurs. Since it forms less perfect crystals, the two peak melting profiles may be due to two types of crystals, or there may be a partial melting/recrystallization followed by complete melting.
Figure 2.
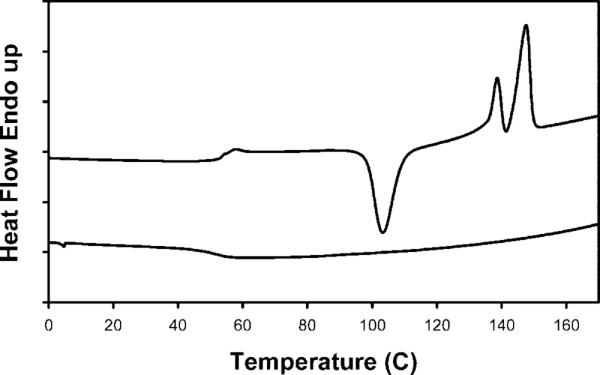
DSC of stPLA32: top trace is heating and lower trace is cooling.
Figure 3.
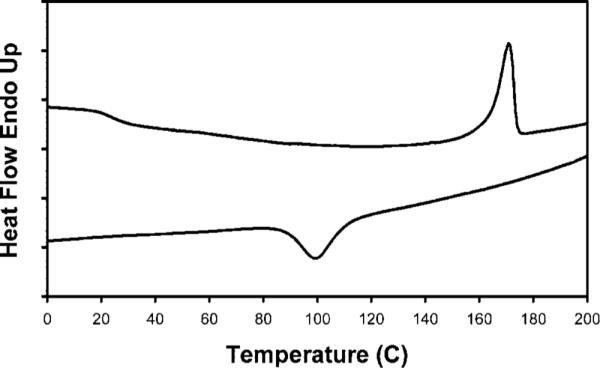
DSC of linear-PLA: top trace is heating and lower trace is cooling.
The DSC of stPLA-PEG32 in Figure 4 shows interesting behavior as well. The heating trace exhibits a large Tm at 54 °C and a small Tm at 97 °C. The cooling trace shows corresponding transitions at lower temperatures. The unusual thermal behavior of stPLA32 was not observed here. The highly crystalline nature of the PEG plays a major role in this case. The sample may contain two types of crystals; one is the PEG crystal and the other is the less perfect PLA crystal. The majority PEG crystal melts at lower temperature and gives the large peak. The smaller amount and less perfect PLA crystals melt at a higher temperature and give a relatively small peak.
Figure 4.
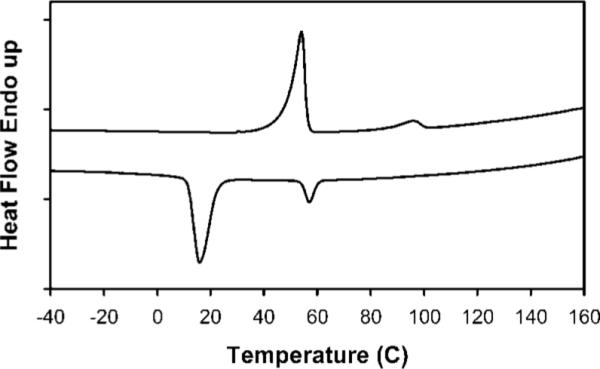
DSC of stPLA-PEG32: top trace is heating and lower trace is cooling.
Micellar Properties of the Star Polymers
The dialysis method was employed to prepare polymeric micelles. Their effective hydrodynamic diameter was measured by DLS. The measurements performed immediately after dialysis revealed the formation of dispersions of star polymer particles with very broad distributions (polydispersity indices were more than 0.3). Removal of the large aggregates by repeated filtration (membranes with pore sizes of 0.45 and 0.22 μm) resulted in the substantial decrease of the concentration of micellar dispersions (ca. 30−50%). The effective diameters of the polymer micelles in solutions prepared by dialysis are presented in Table 2. The micelle size distribution was rather broad in all cases and varied in the range 0.13−0.21, indicating the possible existence of some aggregated species. As an example, a DLS graph of size distribution of stPCL-PEG32 micelles in a polymer solution with a concentration of 15 mg/mL is shown in Figure 5. It is a bimodal distribution with a smaller size component of ∼15.7 nm (85%) and a large size component of ∼134 nm (15%). The size and aggregation stability of the star polymer micelles in aqueous dispersions depended on the water content in the water–organic mixture at the start of the dialysis. Specifically, micelles prepared by dialysis from the water–organic mixtures with higher water content (87.5 vol %) were characterized as having both larger particle size and polydispersity, as well as a lower degree of stability. Formation of aggregates followed by phase separation was observed in these solutions within 2−3 days. The most stable stPCL-PEG32 and stPLA-PEG32 aqueous dispersions were obtained with 67% water content in the initial dialysis mixture. Subsequent measurements of these stPCL-PEG32 and stPLA-PEG32 samples showed that the size of the particles remained practically unchanged for several weeks. Furthermore, it should be noted that the size and size distribution of the micelles formed from the same batch of polymer varied slightly due to variations in the processing parameters such as stirring conditions, rate of water addition, and so forth.
