

Abstract
We have used electron paramagnetic resonance and fluorescence spectroscopy to study the interaction between the kinesin-1 head and its regulatory tail domain. The interaction between the tails and the enzymatically active heads has been shown to inhibit intrinsic and microtubule-stimulated ADP release. Here, we demonstrate that the probe mobility of two different spin-labeled nucleotide analogs in the kinesin-1 nucleotide pocket is restricted upon binding of the tail domain to kinesin-1 heads. This conformational restriction is distinct from the microtubule-induced changes in the nucleotide pocket. Unlike myosin V, this tail-induced restriction occurs independent of nucleotide state. We find that the head-tail interaction that causes the restriction only weakly stabilizes Mg2+ in the nucleotide pocket. The conformational restriction also occurs when a tail construct containing a K922A point mutation is used. This mutation eliminates the tail's ability to inhibit ADP release, indicating that the tail does not inhibit nucleotide ejection from the pocket by simple steric hindrance. Together, our data suggest that the observed head-tail interaction serves as a scaffold to position K922 to exert its inhibitory effect, possibly by interacting with the nucleotide α/β-phosphates in a manner analogous to the arginine finger regulators of some G proteins.
Introduction
The kinesin-1 motor protein translates the energy derived from ATP hydrolysis into the plus end-directed transport of intracellular cargo along microtubules (MTs). The majority of kinesin-1 inside cells exists in a regulated state (1). Regulated kinesin-1 adopts a folded conformation that is tightly ADP-bound and has weak MT affinity (2). This folding can occur in the absence of kinesin light chains, although the light chains confer additional regulatory function (3,4). In the folded conformation, the Hinge II region in the kinesin-1 coiled-coil stalk bends, and an interaction occurs between the neck coiled-coil and the tail coiled-coil (Fig. 1 A) (5–7). This stabilizes the folded conformation and positions the C-terminal regulatory tail domains to directly interact with and inhibit the N-terminal ATPase heads (8,9). Kinetic data show that the tail inhibits intrinsic and MT-stimulated ADP release from kinesin-1 (2,10). A critical lysine residue (K922) in the conserved QIAKPIRP motif of the tail is required for inhibition (2,11). The mechanism by which the tail, specifically the critical K922 residue, inhibits ADP release is unknown.
Figure 1.

EPR probes and kinesin-1 constructs used. (A) K349 and K420 head constructs and the Tail944 tail construct are shown below a schematic of the full-length kinesin heavy chain dimer. Head residues are tan, coiled-coil residues are gray, and predicted globular tail residues are black. These constructs have been described previously (12). (B) Close-up view of the kinesin-1 motor domain (Protein Data Bank (PDB) 1bg2) (19) showing the bound nucleotide and components of the nucleotide pocket. The P-loop, Switch I, and Switch II nucleotide pocket elements are colored orange, red, and cyan, respectively. The G234 residue in Switch II is depicted in spacefill representation. The 2′ and 3′ ADP ribose oxygens (where the 2′,3′-SLATP nitroxide spin label is attached) are indicated by arrows. The figure was generated using Pymol (39); (C) Chemical structures of the nucleotide analogs are shown. 2′,3′-SLATP is derived from ATP via ribose modifications. SSL-NANTP is derived from a substituted phenylring-amino-ethylspacer-triphosphate structure.
We have recently demonstrated by photochemical cross-linking and cryo-electron microscopy (cryo-EM) that the regulatory QIAKPIRP sequence of the tail interacts in the vicinity of the Switch I element of the head, near the kinesin-1 nucleotide pocket (12). Switch I, together with Switch II, form a γ-phosphate-sensing mechanism (Fig. 1 B) that is structurally conserved in the nucleotide pockets of kinesins, myosins, and G proteins (13–16). The nucleotide pocket of kinesin-1 undergoes a conformational change upon binding to MTs, which was observed as a restriction of the mobility of electron paramagnetic resonance (EPR) probes attached to the ribose oxygens of ADP or other diphosphate nucleotides on kinesin-1 and the kinesin family member ncd (17,18). This is thought to correspond to a “closing” of Switch I that promotes the hydrolysis of ATP when the motor binds MTs. A salt bridge forms between Switch I and Switch II to stabilize this “closed” (proximal to the nucleotide) state. In contrast, a cryo-EM structure of a kinesin-1 tail fragment (residues 823–944) complexed with the head on MTs shows an “open” (distal to the nucleotide) Switch I conformation (12), which is associated with the strong nucleotide-binding state seen in crystal structures (19,20). This is consistent with the tail's role in preventing MT-stimulated ADP release.
There are several possible mechanisms for tail-mediated regulation of ADP release and several potential reasons why the K922 residue is critical. Because kinesins and G proteins share multiple structural elements, it has been suggested that they may also be regulated in a similar fashion (10,12). Release of GDP from G proteins is inhibited by binding partners called guanine nucleotide dissociation inhibitors (GDIs). RhoGDIs and RabGDIs are two nonhomologous classes of GDIs that act similarly to inhibit GDP release. Both classes can stabilize specific conformations of the Switch I/Switch II γ-phosphate sensors that are incompatible with nucleotide ejection. RhoGDIs and RabGDIs can also coordinate the Mg2+ ion in the nucleotide pocket, either as a separate mechanism or in concert with the Switch I/Switch II interaction to further stabilize a bound nucleotide (21–23). The third major class of GDIs, known as GoLoco proteins, inhibits GDP release by positioning a critical arginine finger to stabilize the α/β-phosphates of a bound nucleotide (14,24). Based on the homology between kinesins and G proteins, a kinesin-1 head-tail interaction could position the critical K922 tail residue to inhibit ADP release by one or more of these mechanisms: through direct or indirect interactions with the γ-phosphate sensors, Mg2+ ion, α/β-phosphates, ribose oxygens, or coordinating water molecules.
Interestingly, kinesin-1 stands apart from myosins and G proteins because its nucleotide pocket is on the surface of the protein and highly exposed to the aqueous environment (19,25,26). The regulatory tail binds to the kinesin-1 head in the vicinity of Switch I and the nucleotide pocket, leading to the idea that the tail might function by sterically blocking ADP release. The role of the critical K922 in this scenario could be structural and not enzymatic; it may be required for the proper interaction of the tails with the heads so that other elements can inhibit nucleotide release.
