

Abstract
The bacteriorhodopsin transport cycle includes protonation of the retinal Schiff base by Asp96 (M→N reaction) and reprotonation of Asp96 from the cytoplasmic surface (N→N′ reaction). We measured distance changes between pairs of spin-labeled structural elements of interest, and in general observed larger overall structural changes in the N state compared with the N′ state. The distance between the C-D loop and E-F interhelical loops in A103R1/M163R1 increased ∼6 Å in the N state and ∼3 Å in the N′ state. The opposite trend of distance changes in V101R1/A168R1 and L100R1/T170R1 supports counterclockwise rotation of helix F in the N but not the N′ state. Small distance increases were observed in S169R1/S226R1, but little change was seen in G106R1/G155R1. Taking earlier published EPR data into account, we suggest that structural changes of the E-F loop occur first, and then helices F and G begin to move together in the late M state. These motions then reach their maximum amplitude in the N state, evidently to facilitate the release of a proton from Asp96 and the formation of a proton-conduction pathway from Asp96 to the Schiff base. The structural changes reverse their directions and decay in the N′ state.
Abbreviations used: BR, nonilluminated state of bacteriorhodopsin; EPR, electron paramagnetic resonance; DTNB, 5, 5′-dithiobis-(2-nitrobenzoic acid)
Introduction
Bacteriorhodopsin is a light-driven proton pump in the purple membrane of Halobacterium salinarum (1). Photoisomerization of the chromophore, an all-trans retinal bound to Lys216 through the Schiff base, to 13-cis, 15-anti initiates a series of conformational changes in the protein before the initial state recovers (2). During this “photocycle”, a proton is transferred from the cytoplasmic side to the extracellular side of the cell membrane. The reaction cycle can be kinetically described as BR→K→L→M1→M2→M2′→N→N′→O→BR (3,4). In the first half of the photocycle, the retinal Schiff base loses its proton to Asp85, causing the release of a proton to the extracellular surface. In the second half, the proton affinity of Asp96 decreases from pKa >11 in the BR state to ∼7.5 (5) and the Schiff base regains a proton from Asp96. The aspartate anion produced then recruits a proton from the cytoplasmic surface. The intermediates N and N′ are related to the latter two steps of proton translocation: N contains unprotonated Asp96 and N′ contains a protonated one, but the Schiff base and Asp85 are protonated and the retinal is 13-cis, 15-anti in both (6–8).
The cytoplasmic proton channel is lined mostly with hydrophobic residues, and no hydrogen-bonded network is present. It is thus unlike the extracellular channel, where numerous water molecules and charged side chains form a complete network to facilitate proton translocation (9). Because there is no high-resolution crystallographic structure for N, it is not clear how the hydrogen-bonded chain of water molecules detected in N′ (10) between Asp96 and the Schiff base assemble in the previous M2→N reaction to form the pathway for transfer of the proton through the ∼10 Å distance between proton donor and acceptor. The crystal structures of the M (11) and N′ (10) states, before and after N, showed either disorder at the ends of helices F and G, or no significant large-scale structural changes, respectively. Since it is well known that large-scale structural changes can be hindered by the three-dimensional (3D) crystal lattice, these results do not rule out any of the large conformational changes in the M and N states that have been suggested to drive the entry of water into the cytoplasmic region. On the other hand, although the crystal lattice may prevent large conformational changes, evidently it does not hinder the entry of a sufficient number of water molecules (i.e., four) into the cytoplasmic channel to span the distance between the proton donor and acceptor.
Low-resolution structural studies under more physiological conditions than in 3D crystals described significant structural changes around helices F, G, and B that suggested an ∼2 Å outward tilt of helix F and an inward movement of helix G (12–15). Spin-labeling spectroscopy revealed not only extensive changes at the cytoplasmic surface consistent with the tilt (16–18), but also a counterclockwise rotation of helix F (19). However, there are discrepancies in the assignment of these changes to specific intermediates (see Discussion). In an attempt to understand the nature and extent of these movements, we investigated local structure in the N and N′ states by means of the site-directed spin-labeling method.
The relative motion of a particular structural element can be monitored by the distance change between probes at the location of interest and a suitable reference point. Since spin-spin dipolar interaction leads to spectral broadening depending on the interspin distance, the distance between two structural elements, and its changes, can be assessed from the spin-spin interaction of pairs of probes (20,21). Therefore, our strategy was to covalently link spin labels to engineered cysteine residues on different structural elements, produce photostationary states of the V49A mutant that contain the N or N′ state, and analyze the distance changes relative to the BR state.