Table 2.
Characterization of Star Polymer Micelles
| effective diametera, nm | polydispersity index | |
|---|---|---|
| stPCL-PEG32 | 19.0 | 0.19 |
| stPLA-PEG32 | 24.7 | 0.20 |
Effective diameter denotes averaged value calculated from three measurements performed on each sample. The effective diameter values are within ±5 nm. Polymer concentration is 15 mg/mL.
Figure 5.

DLS graph of the stPCL-PEG32 micelle size distribution. Concentration of stPCL-PEG32 was 15 mg/mL.
The morphology of the star polymer micelles was further examined by transmission electron microscopy (TEM) using negative staining. A representative micrograph of the stPCL-PEG32 micelles is presented in Figure 6. The micelles are close to spherical and have a mean number-average diameter of 11.5 ± 0.4 nm. Analysis of TEM images revealed the broad size distribution of the polymer micelles as well as the presence of large aggregates with diameters between 25 and 45 nm (frequency from 6% to 1%). The diameters of the particles determined from the TEM data were less than the effective diameter determined by DLS. This is not surprising, since the DLS determines the hydrodynamic diameter or the “equivalent sphere diameter” in solution, while TEM images were obtained in the absence of the solvent. Nevertheless, both methods exhibited the same trend in the particle sizes of the micelles.
Figure 6.
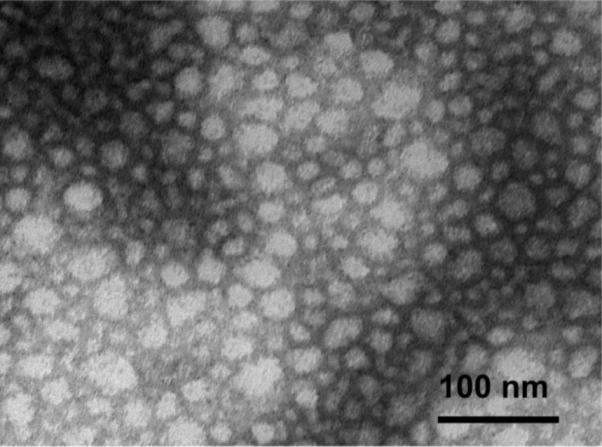
TEM micrograph of stPCL-PEG32 micelles obtained using uranyl acetate staining.
The star polymer micelles were further characterized by fluorescence spectroscopy to determine the partition coefficient of the model hydrophobic probe pyrene between the polymer micelles and the aqueous compartment. Several aspects of the pyrene spectroscopic properties were changed as the polymer concentration increased: the total fluorescence intensity increased, and the vibrational fine structure of the emission spectra changed. This type of behavior is indicative of the partitioning of the pyrene into the more hydrophobic environment. However, the extent of these changes as a function of polymer concentration was drastically different for the polymers studied. For stPCL-PEG32, the total fluorescence intensity increased steadily even in very dilute polymer solutions, and the vibrational fine structure (I1/I3) changed from 1.8 to 1.32. These results suggest that in dilute solutions the intact stPCL-PEG32 molecules formed unimolecular micelles. The calculated value for the partition coefficient (P) for pyrene between stPCL-PEG32 micelles and water at 22 °C was 4150, nearly double than that found for a 16 arm star-PCL-PEG polymer (P = 2410) (16). In contrast, the onset of the changes in the fluorescence spectra of pyrene in the stPLA-PEG32 aqueous dispersions was detected at a polymer concentration of ∼0.01 mg/mL. The partition coefficient for pyrene in this system was found to be ∼600. Furthermore, the stPLA-PEG32 micelle solutions were found to have an I1/I3 ratio of 1.6. This value is higher than the 1.32 value determined for stPCL-PEG32 micelles. These data suggest that the PLA cores of stPLA-PEG32 micelles are more polar compared to the PCL cores. As a result, the environment of the pyrene molecule in the PLA core is less hydrophobic, decreasing the solubilization capacity of stPLA-PEG32 micelles.
Similar studies were conducted for star polymer micelles in phosphate buffer saline (PBS). In the case of stPCL-PEG32, the total fluorescence intensity steadily increased even in the very dilute polymer solutions, suggesting the presence of stPCL-PEG32 molecules as unimolecular micelles. The onset of the changes in the fluorescence spectra of pyrene in the stPLA-PEG32 aqueous dispersions was detected at a polymer concentration of ∼0.067 mg/mL. The calculated values for the partition coefficient for pyrene under these conditions were 1440 for stPCL-PEG32 micelles and 470 for stPLA-PEG32 micelles.
