

Abstract
We apply the astronomical data-analysis technique, Lucky imaging, to improve resolution in single molecule fluorescence microscopy. We show that by selectively discarding data points from individual single-molecule trajectories, imaging resolution can be improved by a factor of 1.6 for individual fluorophores and up to 5.6 for more complex images. The method is illustrated using images of fluorescent dye molecules and quantum dots, and the in vivo imaging of fluorescently labeled linker for activation of T cells.
Introduction
Single molecule fluorescence microscopy has provided many unique insights into complex biological systems that would otherwise be inaccessible by traditional ensemble measurements (1–4). Some of the most recent advances in this field are super-resolution techniques capable of breaking the diffraction limit. These methods allow the position of an individual fluorescent emitter to be resolved with a precision greater than that described by Abbe's Law (5). There are two branches of research that focus on improving imaging resolution. The first is based on structured illumination and includes saturated pattern excitation microscopy (6) and stimulated emission depletion (7), which has been applied to track presynaptic vesicles in live neurons in real time (8) and reveal the structure of mitochondria in vivo (9). Here, we focus on the second branch, which involves locating the center of the point spread function (PSF) of a fluorescent emitter. When imaged, a fluorophore behaves as a point source with an Airy disk PSF. The center of mass of the function, and therefore the position of the molecule, can be obtained by performing a least-squares fit of an appropriate function (such as a Gaussian distribution) to the measured fluorescence intensity profile of the spot.
One of the first successful implementations of high-precision localization imaging was the use of fluorescence imaging with one nanometre accuracy (FIONA) to confirm the hand-over-hand model for myosin V moving on actin (10). This work showed that a localization precision of 1.3 nm can be achieved for a single fluorophore under optimum conditions. The accuracy is principally limited by photon noise with contributions from background noise and pixilation. Thompson et al. (11) suggest that the precision in one dimension can be described by,
| (1) |
Δx is the error in the localization, s is the width of the PSF (described by a 2D Gaussian) and N is the number of photons collected. The first term of the equation is the photon noise, the second is due to the increase in error due to the finite size, a, of the pixels in the image, whereas the third term also takes the background noise, b, into account.
Many methods based on FIONA have been reported (with similarly creative acronyms). These include nanometer localized multiple single molecule (NALMS) (12), point accumulation for imaging in nanoscale topography (PAINT) (13), photoactivatable localization microscopy(PALM) (14,15), spectral position determination microscopy (SPDM) (16), single molecule high resolution imaging with photobleaching (SHRImP) (17), and stochastic optical reconstruction microscopy (STORM) (18,19). Recent reviews have highlighted the usefulness of such tools to study complex biological processes (1–4). The field is growing quickly and with the development of biologically compatible, photoswitchable dyes, these methods are moving away from the physics laboratory and starting to answer relevant biological questions.
In parallel to these improvements in single molecule fluorescence there have been technological advancements in other imaging fields. Our recent application of the astronomical algorithm CLEAN (20,21) led us to examine other astronomical imaging procedures and consider if they might be applied to single molecule imaging. One novel approach to astronomical imaging, Lucky imaging, has attracted media attention because of the high-resolution images that can be obtained (22,23). For example, researchers in the field have utilized fast electron-multiplying charge-coupled device (CCD) cameras and careful data rejection techniques to overcome atmospheric aberrations and other sources of noise, allowing images with a resolution better than that from the Hubble space telescope to be captured from the Earth's surface at a fraction of the cost. The low cost and simplicity of the technique means that it is employed by many amateur astronomers using backyard telescopes with basic CCD detectors, resulting in improved images of planets and deep-sky phenomena.
This work describes the application of Lucky imaging to single molecule fluorescence. Lucky imaging is, in essence, very simple because it involves selective rejection of data. Briefly, large data sets corresponding to an image sequence from fluorescent molecules are recorded using an electron-multiplying CCD camera. Localization methods are used to fit the individual PSFs of each fluorophore into each frame of the resulting video. A selection procedure then removes spots that have localization errors above a defined threshold. Larger errors are caused by noise from various sources, including read-out noise, dark current noise, fluorescent contamination, and focus drift (11). An image is then built up using only the retained fits. This resultant image has a higher resolution than an image that uses Gaussian localization methods alone. Key to this process is the collection of sufficient data (either through fast acquisition, or long image sequences) to enable efficient rejection of low-quality fits. This method can reveal useful information in fluorescent images that would otherwise be unresolved. Here we explore the usefulness of Lucky imaging for single molecule fluorescence from both synthetic dye molecules and quantum dots.