Here, we use EPR spectroscopy to show that the kinesin-1 tail causes a conformational change around the nucleotide pocket that restricts the mobility of spin-labeled nucleotides. The EPR spectra of kinesin-1 heads bound to tails show a decrease in mobility that is qualitatively similar to the spectra of kinesin-1 bound to MTs. However, the tail-induced restriction of probe motility is significantly different from that caused by MT-induced “closing” of Switch I. Importantly, the conformational restriction occurs regardless of whether the Switch I/Switch II γ-phosphate sensor is intact and independent of the regulatory K922 residue. These data support a mechanism for tail-mediated inhibition in which tail residues form interactions in and around the nucleotide pocket, acting as structural supports for head-tail interactions that are not directly involved in inhibition. The supporting structure positions K922 to act as a critical inhibitory agent, possibly by interacting with the nucleotide α/β-phosphates.
Materials and Methods
Cloning and purification of constructs
Untagged cysteine-light monomeric (K349) and dimeric (K420) head constructs of human kinesin-1 heavy chain, as well as the G234A mutant, were received from R. Vale (University of California, San Francisco, CA). These constructs were expressed and purified as described (18). Tail944 was expressed and purified as described (12). The K922A tail mutant was generated using a Quikchange II site-directed mutagenesis kit (Stratagene, Cedar Creek, TX). The primers used were 5′-GCATTCTGCACAGATTGCTGCG CCTATTCGTCCCGGG C-3′ and its complementary sequence.
Exchange of spin-labeled nucleotide analogs into kinesin-1
K349 or K420 heads were dialyzed for 3 h into Labeling buffer (40 mM MOPS, 2 mM MgCl2, 1 mM EGTA, 100 mM NaCl, pH 7.0). The protein was concentrated to ∼200 μM and mixed with 2′,3′ spin-labeled ATP (2′,3′-SLATP) or Spiro spin-labeled 2-[(4-azido-2-nitrophenyl)amino]ethyl triphosphate (SSL-NANTP) at a ∼1:1 molar ratio. These probes are described in Fig. 1 C. 2′,3′-SLATP was synthesized by N. Naber. SSL-NANTP was supplied by J. Grammer, X. Chen, and R. Yount (Washington State University, Pullman, WA). The 2′,3′-SLATP probe was incubated with kinesin-1 for 1 h, and unbound probe was removed by centrifuging the mixture through a Micro Bio-Spin column (Bio-Rad Laboratories, Hercules, CA) equilibrated with Labeling buffer. The EPR spectrum was then recorded. For SSL-NANTP labeling, kinesin-1 was incubated with SSL-NANTP and 0.1 mg/ml myokinase overnight to facilitate the exchange of the spin probe into kinesin-1. Unbound probe was removed as described above, and the EPR spectrum was recorded.
Binding of kinesin-1 tails to heads
Tail944 and TailK922A solutions were concentrated using Centricon centrifugal filters (Millipore, Billerica, MA) and washed several times with a buffer containing 25 mM HEPES, 2 mM MgCl2, 1 mM EGTA, 300 mM NaCl, pH 7.0. The final concentration of the protein was adjusted to ∼250 μM. Additional NaCl was added to labeled kinesin-1 heads to bring the salt concentration to 300 mM NaCl. A fourfold molar excess of tails was added to a solution of labeled heads. The resulting mixture was dialyzed overnight into 25 mM HEPES, 2 mM MgCl2, 1 mM EGTA, 50 mM NaCl, pH 7.0. The EPR spectrum of the resulting solution was then recorded. For experiments involving AlF4, solutions of 2 mM AlCl3, and 10 mM NaF were freshly added to the kinesin-1 mixture before the EPR spectrum was recorded. The time course of nucleotide release experiments was performed by adding 10 mM ADP to the kinesin-1 head-tail solution. The mixture was rapidly mixed by pipetting, inserted into a 25 μl capillary within ∼30 seconds, and placed in the EPR cavity for time-dependent measurements.
EPR spectroscopic measurements
EPR measurements were performed with a Bruker EMX EPR spectrometer from Bruker Instruments, Inc. (Billerica, MA). First derivative, X-band spectra were recorded in a high-sensitivity microwave cavity using 50 s, 100-gauss-wide magnetic field sweeps. The instrument settings were as follows: microwave power, 25 mW; time constant, 164 ms; frequency, 9.83 GHz; modulation, 1 gauss at a frequency of 100 kHz. Each spectrum used in data analysis is an average of 5–50 sweeps from an individual experimental preparation. For nucleotide release experiments, 11 s scans were taken at a 25 gauss field sweep that detects the high-field peak of the free probe in the EPR spectrum, and these were fit to a single exponential function. All experiments were performed at room temperature.
Mant-ADP release assays
Dimeric K420 heads (15–20 μM) were incubated with 100 μM Mant-ADP for 12–60 h at 4°C to allow exchange into the nucleotide pocket. Excess nucleotide was removed by batch binding and elution from Whatman P11 phosphocellulose resin (Whatman, Kent, United Kingdom). Head and tail proteins were dialyzed separately into assay buffer (20 mM HEPES, 30 mM potassium acetate, 20 mM imidazole, 2 mM MgCl2, 0.1 mM EGTA, 5 mM β-mercaptoethanol, pH 7.2) before experiments. Protein concentrations were adjusted to 6 μM K420 and 15 μM tails for all experiments. Steady-state fluorescence measurements were collected on a TimeMaster fluorescence lifetime spectrometer from PTI, Inc. (Birmingham, NJ). The sample was excited at 360 nm, and emission was measured at 450 nm. For the standard nucleotide release experiments, 280 μM ATP was used to initiate Mant-ADP release. For Mg2+ release experiments, a mixture of 7 mM EDTA + 280 μM ATP was used to initiate the reaction. All experiments were performed at room temperature.
Results
Kinesin-1 tails restrict the mobility of EPR probes in the nucleotide pocket
Previous EPR spectroscopy experiments with spin-labeled nucleotides showed that MT binding induces the Switch I element of kinesin-1 to “close” into the nucleotide pocket (17), and these results were later corroborated by high-resolution cryo-EM structures (13,17). We used the same EPR probes on kinesin-1 heads in the presence of truncated tails added in trans to assess whether the tail, like MTs, induces conformational changes in the kinesin-1 nucleotide pocket. For these experiments, we exchanged two spin-labeled nucleotide analogs, 2′,3′-SLATP and SSL-NANTP (Fig. 1 C), into the nucleotide pockets of truncated kinesin-1 monomeric or dimeric heads (residues 1–349, designated K349, or residues 1–420, designated K420; Fig. 1 A). We then measured EPR spectra in the absence and presence of a kinesin-1 tail fragment (residues 823–944, designated Tail944; Fig. 1 A). Due to kinesin-1's intrinsic ATPase activity, the nucleotide analogs were hydrolyzed into their diphosphate forms in the period before measurements were taken.