Materials and Methods
Mutagenesis, expression, and spin labeling
The site-directed spin-labeling method requires substitution of a nitroxide side chain for the native residue at selected sites. This was accomplished by cysteine-substitution mutagenesis of otherwise cysteine-free bacteriorhodopsin, followed by chemical modification of the unique sulfhydryl group with a nitroxide reagent. The site-specific mutants were prepared according to a method developed by M. K. Krebs (22), who generously provided the shuttle vector PBA2 and the host cell MPK409. To aid specific accumulation of the N or N′ intermediate during illumination, all cysteine mutants contained the additional V49A mutation (23). Mutagenesis, expression, and purification of bacteriorhodopsin were performed as previously described (24). All cysteine-containing bacteriorhodopsin samples were kept at −80°C to prevent oxidation of the SH groups.
Spin labeling was performed as previously described (24,25). After the reducing reagent was removed by five centrifugations and washing in 100 mM NaCl, 10 mM phosphate buffer (pH 7.0), the purple membranes were suspended in the same buffer to 1–2 mg/mL concentration, and the spin-label reagent (1-oxyl-2,2,5,5,-tetramethyl pyrroline-3-methyl) methanethiosulfonate (R1), in dimethylsulfoxide, was added immediately. The molar ratio of the reagent to the protein was ∼10:1. In spin dilution experiments, to obtain the EPR spectrum for the state without spin-spin interaction, samples were labeled with a mixture of R1 and its diamagnetic equivalent (R1′, 1-acetyl-2,2,5,5,-tetramethyl-pyrroline-3-methyl) methanethiosulfonate, R1:R1′ = 1:3 molar ratio). The reaction was carried out for 12 h at room temperature. Excess R1 or R1′ reagent was removed by five centrifugations at 4°C. For the purpose of accumulating the N or N′ states by illumination, purple membranes, concentrated to ∼0.1–0.15 mM, were suspended in either solution A (10 mM Tris, 100 mM NaCl, 15% glycerol (w/w), pH 9) or solution B (10 mM phosphate Na, 100 mM NaCl, 15% glycerol (w/w), pH 6).
Characterization of spin-labeled mutants
UV-Vis absorption spectra were recorded on a Shimadzu UV 1601 (Columbia, MD) spectrophotometer. The occupancies of the N or N′ states were determined on an optical spectroscopic multichannel analyzer. The spin-labeled purple membrane (∼5 μL) was loaded into an EPR capillary (1 mm inner diameter) and fixed in an in-house-made holder to which a red laser was connected. To produce a photostationary state, light-adapted purple membranes were illuminated at 4°C by continuous 635 nm light (0.1 mW/mm2), which minimized excitation of the N or N′ intermediate. The occupancies of the intermediates were estimated by fitting the experimental difference spectra by a sum of different proportions of standard difference spectra: N − BR, M − BR, and L − BR (26).
EPR spectroscopy
Illuminated purple membranes in an EPR capillary were prepared as described above and flash-frozen while illuminated in liquid nitrogen. The capillary was transferred to a 120 K cryostat fixed inside the cavity of a Miniscope MS 200 spectrometer (Magnettech, Berlin, Germany). The low temperature (120 ± 1K) was maintained with a temperature controller (model HO2, Magnettech). Continuous-wave EPR spectra were collected with an X microwave band. The basic parameters were as follows: scan width = 200 G, microwave power = 1 mW, and modulation amplitude = 2 G. No sign of power saturation or distortion in the EPR lineshape was found.
After the EPR spectrum of the illuminated sample was collected, the capillary was warmed to 293 K and equilibrated for 15 min to fully reconvert the sample to the nonilluminated state that contains no photocycle intermediates. The capillary was again flash-frozen by a strong flow of cold nitrogen gas and kept at 120 K to collect the nonilluminated spectrum. Collection of data from the same capillary, without any geometry change inside the cavity, minimized errors of baseline fluctuation, peak drift, and intensity variation, which may induce artifacts with lineshape changes between nonilluminated and illuminated states.