We also demonstrate the application of Lucky imaging to the in vivo localization of a fluorescently labeled derivative of the linker for activation of T cells (LAT) protein. The activation of antigen-specific T cells is a key step in the host-adaptive immune response to pathogens and involves antigen recognition and signaling through the T-cell receptor (TCR). These events are relayed by a large number of kinases and adaptor molecules that associate at the plasma membrane to form spatially restricted signaling foci. Study of the spatiotemporal dynamics of TCR signaling components has provided many new insights into mechanisms governing both TCR triggering as well as downstream signaling events (24). The adaptor molecule LAT is a key nucleator of TCR signaling assemblies (25). As such, the ability to accurately determine its spatial distribution in real time will be particularly useful to further our understanding of TCR signaling dynamics.
Materials and Methods
Data were collected using an inverted microscope (TE-2000/Eclipse Ti-U; Nikon Instruments Europe B.V., Surrey, England) with through-objective total internal reflection (TIR) illumination (100x Plan Apo N.A. 1.45, Nikon Instruments) using either 488 nm (5 mW Ar ion, Spectra Physics, Mountain View, CA) or 532 nm (15 mW Compass 215M; Coherent Inc, Santa Clara, CA) laser light. The emitted fluorescence was collected through the same objective, transmitted through dichroic (Q495LP or Q565LP) and band-pass (HQ525/50 or HQ580/60) filters (Chroma Technology Corp, Rockingham, VT), and imaged using a 128 × 128 pixel frame-transfer electron-multiplying CCD detector (iXon DU-860; Andor Technology PLC, Belfast, UK). Image sequences were converted to 16-bit tagged image file format bitmap stacks.
The Lucky method was implemented in Igor Pro (Wavemetrics; Portland, OR). Single molecule detection and localization is achieved using the astronomical algorithm CLEAN (20), as previously described by our group (21). Briefly, the brightest pixel in the first frame of the image is located. A region of interest centered on this pixel (typically 8 × 8 pixels) is then extracted from the image. A 2D Gaussian function is fitted to the region of interest and the fitting parameters are recorded. CLEAN then iterates to find the next brightest pixel in the image and the procedure continues in this manner until a defined noise threshold is reached, after which the algorithm proceeds to the next image frame and repeats. The result is an array containing the x and y coordinates of each fluorescent spot, along with the fitting parameters and their respective errors. In the case of Lucky imaging, a filter is applied to this array, rejecting any spots that have localization errors above a defined value. The spots that are retained after filtering are drawn to a new image stack using the parameters obtained from the localization of the PSF. The final image reported here is built up by summing the image frames together (Figs. 1 and 2). For moving objects, acceptance of a particular spot is dependent only on the localization precision in an individual frame, and not on the summed image.
Figure 1.
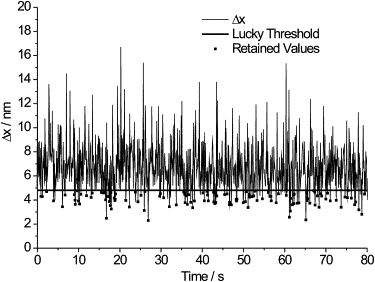
Illustration of the implementation of Lucky imaging for single molecule fluorescence. Δx, the precision in the 2D Gaussian fitting of the PSF for a Cy3B fluorophore, is plotted as a function of time where each of the time points is an image frame from a TIRF image stack recorded with an exposure time of 0.08 s per frame. The bold line shows the Lucky threshold applied to the data at 4.80 nm. The square data points show the retained data points.
Figure 2.
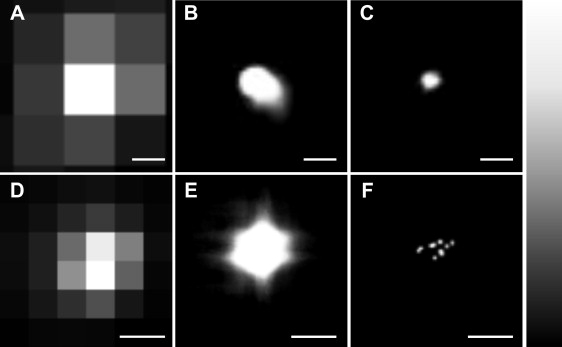
The upper panels show data accumulated by TIRF imaging a single Cy3B molecule dried onto a glass coverslip. (A) The sum of 1000 raw image frames recorded with an exposure time of 0.08 s per frame. B shows the output after Gaussian fitting, whereas image C is the sum of the frames with values for Δx below 4.80 nm (solid line in Fig. 1),;the scale bars in A, B, and C are 100 nm. The lower panels show TIRF images of quantum dot aggregates dried onto a glass coverslip composed of 5000 summed frames recorded with an exposure time of 0.08s in D the raw diffraction limited format, E after Gaussian fitting and F with a Lucky threshold of 9.4 nm applied to the localization data. The scale bars are 250 nm in these three images. The panel to the far right shows the look-up table for the images ranging, from black at low intensity to white at high intensity.