Both 2′,3′-SLADP and SSL-NANDP probes are partially immobilized by binding to the kinesin-1 nucleotide pocket (Fig. 2). The splittings of the immobilized components of 2′,3′-SLADP-bound K349 are 43.3 ± 0.1 gauss, consistent with previously reported data (17). Griffith and Jost (27) have shown that the EPR spectra of nitroxide spin labels can be modeled as rapid, subnanosecond mobility within a cone, where the angle of the cone approximates the steric restriction in mobility caused by the protein surface adjacent to the probe. The spectra observed for kinesin-1 correspond to a wide cone angle of 133.2 ± 0.2° within which the probe can diffuse, indicating that the ribose oxygens on nucleotides bound to kinesin-1 are remarkably open to the aqueous environment. In contrast, the SSL-NANDP spin-label ring is attached to the 2′ carbon atom of NANTP, which corresponds to the 5′ carbon position of ADP based on the crystal structure of NANTP-based analogs bound to myosin (28). This positions the probe deeper inside the nucleotide pocket than the spin-label ring on the ribose of 2′,3′-SLADP (17). Additionally, the spiro linkage to the SSL-NANDP spin moiety is more restrictive to motion than the ester linkage of 2′,3′-SLADP. Thus, larger splittings are seen when SSL-NANDP is bound to kinesin-1 in solution, 60.3 ± 0.1 gauss, corresponding to a narrower 77.2 ± 0.2° cone angle through which the probe can diffuse (Fig. 2). Although our probes' true region of diffusion is unlikely to be a geometric cone, it is nonetheless constructive to compare the spectra observed here with the simulations of Griffith and Jost (27) to quantify the degree of probe mobility.
Figure 2.
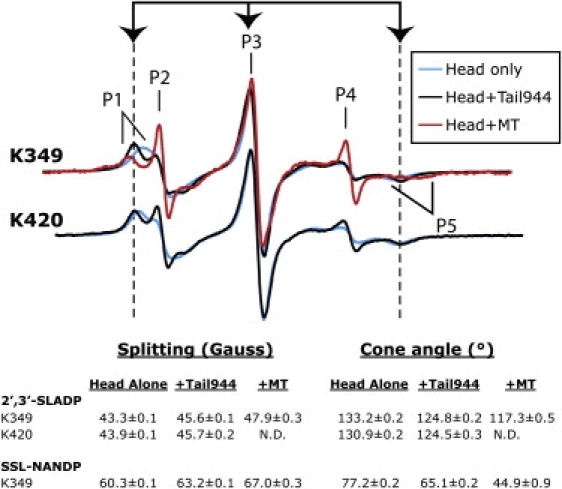
Kinesin-1 tails induce a conformational restriction of the nucleotide pocket that differs from the conformational changes that occur upon microtubule binding. Spectra of 2′,3′-SLADP-bound kinesin-1 are shown in the absence of tail (cyan), in the presence of tail (black), and in the presence of MTs (red). Spectra of SSL-NANDP are not shown because of a large spectral component corresponding to free probe. Arrows and dashed lines mark the EPR spectral splittings of kinesin-1 heads bound to the nucleotide spin probe in the presence of tails. The splittings between the low field peak (P1) and high field dip (P5) of the immobilized components, and full cone angles corresponding to these splittings, are shown below the spectra.
In the presence of Tail944, the mobility of both EPR probes is restricted. The splittings of 2′,3′-SLADP increase to 45.6 ± 0.1 gauss. These values correspond to a decrease in probe mobility from a cone angle of 133.2 ± 0.2° for 2′,3′-SLADP on heads alone to 124.8 ± 0.2° in the tail-bound state. With SSL-NANDP-bound K349, the splittings also increase after addition of Tail944 to 63.2 ± 0.1 gauss (65.1 ± 0.2° cone angle). For all these measurements, we used ∼20 μM heads and ∼80 μM tails. The change in EPR signal due to the tails was saturated such that increasing tail concentration did not significantly change the EPR spectrum, and this concentration is in large excess of the reported Kd (<0.1 μM) for a head-tail interaction (10). Thus, we anticipate that all heads are tail bound under these conditions.
The observation that a decrease in mobility occurs with two different analogs that place probes at different positions in the nucleotide site indicates that the changes in mobility due to the kinesin-1 tail are not due to a simple local change in structure, but reflect a more global restriction of the area around the nucleotide pocket. We next sought to explore the role of this tail-induced restriction in kinesin-1's regulatory mechanism.
The tail-induced conformational restriction is distinct from the changes observed in the nucleotide pocket upon MT binding
The spectra of 2′,3′-SLADP and SSL-NANDP exhibit two major components in the presence of MTs: a highly mobile component corresponding to free probe and a more immobilized component corresponding to probe that is bound to kinesin-1 on MTs (17). The large amount of free probe reflects kinesin-1's weak affinity for these probes, and diphosphate nucleotides in general, when it is bound to MTs (29). Nevertheless, at the high protein concentrations used, we were able to compare immobilized spectral components of MT-bound or tail-bound heads containing spin-labeled nucleotides. The values we measured for the EPR spectral splittings of kinesin-1 on MTs agree with previously reported results (17).
Similar to MTs, the tail restricts the nucleotide pocket. However, our results suggest that tails and MTs restrict the nucleotide pocket in different ways. The splitting of the immobilized components of 2′,3′-SLADP-bound K349 or K420 in the presence of Tail944 is 45.6 ± 0.1 gauss (124.8 ± 0.2° cone angle), which differs from that of MT-bound K349 (47.9 ± 0.3 gauss, 117.3 ± 0.5° cone angle; Fig. 2). The difference is more prominent in K349 containing SSL-NANDP, which has a splitting of 63.2 ± 0.1 gauss (65.1 ± 0.2° cone angle) in the presence of Tail944 and 67.0 ± 0.3 gauss (44.9 ± 0.9° cone angle) in the presence of MTs.