Encapsulation and Release of Etoposide in Copolymer Micelles
Two general drug-loading methods were explored for encapsulating etoposide in stPCL-PEG32 micelles. In the first method (dialysis), etoposide (2 mg) was dissolved in acetonitrile and then mixed with stPCL-PEG32 polymer (5 mg) dissolved in DMF (1 mL). Next, distilled water was slowly added dropwise to the mixture (25 μL of water per 2−3 min) to achieve a water content of 67 vol %. Drug precipitation was not observed upon mixing water with the drug–polymer solution. The obtained water–organic solvent mixture containing etoposide and polymer was then dialyzed against water. No etoposide was detected in the final dispersions. Therefore, this technique is not applicable, possibly due to the water solubility of etoposide (ca. 0.12 mg/mL).
In the second method, micelle extraction (2) was used to encapsulate etoposide into the star micelles. Briefly, an aliquot of etoposide stock solution in acetonitrile was added to the empty vial, the solvent was evaporated to dryness, and 1 mL micelle solution was added to the vial and agitated for 2 days. Polymer concentrations ranging from 2 mg/mL to 15 mg/mL were used. The solution was then centrifuged to remove insoluble drug, and the etoposide concentration in the solution was determined by HPLC. The loading levels of etoposide into the copolymer micelles are presented in Table 3. The maximum etoposide loading (7.8 w/w % for stPCL-PEG32 and 4.3 w/w % for stPLA-PEG32, respectively) was achieved at a polymer concentration of ca. 3 mg/mL. Further increase in the polymer concentration resulted in a decrease in the drug loading level. Such complex solubilization behavior is typical of systems where there is a strong interaction between the drug and the micelle core (3, 18, 19). In this case, the solubilization capacity might increase to a saturation level with increasing polymer concentration. As a result, the relative drug loading in the micelles will decrease. It is noteworthy that the incorporation of drug did not affect the size of the star polymer micelles. The etoposide/star polymer dispersions remained stable over several days. Furthermore, they could be stored at 4 °C for at least 24 h without precipitation of etoposide.
Table 3.
Etoposide Loading in stPCL-PEG32 and stPLA-PEG32 Polymers
| polymer | polymer conc. (mg/mL) | etoposide (mg/mL)a | loading in polymer (w/w %) |
|---|---|---|---|
| stPCL-PEG32 | 2.4 | 0.12 | 5.0 |
| 3.0 | 0.23 | 7.8 | |
| 5.0 | 0.29 | 5.9 | |
| 8.0 | 0.31 | 3.9 | |
| 15 | 0.50 | 3.4 | |
| stPLA-PEG32 | 2.4 | 0.05 | 2.2 |
| 3.0 | 0.13 | 4.3 | |
| 5.0 | 0.16 | 3.2 | |
| 8.0 | 0.19 | 2.4 | |
| 15 | 0.43 | 2.8 |
The etoposide concentration values are within ± 3%.
The drug release from the etoposide/stPCL-PEG32 system (polymer concentration 5 mg/mL) was evaluated in phosphate buffered saline (pH 7.4, 0.14 M NaCl) by the dialysis method. About 60% of the loaded etoposide (an averaged value calculated based on three experiments) was released from the micelles during the first 24 h. Overall, these studies suggested that etoposide can be loaded and efficiently released from star polymer micelles.
Concluding Remarks
In summary, we have successfully synthesized two types of amphiphilic star-polymers with PCL and PLA as the lipophilic components. The star polymer structures were confirmed by SEC, spectroscopic, and thermal characterizations. The micelle properties of the polymers were investigated by various techniques. The formation of unimolecular micelles from the stPCL-PEG32 has been demonstrated by pyrene fluorescence studies. The results from fluorescence, DLS, and TEM experiments have shown that unimolecular micelles and aggregated multimolecular micelles coexist in the water solution. A systematic study revealed the optimal condition for etoposide encapsulation by the polymer micelles. The stPCL-PEG32 has a higher etoposide loading capacity than stPLA-PEG32. This may be due to the more lipophilic properties of the PCL core. The overall etoposide loading capacity of the 32 arm star polymer is lower than the 16 arm star polymer we reported previously (16). One possible reason for this may be due to the higher PCL polymer density in the 32 arm star polymer micelle interior, therefore leaving less space for drug loading. The in vivo testing for tumor treatment by the etoposide/star polymer formulation will be report in separate paper.
ACKNOWLEDGMENT
Funding of this work by the NIH (5R44EB000551) is gratefully acknowledged. The authors thank Mr. Monsy Jacob and Mr. Sangrama K. Sahoo for help in obtaining 1H NMR spectra.