Images of Cy3B (GE Healthcare Life Sciences, Amersham, UK) were obtained by drying a 20-pM solution of the dye onto an O2 plasma cleaned coverslip and illuminating with the 532-nm laser. Similarly a ∼10-nM solution of quantum dots (Evitag, Catskill Green, Evident Technologies, Troy, NY) was dried onto a clean coverslip and illuminated with the 488-nm laser. Quantum dot aggregation was achieved by diluting stock solutions (12 M) in ultrapure water (MilliQ 18 MΩ double distilled, 0.2 μm filtered) for 24 h at 25°C. Subsequent sonication with a small amount of subsequent sonication was shown to reverse this aggregation.
JCaM2 leukemic T cells stably expressing LAT-yellow fluorescent protein (YFP) fusion protein (26) were adhered to CD3-antibody–coated glass coverslips, as described by Douglass and Vale (27). After 10 min, the cells were visualized with 488-nm TIR fluorescence (TIRF) illumination.
Results
In the first instance, fluorescent dye molecules were used to test the effectiveness of the Lucky method. Cy3B was dried onto a glass coverslip and imaged as described. Fig. 1 illustrates the effect of Lucky imaging on an individual fluorescent spot taken from such an image. The precision in the Gaussian fit in the x dimension, Δx, is plotted as a function of time where each of the time points is an image frame from a stack recorded with an exposure time of 0.08 s per frame. The raw image, a sum of each of the frames, is shown in Fig. 2 A. After Gaussian fitting, the average value of Δx is 6.71 ± 0.06 nm and the resulting image of the spot, again a sum of each of the frames, is shown in Fig. 2 B. The bold line at 4.80 nm in Fig. 1 illustrates the Lucky threshold applied to the Gaussian localized data, and fits with a Δx value greater than this bold line are rejected. The data points below the line, indicated by solid squares, are the spots that are retained after filtering. The mean value of Δx is now improved to 4.05 ± 0.05 nm and the resulting image (which has been similarly filtered in the y direction) is shown in Fig. 2 C. The improvement in image resolution is clear from this one spot.
Fig. 3 A shows the relationship between the applied Lucky threshold and the resulting root mean-squared Δx data for one fluorescent spot imaged for 1000 frames at 0.08 s. At high thresholds, the Δxrms value is unaffected but, as expected, as the threshold is lowered, the Δxrms value decreases as Gaussian fits with large errors are removed. Once the threshold is <10 nm, Δxrms appears to decrease linearly with the threshold until the resolution limit, 3 nm in this case, is reached. Fig. 3 B shows the root mean-squared Δx for one fluorescent spot imaged for 300 frames at a variety of exposure times. At very fast exposure times, Eq. 1 is limited by low values of N that is, the intensity of the spot is low. As the exposure time increases, so does N, but at long exposure times, the background noise term, b, is also seen to increase. A balance between these two effects must be considered. Since the Lucky technique relies on image rejection, there is an incentive to record images at fast exposure times to maximize the number of data points that are retained. An exposure time of 0.08 s was therefore chosen as a compromise between fluorophore imaging intensity and noise accumulation.
Figure 3.