The Fourier deconvolution method assumes that the dipolar interaction EPR spectrum containing a spin pair can be represented as the convolution of the noninteracting EPR spectrum with a broadening Pake function (27). By simulation of this deconvoluted Pake pattern, reliable distance distributions from 8 to 25 Å were generated (28). The reliability of extracting the distance distribution for the N or N′ state from illuminated samples was limited mostly by the intrinsic uncertainty (1–2 Å) (27) of the Fourier deconvolution method, the amplitudes of the differences between illuminated and nonilluminated spectra, and the occupancies of the N or N′ state in the illuminated samples. The deconvolution methods produced usable distance changes for A103R1/M163R1, but not in the other four mutants. However, since spin-spin dipolar interaction leads to spectral broadening to a degree dependent on the interspin distance, the distance changes and their directions between the BR and N or N′ states could be qualitatively assessed from the shape of the difference EPR spectra of illuminated minus nonilluminated samples. Since bacteriorhodopsin forms trimers in the purple membrane, the spin-spin interaction spectra may contain both intra- and intermolecular distance information. However, the spin dilution results (see Fig. 3) and structural analysis of the trimers (not shown) both suggest that intermolecular spin-spin interactions contribute virtually none of the EPR signals.
Figure 3.
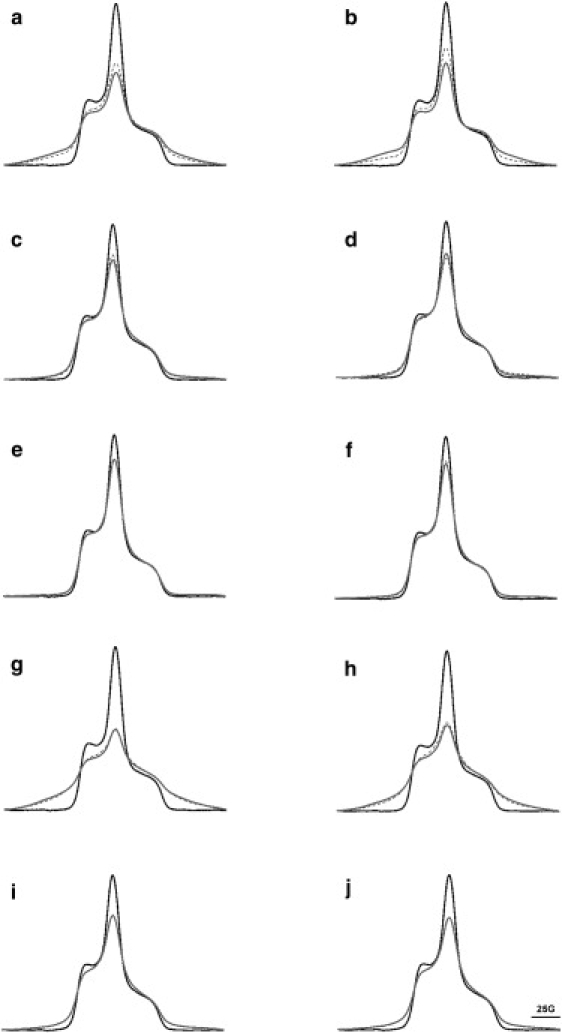
EPR spectra without and during illumination, at pH 6 and 9, and 120 K. Panels a and b are A103R1/M163R1 at pH 6 and pH 9; c and d are V101R1/A168R1 at pH 6 and pH 9; e and f are L100R1/T170R1 at pH 6 and pH 9; g and h are S169R1/S226R1 at pH 6 and pH 9; and i and j are G106R1/G155R1 at pH 6 and pH 9, respectively. Gray, solid lines are the observed spin-interacting spectra of nonilluminated, doubly labeled mutants. Black, dashed lines are the observed spin-interacting spectra of illuminated samples. Black, solid lines are the expected noninteracting spectra, measured with the spin dilution method. For each mutant, the spectra are superimposed and normalized to the same spin number.
Results
Functional characterization of spin-labeled mutants
We used five double cysteine mutants and attached spin labels at the cytoplasmic surface where they are located to monitor distance changes between structural elements of interest, as shown in Fig. 1. Ala103 and Met163 are on the C-D and E-F loops, respectively. Leu100 and Val101 are both at the end of helix C, but with different side-chain orientations, pointing toward helices G and E, respectively. Ala168, Ser169, and Thr170 are all at the end of helix F, but oriented toward helix E, the lipid phase, and helix G, respectively. Gly106, Gly155, and Ser226 are located on the end of helices D, E, and G, respectively, and face outward from the helical bundle. Thus, the spin-labeled double cysteine mutant A103R1/M163R1 was intended to measure distance changes between loops C-D and E-F, and both V101R1/A168R1 and L100R1/T170R1 were for changes in the distance between helices C and F but with orientations at either side of the helices. S169R1/S226R1 and G106R1/G155R1 were intended to determine distance changes between helices F and G, and helices D and E, respectively.
Figure 1.