Head-tail binding and the observed conformational changes do not specifically require ADP in the kinesin-1 nucleotide pocket
The significant structural homology between G proteins, kinesins, and myosins led us to postulate that the kinesin-1 head-tail interaction could share similarities with interactions seen between the myosin V head and tail as well as between GDIs and G proteins. However, there is a significant difference between the regulatory mechanisms of myosin V and G proteins. Whereas GDIs seem to bind with nearly equal affinity to their cognate G proteins in GDP and GTP states (30), head-tail interactions in myosin V appear to be nucleotide dependent. Nucleotide-linked conformational changes in the head can promote tail binding, and tail-induced inhibition of actin binding is observed more prominently when ADP is in the nucleotide pocket (31).
To test whether the observed kinesin-1 head-tail interaction depends on nucleotide state, we added 2 mM AlCl3 and 10 mM NaF to 2′,3′-SLADP-bound K349, to generate 2′,3′-SLADP•AlF4 bound to heads, which is an ATP analog state (32–34). The EPR spectrum of 2′,3′-SLADP-bound K349 did not change after addition of AlF4. Likewise, the spectrum resulting from tail-induced immobilization of the probe was identical in the absence and presence of AlF4 (Fig. 3). In a complementary experiment, we added 2′,3′-SLATP to a G234A mutant of K349. The G234A mutation results in a motor that cannot form the Switch I/Switch II salt bridge that serves as kinesin-1's γ-phosphate sensor (33). G234A kinesin-1 is also unable to hydrolyze ATP, thus a 2′,3′-SLATP probe exchanged into its nucleotide pocket remains in a triphosphate state. G234A K349 containing 2′,3′-SLATP showed the same spectral shift as wild-type K349 containing 2′,3′-SLADP after addition of Tail944 (Fig. 3). Together, the data suggest that the head-tail interaction can occur independent of an intact γ-phosphate sensing mechanism, and regardless of the presence or absence of the nucleotide γ-phosphate. This is consistent with how GDIs bind their partner G proteins in both nucleotide states, but effectively rules out a mechanism that involves the tail targeting the γ-phosphate sensors.
Figure 3.
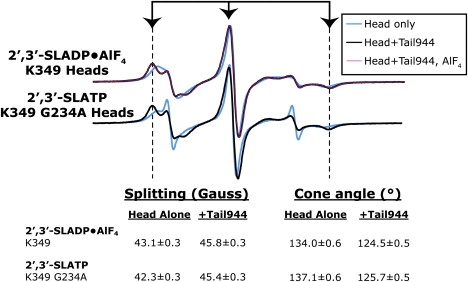
The tail-induced conformational change occurs independent of nucleotide state. Spectra of kinesin-1 bound to 2′,3′-SLADP or 2′,3′-SLATP are shown in the absence of tail (cyan), in the presence of tail (black), and in the presence of tail and 2 mM AlCl3 + 10mM NaF (pink). EPR spectral splittings are indicated as in Fig. 2. G234A kinesin-1 does not hydrolyze ATP, so the probe in the nucleotide pocket remains 2′,3′-SLATP (not 2′,3′-SLADP). 2 mM AlCl3 + 10 mM NaF induces an ADP•AlF4 triphosphate mimic state. The tail-bound spectral splittings are identical in the absence and presence of AlF4 or the G234A mutation, indicating that the tail-induced conformational restriction can occur with ADP or ATP in the pocket.
The tail-induced conformational restriction only weakly stabilizes Mg2+ in the nucleotide pocket
Mg2+ is required for tight binding of nucleotides to kinesin motors, and chelation of Mg2+ efficiently strips away the bound nucleotide (35). Mg2+ is also a target for some guanine nucleotide exchange factors (GEFs) that remove GDP from G proteins by disrupting Mg2+ binding (36). We tested the possibility that the tail could stabilize a bound nucleotide by coordinating Mg2+ in the nucleotide pocket.
We performed Mant-ADP-release experiments using EDTA to chelate Mg2+ out of the K420 nucleotide pocket. Our data show that Tail944 only marginally affected EDTA-induced removal of the Mg2+ ion (release rate of 0.073 ± 0.006 s−1 for K420 alone vs. 0.055 ± 0.005 s−1 for K420 in the presence of Tail944; Fig. 4). This trend is consistent with results from Hackney and Stock (10), who showed that the tail weakly inhibits initial Mg2+ release (reported rates of 0.037 s−1 for heads alone and 0.025 s−1 for heads in the presence of a tail construct). They concluded that most, if not all, of their observed decrease in rate could be explained by the inhibition of the intact Mg•ADP complex occurring simultaneously. In our experiments, EPR spectra of 2′,3′-SLADP-bound heads in the presence of Tail944 very quickly lost any immobilized component from bound nucleotide after addition of EDTA (faster than the time required to measure spectra). Thus, the tail-induced effect may not be significant, in which case we conclude that the tail does not bind either directly or indirectly to stabilize Mg2+ in the nucleotide pocket. Even if the effect is significant, tail stabilization of Mg2+ does not appear to be the major contributing factor to inhibition of nucleotide release in solution. However, the possibility exists that the tail could inhibit specific MT-induced mechanisms for triggering Mg2+ release, as our experiments did not assess MT-stimulated Mg2+ release explicitly.
Figure 4.
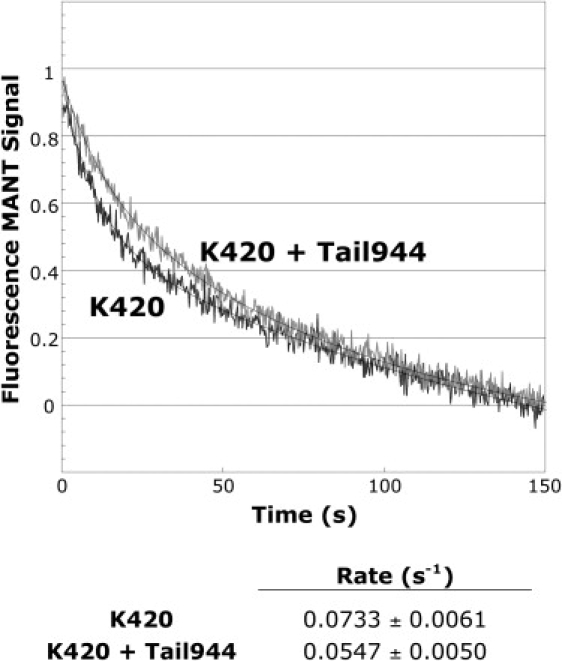
The tail only weakly inhibits release of Mg2+ from the nucleotide pocket. Removal of Mg2+ from the nucleotide pocket is followed sequentially by rapid release of bound nucleotide. Normalized time-dependent traces of Mant-ADP release from K420 occurring after addition of EDTA + ATP are shown. Smooth lines are weighted first-order fits to the normalized average traces. Rates and standard deviations are shown below the curves (n = 5 for both samples). The rate of Mant-ADP release from EDTA-treated K420 was only marginally different in the absence and presence of Tail944.