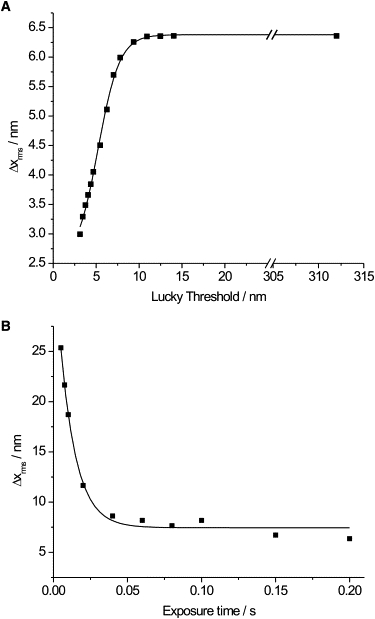
(A) Data from a TIRF experiment of a single Cy3B molecule imaged for 1000 frames with an exposure time of 0.08 s per frame. The sigmoidal relationship between Δxrms and the applied Lucky threshold is shown. (B) Δxrms data for one fluorescent spot imaged for 300 frames at a variety of exposure times to assess the optimum exposure time for our apparatus. All the data are from the same Cy3B molecule. Δxrms decays exponentially (τ = 0.011) with exposure time.
To illustrate this method, images of aggregated quantum dots were analyzed using the Lucky method. Fig. 2 D–F each show a small section of one such experiment (5000 frames summed) for the raw image, the image following Gaussian fitting, Δxrms = 32.60 ± 0.49 nm, and with a Lucky threshold of 9.4 nm applied, Δxrms = 5.78 ± 0.39 nm, respectively. The benefit of the Lucky method is clear; in Fig. 2 D there appears to be one source of fluorescence, whereas in Fig. 2 F, at least seven fluorescent emitters can be seen.
Fig. 4 shows TIRF images of a live Jurkat T cell stimulated on CD3-antibody–coated glass coverslips. The images are each a sum of 3000 frames, with the original data recorded at an exposure time of 0.1 s per frame. Again, images are shown before (Fig 4 A and D) and after (Fig 4 B and E) Gaussian fitting and with an applied Lucky threshold of 15.6 nm (Fig. 4 C and F). The difference between the raw and fitted image is striking. Using Gaussian localization allows visualization of the centripetal migration of fluorescent LAT-YFP clusters in the cell. This behavior has been observed previously using confocal microscopy (28). In the case of the fitted image shown in Fig. 4 B, Δxrms = 42.86 ± 0.48 nm. Further improvement is seen when Lucky imaging is used and Δxrms is reduced to 8.82 ± 0.08 nm. The lower panels show magnified images of the section highlighted in Fig. 4 A; it is clear that the Lucky technique provides improved resolution.
Figure 4.
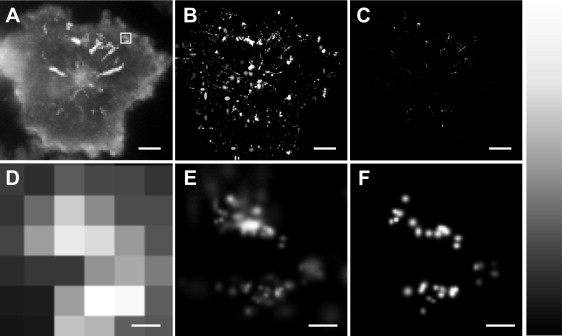
TIRF images of a live T cell in which the protein LAT has been labeled with enhanced YFP. The image was recorded with an exposure time of 0.1 s and is shown in the raw diffraction limited format (A and D, plotted as the maximum pixel intensity for 3000 frames), after Gaussian fitting (B and E, the sum of 3000 frames), and with a Lucky threshold of 15.6 nm applied (C and F, the sum of 3000 frames). The scale bars in the upper panels are 2 μm. The lower panels show the section of the image that is highlighted by the white box in A and the scale bars are 150 nm. The panel to the far right shows the look-up table for the images, ranging from black at low intensity to white at high intensity.
Discussion
We have shown that Lucky imaging can improve the resolution of single molecule microscopy. Much like its astronomical counterpart, it provides a remarkably simple and quick way to capture high-resolution images. The results reported here are illustrative of the improvement of resolution that can be achieved under many different experimental conditions. The analysis is straightforward, involving a balance between the threshold for precision required and the number of fits that are retained after filtering. The method does not require any specialist equipment or optical arrangement, so it can be quickly applied to many single molecule experiments. The technique is principally restricted by the fundamental resolution limits of the imaging system, as described by Thompson et al. (11); i.e., from Eq. 1 the precision available is determined by the number of photons emitted by an individual fluorophore, N, and the pixel noise, a. Lucky imaging essentially reduces contributions from the background noise, b, and also rejects instances where N is low, leading to otherwise inaccurate fits. Key to efficient implementation of Lucky imaging is a large enough data set, in which aggressive rejection of data points from individual single-molecule trajectories can be discarded. This can be achieved through either fast data acquisition or long image sequences. Faster acquisition is limited by the signal/noise ratio from an individual fluorophore, whereas longer image sequences are limited by photobleaching.