LITERATURE CITED
- 1.Kwon GS, Kataoka K. Block copolymer micelles as long-circulating drug vehicles. Adv. Drug Delivery Rev. 1995;16:295. [Google Scholar]
- 2.Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf., B: Biointerfaces. 1999;16:3. [Google Scholar]
- 3.Kabanov AV, Alakhov V.Yu. Pluronic block copolymers in drug delivery: From micellar microcontainers to biological response modifiers. Crit. Rev. Ther. Drug Carrier Syst. 2002;19:1. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.10. [DOI] [PubMed] [Google Scholar]
- 4.Torchilin VP. Polymeric micelles in diagnostic imaging. Colloids Surf., B: Biointerfaces. 1999;16:305. [Google Scholar]
- 5.Wilhelm M, Zhao C, Wang Y, Xu R, Winnik MA, Mura J, Reiss G, Croucher MD. Poly(styrene-ethylene oxide) block copolymer micelle formation in water: a fluorescence probe study. Macromolecules. 1991;24:1033. [Google Scholar]
- 6.Kwon GS, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Micelles based on AB block copolymers of poly(ethylene oxide) and poly( -benzyl L-aspar-tate). Langmuir. 1993;9:945. [Google Scholar]
- 7.Kabanov AV, Chekhonin VP, Alakhov V. Yu., Batrakova EV, Lebedev AS, Melik-Nubarov NS, Arzhakov SA, Levashov AV, Morozov GV, Severin ES, Kabanov VA. The neuroleptic activity of haloperidol increases after its solubilization in surfactant micelles. FEBS Lett. 1989;258:343. doi: 10.1016/0014-5793(89)81689-8. [DOI] [PubMed] [Google Scholar]
- 8.Hagan SA, Coombes AGA, Garnett MC, Dunn SE, Davies MC, Illum L, Davis SS, Harding SE, Purkiss S, Gellert PR. Polylactide-poly(ethylene glycol) copolymers as drug delivery system. 1. Characterization of water dispersible micelle-forming systems. Langmuir. 1996;12:2153. [Google Scholar]
- 9.Kataoka K. Block copolymer micelles as vehicles for drug delivery. J. Controlled Release. 1993;24:119. [Google Scholar]
- 10.Iijima M, Nagasaki Y, Okada T, Kato M, Kataoka K. Core-polymerized reactive micelles from heterotelechelic amphiphilic block copolymers. Macromolecules. 1999;32:1140. [Google Scholar]
- 11.Kim J-H, Emoto K, Iijima M, Nagasaki Y, Aoyagi T, Okano T, Sakurai Y, Kataoka K. Core-stabilized polymeric micelle as potential drug carrier: increased solubilization of taxol. Polym. AdV. Technol. 1999;10:647. [Google Scholar]
- 12.Liu H, Jiang A, Guo J, Uhrich KE. Unimolecular micelles: synthesis and characterization of amphiphilic polymer systems. J. Polym. Sci., Part A: Polym. Chem. 1999;37:703. [Google Scholar]
- 13.Kim KH, Cui GH, Lim HJ, Huh J, Ahn C, Jo WH. Synthesis and micellization of star-shaped poly-(ethylene glycol)-block-poly(ε-caprolactone). Macromol. Chem. Phys. 2004;205:1684. [Google Scholar]
- 14.Deng M, Chen X, Piao L, Zhang X, Dai Z, Jing X. Synthesis of four-armed poly(ε-caprolactone)-block-poly(ethylene oxide) by diethylzinc catalyst. J. Polym. Sci., Part A: Polym. Chem. 2004;42:950. [Google Scholar]
- 15.Zeng F, Lee H, Chidiac M, Allen C. Synthesis and characterization of six-arm star poly(δ-valerolactone)-block-methoxy poly(ethylene glycol) copolymer. Biomacromolecules. 2005;6:2140. doi: 10.1021/bm050124+. [DOI] [PubMed] [Google Scholar]
- 16.Wang F, Bronich TK, Kabanov AV, Rauh RD, Roovers J. Synthesis and evaluation of a star amphiphilic block copolymer from poly(-caprolactone) and poly(ethylene glycol) as a potential drug delivery carrier. Bioconjugate Chem. 2005;16:397. doi: 10.1021/bc049784m. [DOI] [PubMed] [Google Scholar]
- 17.Roovers J, Toporowski PM, Douglas J. Thermodynamic properties of dilute and semidilute solutions of regular star polymers. Macromolecules. 1995;28:7064. [Google Scholar]
- 18.Xing L, Mattice WL. Strong solubilization of small molecules by triblock-copolymer micelles in selective solvents. Macromolecules. 1997;30:1711. [Google Scholar]
- 19.Gadelle F, Koros WJ, Schechter RS. Solubilization of aromatic solutes in block copolymers. Macromolecules. 1995;28:4883. [Google Scholar]