The K922 residue is critical for inhibition but not required for inducing the conformational restriction observed during head-tail interactions
Results by several groups have identified the tail K922 residue as critical to regulation (2,11). The mutation K922E in fungal kinesin results in an unregulated motor that accumulates at hyphal tips (11), and deletion of residues 920–922 (ΔIAK) abolishes regulation in human kinesin-1 (2,4). The reason why K922 is critical is not known. It could be a structural lynchpin that is required for the proper interaction of the tails with the heads to inhibit ADP release by a steric blocking mechanism. In this case, we would expect a K922A mutation to disrupt proper binding of tails to Switch I and the nucleotide pocket. Alternatively, other elements in the tail may interact with the head to properly position K922 for inhibition by binding to the nucleotide or coordinating nucleotide-sensing elements into a tightly bound configuration. If this is the case, we expect that a K922A mutation would abolish regulation without having a significant effect on the structure of the tail bound to the head.
To distinguish between the possibilities discussed above, we tested whether the characteristic conformational restriction of the nucleotide pocket occurs in the presence of Tail944 containing a K922A point mutation (TailK922A). We found that TailK922A induces an identical restriction of 2′,3′-SLADP and SSL-NANDP EPR probes as wild-type Tail944 (Fig. 5). Next, we measured rates of 2′,3′-SLADP release from K420 in the presence of Tail944 or TailK922A (Fig. 6, A and C). K420 alone released 2′3′-SLADP at a rate of 0.0177 ± 0.0004 s−1. In the presence of Tail944, the release rate was slowed to 0.0101 ± 0.0004 s−1. Whereas Tail944 inhibited the rate of 2′,3′-SLADP release in solution, TailK922A had no effect (release rate of 0.0189 ± 0.0013 s−1) despite the fact that it induces an identical restriction of EPR probe mobility as Tail944. Because the accuracy of rates measured by EPR is limited by low time resolution and difficulties in mixing, we confirmed our results in an independent assay by measuring K420 Mant-ADP release rates. Similarly, the rate of ADP release was inhibited by Tail944 but not TailK922A (Fig. 6, B and C). The rates determined by fluorescence spectroscopy are different by a factor of 2–3 from those determined by EPR. This is likely due to the structural differences between the Mant-ADP reporter and the 2′,3′-SLADP probe. The data reinforce the fact that K922 is critical for inhibition, but it does not participate in the head-tail interaction that causes a conformational restriction of the nucleotide pocket. As a corollary, we conclude that restriction of the nucleotide pocket alone does not inhibit ADP release. Therefore, the tails do not appear to prevent nucleotide release via a steric blocking mechanism.
Figure 5.
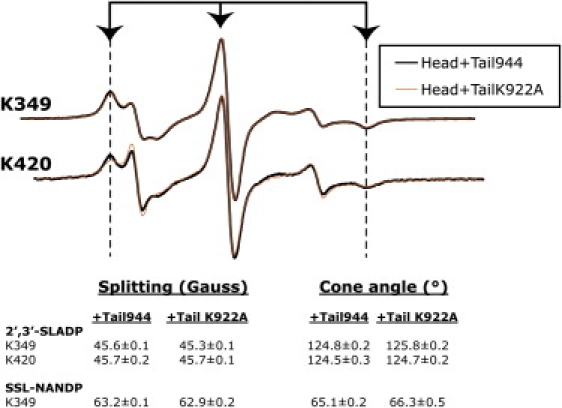
K922A tails induce similar structural changes as wild-type tails. Spectra of 2′,3′-SLADP-bound kinesin-1 are shown in the presence of Tail944 (black) and in the presence of TailK922A (orange). Only the 2′,3′-SLADP spectra are shown because of the large free component in SSL-NANDP spectra. EPR spectral splittings are indicated as in Fig. 2. The 2′,3′-SLADP and SSL-NANDP splittings of monomeric (K349) and dimeric (K420) head spectra are the same with Tail944 or TailK922A. The Tail944 and TailK922A spectra are hard to distinguish because they are almost perfectly superimposed on each other.
Figure 6.
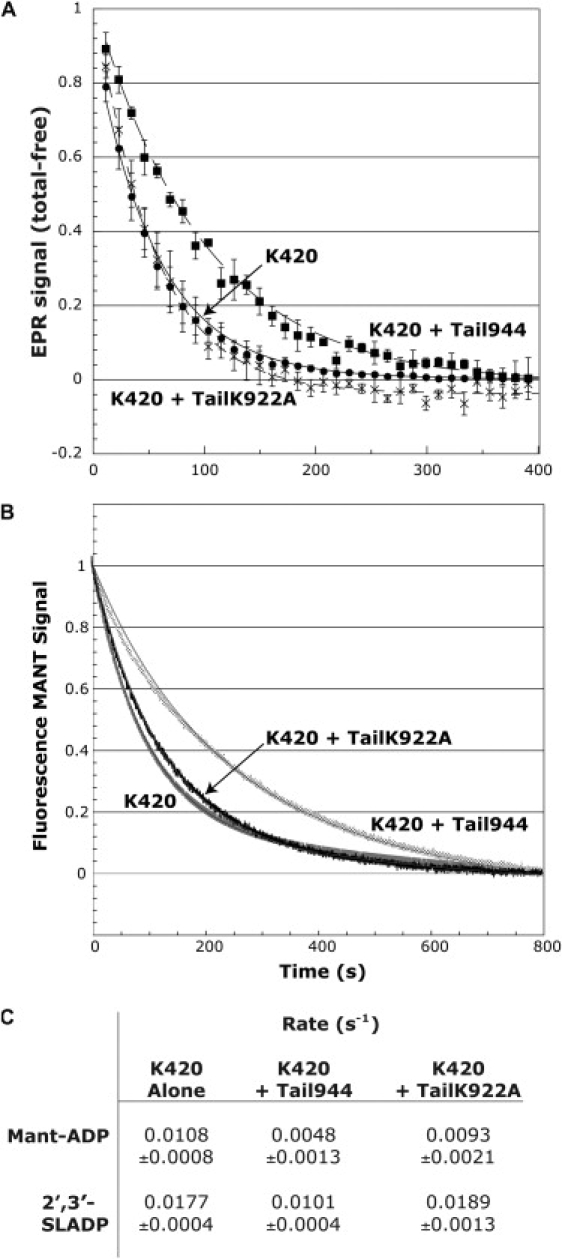
Wild-type tails inhibit nucleotide release, whereas K922A tails have no effect. Normalized time-dependent traces of nucleotide release from K420 occurring after addition of ATP are shown. Smooth lines are weighted first-order fits to the normalized average traces. (A) Release of 2′,3′-SLADP. Data points shown are the averages ± standard error of 4–6 measurements. (B) Release of Mant-ADP. Data points shown are averages of 4–8 measurements. Standard errors in these traces are too small to display. (C) Calculated rates for 2′,3′-SLADP and Mant-ADP. Rates and errors are given by the weighted first-order exponential fits shown in (A) and (B). The value for Mant-ADP release from K420 alone is consistent with published data on Mant-ADP release rates from kinesin-1 heads (40). The 2′,3′-SLADP data measured by EPR are different due to the lower affinity of kinesin-1 for the spin-labeled nucleotides. Both experiments demonstrate that the K922A Tail-944 is unable to inhibit nucleotide release from K420.