Lucky imaging has been used to improve the resolution of an image of aggregated quantum dots to reveal the number of particles in the aggregate. The stoichiometry of similarly diffraction-limited dye molecule complexes can be deduced by studying their stepwise photobleaching behavior, observed as stepped decreases in fluorescence intensity (29). However, because of the photoblinking behavior of quantum dots, the fluorescence intensity of aggregates remains constant with time, so that photobleaching studies are not feasible. Lucky imaging offers a method to confirm the number of quantum dots in a diffraction limited spot that is applicable labeling studies (30). Fig. 2 shows that by Gaussian fitting alone (Fig 2 E), there is no way of distinguishing the number of fluorescent emitters present. Once a lucky threshold has been applied, Fig. 2 F reveals that there are at least seven sources of fluorescence in the aggregate.
Another recently reported localization technique, STORM (18), makes use of photoswitchable fluorophores to image a stochastically different subset of molecules in each image frame before combining all the images into a high-resolution composite. The photoblinking behavior of quantum dots is similar to switchable systems in that these particles switch between a bright and dark state with time. It is likely that the simple experiment described here can be expanded to all switchable systems and those methods, such as STORM and PALM (14), can benefit. Previous methods make use of data rejection techniques based on the width of the fitted Gaussian, whereas Lucky imaging directly rejects fits with large localization errors, providing higher resolution.
To fully understand many functional cellular processes, it is necessary to obtain structural information with an appropriately high spatial resolution. By analyzing a live cell image, we demonstrate that the Lucky technique can be practical for in vivo single molecule studies. This suggests that it may be applied to a range of future experiments, such as observing the nanometer resolution of clustering of receptors on the cell surface (19), determining the stoichiometry of biologically interesting complexes, or counting molecules on the cell membrane or RNA transcripts. Investigations of viral entry and endocytosis, which rely on particle tracking and localization, could also benefit from Lucky thresholding (31,32). Additionally, it may be feasible to access cellular structural information that has previously been limited by the available resolution, for example, individual expression domains in genetically active and inactive sites, the arrangement of the polyribosomes, nuclear pore complex distribution, or the nuclear distribution of replication factories.
Acknowledgments
The authors thank Katherine Emmett, for her initial contributions to the localization procedures, and James Thompson, for useful discussion.
Financial support is from the Biotechnology and Biological Sciences Research Council and Nikon under the Nikon Oxford Molecular Imaging Centre project. M.I.W holds a Royal Society University Research Fellowship.
References
- 1.Hell S.W. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 2.Moerner W.E. New directions in single-molecule imaging and analysis. Proc. Natl. Acad. Sci. USA. 2007;104:12596–12602. doi: 10.1073/pnas.0610081104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walter N.G., Huang C.Y., Manzo A.J., Sobhy M.A. Do-it-yourself guide: how to use the modern single-molecule toolkit. Nat. Methods. 2008;5:475–489. doi: 10.1038/nmeth.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenleaf W.J., Woodside M.T., Block S.M. High-resolution, single-molecule measurements of biomolecular motion. Annu. Rev. Biophys. Biomol. Struct. 2007;36:171–190. doi: 10.1146/annurev.biophys.36.101106.101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michalet X., Weiss S. Using photon statistics to boost microscopy resolution. Proc. Natl. Acad. Sci. USA. 2006;103:4797–4798. doi: 10.1073/pnas.0600808103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heintzmann R., Jovin T.M., Cremer C. Saturated patterned excitation microscopy–a concept for optical resolution improvement. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2002;19:1599–1609. doi: 10.1364/josaa.19.001599. [DOI] [PubMed] [Google Scholar]