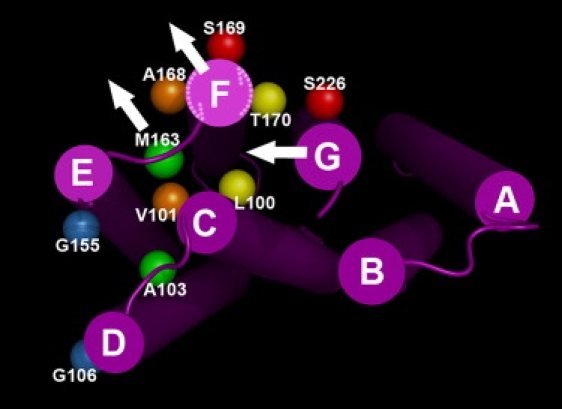
Bacteriorhodopsin model (PDB code 1C3W) viewed from the cytoplasmic side. Side chains replaced by cysteine and modified by spin labels are shown as balls at the coordinates of the respective CB atoms. Pairs of balls in the same color show double cysteine mutants to which two spin labels are attached. The three white arrows represent the possible motional reorientation of helices F and G, and the E-F loop (at Met163). The two dashed-line arrows show the possible counterclockwise rotation of helix F.
Because both M and N intermediates of wild-type BR decay in a few milliseconds, the N state is poorly accumulated under illumination and contaminated by significant amounts of the M intermediate. The mutant V49A has a structure similar to that of wild-type bacteriorhodopsin (10) but a ∼10-fold slower N decay (23), and it accumulates ∼6-fold less M (29) than the wild-type. Therefore, all cysteine mutants described above were constructed with the V49A mutation as background, with the expectation that N would be accumulated with little interference from M. Since the pKa of Asp96 is lowered in the photocycle to ∼7.5, as in the wild-type (5), the N state accumulated at pH 6 will contain protonated Asp96 (i.e., the N′ intermediate), whereas the N state accumulated at pH 9 will contain deprotonated Asp96 (i.e., the N intermediate). This is supported by Fourier transform infrared spectra of the V49A photocycle at low and high pH (23), and was the rationale used in a previous crystallographic study of N′ (10). Fig. 2 shows an example of the photostationary state of the V49A/S169R1/S226R1 mutant at pH 6. Continuous 635 nm light caused the accumulation of a slightly blue-shifted photoproduct with decreased absorption (Fig. 2 a), as expected for the N and N′ states, which do not differ significantly in their absorption spectra. In Fig. 2 b, the unique depletion shape of the difference spectrum, which contains no positive peaks at 410 nm, 640 nm, or 500 nm, rules out significant contributions from the other possible photoproducts (M, O, or L) (26). We tested the absorption spectra and estimated the occupancies of the N and N′ states in all mutants used.
Figure 2.
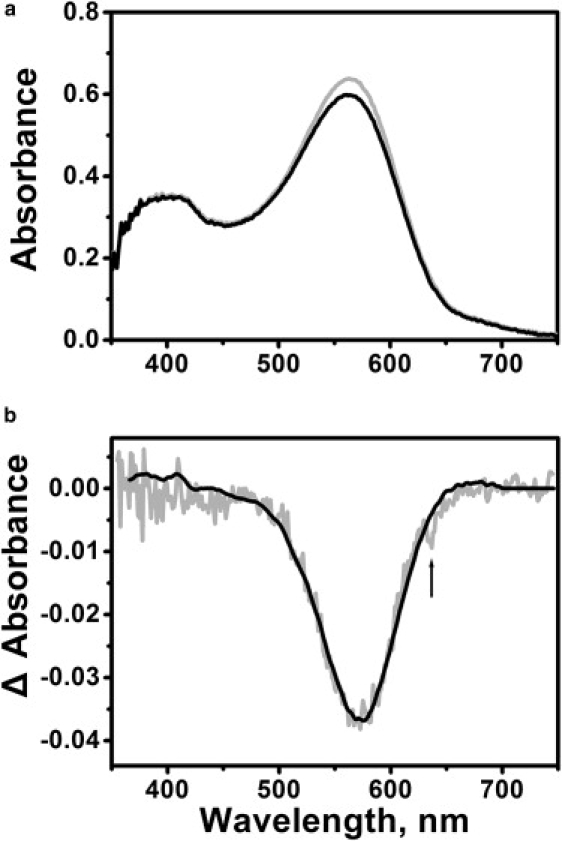
(a) Absorption spectra of V49A/S169R1/S226R1 without (gray) and during (black) illumination in pH 6.0 buffer (10 mM Pi/Na, 100 mM NaCl, 15% (w/w) glycerol). The decreased absorption in the photostationary state indicates accumulation of the N state. (b) The characteristic N − BR spectrum (gray) obtained by subtracting black spectra from gray spectra in panel a. This difference spectrum was fitted with a standard N − BR spectrum (black) (26) to calculate occupancy of the N intermediate. The black arrow shows 635 nm light from the laser scattered by the membranes.