The combined data lead us to believe that the tail-induced conformational changes we observe in the nucleotide pocket may be a stabilizing interaction between the tail and the head that is not directly involved in the tail's regulatory function. This is the first evidence of a stabilizing interaction between the kinesin-1 tail and the nucleotide pocket in the head. It is possible that the tail-induced restriction of the nucleotide pocket serves to increase the binding affinity of the kinesin-1 tail and head, and to specifically position the regulatory K922 residue to inhibit nucleotide release.
Discussion
We have shown that the kinesin-1 tail induces a specific conformational restriction of the nucleotide pocket. This conformational restriction is not caused by the “closing” of Switch I that has been observed when kinesin-1 binds MTs (13,17). In fact, it may have the opposite effect on Switch I. Dietrich et al. (12) reported that the tail induces a conformation of Switch I that is similar to the conformation seen in crystal structures, in which ADP is very tightly bound. The tail-induced restriction of the nucleotide pocket observed here may be due to stabilizing interactions between the tail and the head that hold the tail in place to induce tight binding of the head to the nucleotide and the corresponding “open”, or solution-like conformation of Switch I.
As the spin label on 2′,3′-SLADP is directly attached to the 2′/3′ ribose oxygens, it serves as a direct sensor for interactions that might involve them. Because the TailK922A fragment restricts the mobility of our EPR probes exactly like Tail944, we rule out the possibility that K922 interacts with the ribose oxygens of the bound nucleotide. Because TailK922A is incapable of inhibiting nucleotide release, this also tells us that the tail does not block nucleotide exit by a steric hindrance mechanism involving the conformational restriction of the nucleotide pocket that we observe. However, the data remain consistent with a possible interaction of the K922 residue in the vicinity of the α/β-phosphates. Such an interaction would not necessarily hinder the mobility of 2′,3′-SLADP or SSL-NANDP probes, as the spin labels are somewhat distal to the phosphates. The observed mobility shifts likely occur as a result of positioning K922, which although critical to the tail's activity, is not required for the tail to bind to the nucleotide site.
A comparison of the kinesin-1 head-tail interaction with GDI/G protein interactions leads us to a possible model for tail-mediated inhibition. It is known that the GoLoco class of GDIs inhibit GDP release by inserting an arginine finger into the nucleotide binding site to coordinate the α/β-phosphates (14). Interestingly, a R516A mutation of the critical arginine in the RGS14 GDI results in a tenfold reduction in GDI activity, and an R516F mutation completely abrogates activity (24). However, the mutations do not decrease the ability of the GDI to complex with its partner G protein. This is the phenomenon observed between the kinesin-1 head and tail. Furthermore, when the structures of the GoLoco-G protein complex and kinesin-1 are aligned using the conserved P-loop (GxxxxGKS/T) and Switch II (DxxG) motifs, we see that the inhibitory GoLoco peptide is positioned in the same area we predict the kinesin-1 tail to be based on our previous cross-linking data (Fig. 7, A–D). In the superimposed structure, the critical GoLoco arginine is perfectly positioned to coordinate the α/β-phosphates of kinesin-1's bound ADP.
Figure 7.
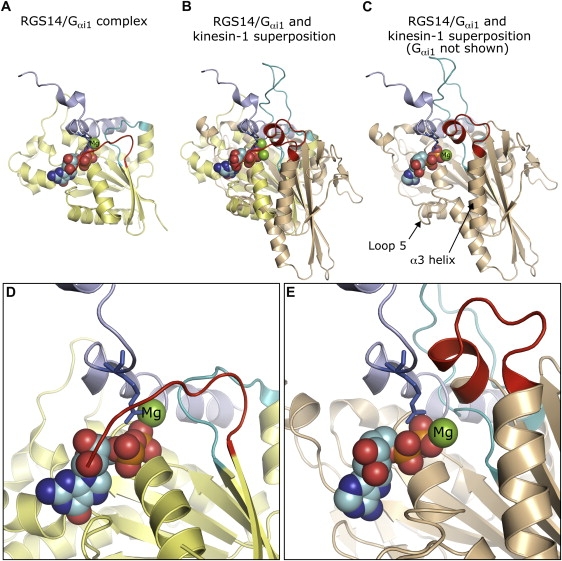
A possible role of the tail K922 residue in kinesin-1 inhibition. (A) Structure of the RGS14 GDI GoLoco region complexed to the Ras-like domain of Gαi1 (PDB 1kjy) (24). The RGS14 GoLoco region that contains the critical regulatory arginine finger is shown in purple. Switch I and Switch II in Gαi1 are indicated in red and cyan, respectively. (B) The RGS14 GDI GoLoco motif superimposed onto the kinesin-1 crystal structure (PDB 1bg2) (19). The same superposition is shown in (C) without Gαi1. Kinesin-1 was aligned with the Gαi1 Ras-like domain using the P-loop (GxxxxGKS/T) and Switch II (DxxG) motifs that are conserved between G proteins and motor proteins. Switch I and Switch II are indicated as above. The kinesin-1 tail interacts with Switch I in approximately the same area that RGS14 is positioned in the structure, as shown by chemical cross-linking (12). In a manner analogous to RGS14 and Gαi1, scaffolding interactions could position K922 to coordinate the nucleotide α/β-phosphates as a “lysine finger”. Close-up views of the bound nucleotide and RGS14 regulatory arginine are shown in approximately the same orientation as (A–C) for Gαi1 (D) and kinesin-1 (E). Figures were generated with Pymol (39).