- 7.Hell S.W., Wichmann J. Breaking the diffraction resolution limit by stimulated-emission - stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 8.Westphal V., Rizzoli S.O., Lauterbach M.A., Kamin D., Jahn R. Video-rate far-field optical nanoscopy dissects synaptic vesicle movement. Science. 2008;320:246–249. doi: 10.1126/science.1154228. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt R., Wurm C.A., Jakobs S., Engelhardt J., Egner A. Spherical nanosized focal spot unravels the interior of cells. Nat. Methods. 2008;5:539–544. doi: 10.1038/nmeth.1214. [DOI] [PubMed] [Google Scholar]
- 10.Yildiz A., Forkey J.N., McKinney S.A., Ha T., Goldman Y.E. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]
- 11.Thompson R.E., Larson D.R., Webb W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qu X., Wu D., Mets L., Scherer N.F. Nanometer-localized multiple single-molecule fluorescence microscopy. Proc. Natl. Acad. Sci. USA. 2004;101:11298–11303. doi: 10.1073/pnas.0402155101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharonov A., Hochstrasser R.M. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc. Natl. Acad. Sci. USA. 2006;103:18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Betzig E., Patterson G.H., Sougrat R., Lindwasser O.W., Olenych S. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 15.Biteen J.S., Thompson M.A., Tselentis N.K., Bowman G.R., Shapiro L. Super-resolution imaging in live Caulobacter crescentus cells using photoswitchable EYFP. Nat Methods. 2008 doi: 10.1038/NMETH.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemmer P., Gunkel M., Baddeley D., Kaufmann R., Urich A. SPDM: light microscopy with single-molecule resolution at the nanoscale. Appl. Phys. B. 2008;93:1–12. [Google Scholar]
- 17.Gordon M.P., Ha T., Selvin P.R. Single-molecule high-resolution imaging with photobleaching. Proc. Natl. Acad. Sci. USA. 2004;101:6462–6465. doi: 10.1073/pnas.0401638101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rust M.J., Bates M., Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat. Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang B., Wang W., Bates M., Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hogbom J.A. Aperture synthesis with a non regular distribution of interferometer baselines. Astron. Astrophys. Suppl. Ser. 1974;15:417–426. [Google Scholar]
- 21.Thompson J.R., Heron A.J., Santoso Y., Wallace M.I. Enhanced stability and fluidity in droplet on hydrogel bilayers for measuring membrane protein diffusion. Nano Lett. 2007;7:3875–3878. doi: 10.1021/nl071943y. [DOI] [PubMed] [Google Scholar]
- 22.Baldwin J.E., Warner P.J., Mackay C.D. The point spread function in Lucky Imaging and variations in seeing on short timescales. Astron. Astrophys. 2008;480:589–597. [Google Scholar]
- 23.Coffey V.C. Astronomical imaging - ‘Lucky’ camera creates ground-based images better than Hubble's. Laser Focus World. 2007;43:42. [Google Scholar]
- 24.Seminario M.C., Bunnell S.C. Signal initiation in T-cell receptor microclusters. Immunol. Rev. 2008;221:90–106. doi: 10.1111/j.1600-065X.2008.00593.x. [DOI] [PubMed] [Google Scholar]
- 25.Zhang W.G., Samelson L.E. The role of membrane-associated adaptors in T cell receptor signalling. Semin. Immunol. 2000;12:35–41. doi: 10.1006/smim.2000.0205. [DOI] [PubMed] [Google Scholar]
- 26.Hartgroves L.C., Lin J., Langen H., Zech T., Weiss A. Synergistic assembly of linker for activation of T cells signaling protein complexes in T cell plasma membrane domains. J. Biol. Chem. 2003;278:20389–20394. doi: 10.1074/jbc.M301212200. [DOI] [PubMed] [Google Scholar]
- 27.Douglass A.D., Vale R.D. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell. 2005;121:937–950. doi: 10.1016/j.cell.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bunnell S.C., Singer A.L., Hong D.I., Jacque B.H., Jordan M.S. Persistence of cooperatively stabilized signaling clusters drives T-cell activation. Mol. Cell. Biol. 2006;26:7155–7166. doi: 10.1128/MCB.00507-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das S.K., Darshi M., Cheley S., Wallace M.I., Bayley H. Membrane protein stoichiometry determined from the step-wise photobleaching of dye-labelled subunits. ChemBioChem. 2007;8:994–999. doi: 10.1002/cbic.200600474. [DOI] [PubMed] [Google Scholar]
- 30.Dahan M., Levi S., Luccardini C., Rostaing P., Riveau B. Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science. 2003;302:442–445. doi: 10.1126/science.1088525. [DOI] [PubMed] [Google Scholar]
- 31.Merrifield C.J. Seeing is believing: imaging actin dynamics at single sites of endocytosis. Trends Cell Biol. 2004;14:352–358. doi: 10.1016/j.tcb.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Seisenberger G., Ried M.U., Endress T., Buning H., Hallek M. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science. 2001;294:1929–1932. doi: 10.1126/science.1064103. [DOI] [PubMed] [Google Scholar]