In crystal structures of nucleotide-free G proteins complexed with GEFs, the P-loop lysine, which formerly contacted negative charges on the α/β-phosphates, is rotated away to interact with acidic residues either on the G protein or a glutamic acid finger on the GEF (36). Although no causality is implied between the loss of this lysine-phosphate interaction and nucleotide release, we note that the presence of the tail K922 residue may serve to preserve a stabilizing interaction for the nucleotide α/β-phosphates. Analysis of the G protein/GEF complexes also reveals that the position of Switch I is dramatically altered in these crystal structures. In particular, coordination of the Mg2+ ion by a critical hydroxyl is lost. It is possible that MTs, acting as a nucleotide exchange factor, may likewise remove Switch I from the Mg2+ ion, resulting in sequential ADP release. This mechanism has been proposed for the actin-myosin V system (37,38). Indeed, if this were also the mechanism for MT-stimulated ADP release from kinesin-1, then it makes sense that our previously published cryo-EM structure of the kinesin-1 head-tail-MT complex shows Switch I in a solution-like position (12); the tail prevents MTs from moving Switch I. In solution, however, this does not seem to be the predominant mechanism. As mentioned, the tail only marginally affects release of Mg2+ from the nucleotide pocket. Additionally, Hackney and Stock (10) have shown that the tail can still strongly inhibit Mg-free nucleotide release. Thus, there may be multiple routes for nucleotide release in solution and on MTs, and K922 may inhibit some of these.
Based on the work presented here, we propose a model wherein the tail K922 may act as a GoLoco-like “lysine finger” by interacting directly with the nucleotide α/β-phosphates, which explains our observed lack of dependence on the nucleotide γ-phosphate. In this scenario, the tail K922 keeps the nucleotide in the pocket by the K922-phosphate interactions coupled with the head-tail stabilizing interactions. This would appear to be the likely mechanism in solution. Upon MT binding, K922 may work in conjunction with a separate tail-induced mechanism, possibly the stabilization of Switch I in an “open” conformation, to prevent MT-stimulated Mg2+ and ADP release. We await a complete structural view of the head-tail complex to verify our proposed model.
Acknowledgments
The authors thank R. Vale for constructs; J. Grammer, X. Chen, and R. Yount for the generous gift of the SSL-NANTP probe; and E. Pate for the critical reading of the manuscript.
This work was supported by grants 1 R01GM072656 (Y.L.W. and S.E.R.), T32 GM008382 (K.A.D.) and Program Project AR 42895 (N.N. and R.C.) from the National Institutes of Health.
References
- 1.Hollenbeck P.J. The distribution, abundance and subcellular localization of kinesin. J. Cell Biol. 1989;108:2335–2342. doi: 10.1083/jcb.108.6.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hackney D.D., Stock M.F. Kinesin's IAK tail domain inhibits initial microtubule-stimulated ADP release. Nat. Cell Biol. 2000;2:257–260. doi: 10.1038/35010525. [DOI] [PubMed] [Google Scholar]
- 3.Verhey K.J., Lizotte D.L., Abramson T., Barenboim L., Schnapp B.J. Light chain-dependent regulation of Kinesin's interaction with microtubules. J. Cell Biol. 1998;143:1053–1066. doi: 10.1083/jcb.143.4.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cai D., Hoppe A.D., Swanson J.A., Verhey K.J. Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J. Cell Biol. 2007;176:51–63. doi: 10.1083/jcb.200605097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman D.S., Vale R.D. Single-molecule analysis of kinesin motility reveals regulation by the cargo-binding tail domain. Nat. Cell Biol. 1999;1:293–297. doi: 10.1038/13008. [DOI] [PubMed] [Google Scholar]
- 6.Stock M.F., Guerrero J., Cobb B., Eggers C.T., Huang T.G. Formation of the compact confomer of kinesin requires a COOH-terminal heavy chain domain and inhibits microtubule-stimulated ATPase activity. J. Biol. Chem. 1999;274:14617–14623. doi: 10.1074/jbc.274.21.14617. [DOI] [PubMed] [Google Scholar]
- 7.Bathe F., Hahlen K., Dombi R., Driller L., Schliwa M. The complex interplay between the neck and hinge domains in kinesin-1 dimerization and motor activity. Mol. Biol. Cell. 2005;16:3529–3537. doi: 10.1091/mbc.E04-11-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hackney D.D., Levitt J.D., Suhan J. Kinesin undergoes a 9 S to 6 S conformational transition. J. Biol. Chem. 1992;267:8696–8701. [PubMed] [Google Scholar]
- 9.Hisanaga S., Murofushi H., Okuhara K., Sato R., Masuda Y. The molecular structure of adrenal medulla kinesin. Cell Motil. Cytoskeleton. 1989;12:264–272. doi: 10.1002/cm.970120407. [DOI] [PubMed] [Google Scholar]
- 10.Hackney D.D., Stock M.F. Kinesin tail domains and Mg2+ directly inhibit release of ADP from head domains in the absence of microtubules. Biochemistry. 2008;47:7770–7778. doi: 10.1021/bi8006687. [DOI] [PubMed] [Google Scholar]
- 11.Seiler S., Kirchner J., Horn C., Kallipolitou A., Woehlke G. Cargo binding and regulatory sites in the tail of fungal conventional kinesin. Nat. Cell Biol. 2000;2:333–338. doi: 10.1038/35014022. [DOI] [PubMed] [Google Scholar]
- 12.Dietrich K.A., Sindelar C.V., Brewer P.D., Downing K.H., Cremo C.R. The kinesin-1 motor protein is regulated by a direct interaction of its head and tail. Proc. Natl. Acad. Sci. USA. 2008;105:8938–8943. doi: 10.1073/pnas.0803575105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sindelar C.V., Downing K.H. The beginning of kinesin's force-generating cycle visualized at 9-A resolution. J. Cell Biol. 2007;177:377–385. doi: 10.1083/jcb.200612090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willard F.S., Kimple R.J., Siderovski D.P. Return of the GDI: the GoLoco motif in cell division. Annu. Rev. Biochem. 2004;73:925–951. doi: 10.1146/annurev.biochem.73.011303.073756. [DOI] [PubMed] [Google Scholar]
- 15.Kull F.J., Endow S.A. Kinesin: switch I & II and the motor mechanism. J. Cell Sci. 2002;115:15–23. doi: 10.1242/jcs.115.1.15. [DOI] [PubMed] [Google Scholar]
- 16.Himmel D.M., Gourinath S., Reshetnikova L., Shen Y., Szent-Gyorgyi A.G. Crystallographic findings on the internally uncoupled and near-rigor states of myosin: further insights into the mechanics of the motor. Proc. Natl. Acad. Sci. USA. 2002;99:12645–12650. doi: 10.1073/pnas.202476799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naber N., Minehardt T.J., Rice S., Chen X., Grammer J. Closing of the nucleotide pocket of kinesin-family motors upon binding to microtubules. Science. 2003;300:798–801. doi: 10.1126/science.1082374. [DOI] [PubMed] [Google Scholar]
- 18.Naber N., Rice S., Matuska M., Vale R.D., Cooke R. EPR spectroscopy shows a microtubule-dependent conformational change in the kinesin switch 1 domain. Biophys. J. 2003;84:3190–3196. doi: 10.1016/S0006-3495(03)70043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kull F.J., Sablin E.P., Lau R., Fletterick R.J., Vale R.D. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kozielski F., Sack S., Marx A., Thormahlen M., Schonbrunn E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 21.Scheffzek K., Stephan I., Jensen O.N., Illenberger D., Gierschik P. The Rac-RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nat. Struct. Biol. 2000;7:122–126. doi: 10.1038/72392. [DOI] [PubMed] [Google Scholar]
- 22.Hoffman G.R., Nassar N., Cerione R.A. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell. 2000;100:345–356. doi: 10.1016/s0092-8674(00)80670-4. [DOI] [PubMed] [Google Scholar]
- 23.Rak A., Pylypenko O., Durek T., Watzke A., Kushnir S. Structure of Rab GDP-dissociation inhibitor in complex with prenylated YPT1 GTPase. Science. 2003;302:646–650. doi: 10.1126/science.1087761. [DOI] [PubMed] [Google Scholar]
- 24.Kimple R.J., Kimple M.E., Betts L., Sondek J., Siderovski D.P. Structural determinants for GoLoco-induced inhibition of nucleotide release by Galpha subunits. Nature. 2002;416:878–881. doi: 10.1038/416878a. [DOI] [PubMed] [Google Scholar]
- 25.Noel J.P., Hamm H.E., Sigler P.B. The 2.2 A crystal structure of transducin-alpha complexed with GTP gamma S. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- 26.Ford B., Skowronek K., Boykevisch S., Bar-Sagi D., Nassar N. Structure of the G60A mutant of Ras: implications for the dominant negative effect. J. Biol. Chem. 2005;280:25697–25705. doi: 10.1074/jbc.M502240200. [DOI] [PubMed] [Google Scholar]
- 27.Griffith O.H., Jost P.C. Lipid spin labels in biological membranes. In: Berliner L.J., editor. Spin Labeling Theory and Applications. Academic Press; NY: 1976. pp. 454–523. [Google Scholar]
- 28.Gulick A.M., Bauer C.B., Thoden J.B., Pate E., Yount R.G. X-ray structures of the Dictyostelium discoideum myosin motor domain with six non-nucleotide analogs. J. Biol. Chem. 2000;275:398–408. doi: 10.1074/jbc.275.1.398. [DOI] [PubMed] [Google Scholar]
- 29.Ma Y.Z., Taylor E.W. Interacting head mechanism of microtubule-kinesin ATPase. J. Biol. Chem. 1997;272:724–730. doi: 10.1074/jbc.272.2.724. [DOI] [PubMed] [Google Scholar]
- 30.Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell. Signal. 1999;11:545–554. doi: 10.1016/s0898-6568(98)00063-1. [DOI] [PubMed] [Google Scholar]
- 31.Olivares A.O., Chang W., Mooseker M.S., Hackney D.D., De La Cruz E.M. The tail domain of myosin Va modulates actin binding to one head. J. Biol. Chem. 2006;281:31326–31336. doi: 10.1074/jbc.M603898200. [DOI] [PubMed] [Google Scholar]
- 32.Muller J., Marx A., Sack S., Song Y.H., Mandelkow E. The structure of the nucleotide-binding site of kinesin. Biol. Chem. 1999;380:981–992. doi: 10.1515/BC.1999.122. [DOI] [PubMed] [Google Scholar]
- 33.Rice S., Lin A.W., Safer D., Hart C.L., Naber N. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- 34.Shibuya H., Kondo K., Kimura N., Maruta S. Formation and characterization of kinesin.ADP.fluorometal complexes. J Biochem (Tokyo) 2002;132:573–579. doi: 10.1093/oxfordjournals.jbchem.a003259. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu T., Sablin E., Vale R.D., Fletterick R., Pechatnikova E. Expression, purification, ATPase properties, and microtubule-binding properties of the ncd motor domain. Biochemistry. 1995;34:13259–13266. doi: 10.1021/bi00040a042. [DOI] [PubMed] [Google Scholar]
- 36.Vetter I.R., Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 37.Rosenfeld S.S., Houdusse A., Sweeney H.L. Magnesium regulates ADP dissociation from myosin V. J. Biol. Chem. 2005;280:6072–6079. doi: 10.1074/jbc.M412717200. [DOI] [PubMed] [Google Scholar]
- 38.Coureux P.D., Wells A.L., Menetrey J., Yengo C.M., Morris C.A. A structural state of the myosin V motor without bound nucleotide. Nature. 2003;425:419–423. doi: 10.1038/nature01927. [DOI] [PubMed] [Google Scholar]
- 39.Delano, W.L. 2002. The PyMOL Molecular Graphics System. http://www.pymol.org. Accessed August 19, 2008.
- 40.Ma Y.Z., Taylor E.W. Kinetic mechanism of kinesin motor domain. Biochemistry. 1995;34:13233–13241. doi: 10.1021/bi00040a039. [DOI] [PubMed] [Google Scholar]