The results in Table 1 show that the spectra and photocycle of V49A are nearly unperturbed by cysteine mutagenesis and spin labeling. In all mutants, the absorption maxima were blue-shifted, but by only 1–3 nm, and labeling caused only small changes. This suggests that the overall structures of these spin-labeled mutants are similar to that of V49A. The occupancies of N and N′ accumulated in L100R1/T170R1 and S169R1/S226R1 were almost the same as with the V49A background mutation. Only a slight amount of M (2%, or less than 1/10 of N or N′) was accumulated in A103R1/M163R1 and V101R1/A168R1 at pH 9. This amount of M may be neglected when compared with the wild-type system, in which M occupancies of spin-labeled mutants (e.g., V167R1/I222R1 and V173R1/I222R1) can reach as high as 24% (19). The mutant G106R1/G155R1 is an exception, with 6% L and 9% M accumulated additionally at pH 6 and pH 9, respectively. The EPR data for this location may contain structural information partly for L or M as well.
Table 1.
Characteristic absorption properties of bacteriorhodopsin mutants before and after spin labeling
| Abs. max. (nm) before spin labeling | Abs. max. (nm) after spin labeling | Intermediates and occupancies, pH 6 | Intermediates and occupancies, pH 9 | |
|---|---|---|---|---|
| V49A | 565 nm | N/A | N′ (22%) | N (21%) |
| A103R1/M163R1 | 565 nm | 564 nm | N′ (28%) | N (26%);M (2%) |
| V101R1/A168R1 | 565 nm | 563 nm | N′ (20%) | N (25%);M (2%) |
| L100R1/T170R1 | 564 nm | 563 nm | N′ (19%) | N (22%) |
| S169R1/S226R1 | 565 nm | 564 nm | N′ (22%) | N (20%) |
| G106R1/G155R1 | 564 nm | 562 nm | N′ (23%);L (6%) | N (15%);M (9%) |
All mutants contained the V49A residue change as background. The occupancies of N, N′, L, or M intermediates are shown for the spin-labeled mutants under continuous illumination with 635 nm light.
Distance change between the C-D and the E-F loops (A103R1/M163R1)
Previous studies of A103R1/M163R1 suggested a 6.5 Å distance increase between the C-D and E-F loops in the N state (18). At the pH used, this referred to a mixture of N and N′. By modulating the protonated state of Asp96 so as to accumulate the N or N′ state, we can calculate the distance changes between these two substates. In Fig. 3, a and b, the EPR spectra at pH 6 and pH 9 of nonilluminated and illuminated samples show significant lineshape broadening and a large decrease of the central peak amplitude when compared with the spectrum expected for two noninteracting labels. We conclude that there is strong dipolar interaction, but less in the illuminated samples. In Fig. 3 a, where the N′ state was accumulated in the sample illuminated at pH 6, the decrease of spin-spin interaction (indicated by the amplitude increase at the central peak as well as the amplitude decrease at the side region of the spectrum) clearly shows that the distance between the C-D and E-F loops increases in the N′ state relative to the nonilluminated BR state. In Fig. 3 b, where the N state was accumulated in the illuminated sample (at pH 9), the similar spectral features show that the distance increases in the N state also, but with an apparently larger change of amplitude than in the N′ state.
Fourier deconvolution revealed the magnitude of distance changes between the BR state and N or N′ states. In the nonilluminated state at pH 6 (Fig. 4 a), the distance distribution contains a dominant population at ∼7 Å and a broad tail that extends from ∼11 to 17 Å. The mean distance of ∼10 Å is consistent with the distance of 10.4 Å between the CB atoms of A103C and M163C in the high-resolution crystal structure. In the N′ state (Fig. 4 b), the distance distribution contains two small populations at ∼7 Å and 9 Å, with a tail of increased amplitude between 12 and 19 Å. The mean distance is ∼13 Å. The 3 Å distance increase suggests that the E-F loop moves away from the C-D loop in the N′ state. The distance distribution in the BR state at pH 9 (Fig. 4 c) contains a dominant population at ∼7 Å and a tail between 13 and 19 Å, similar to the distribution at pH 6. The mean distance is ∼10 Å, as at lower pH. In the N state (Fig. 4 d), the distance distribution contains only one wide population, between 12 and 19 Å. The mean distance is increased to ∼16 Å. This 6 Å distance increase in the N state, consistent with the earlier report of 6.5 Å, suggests that the E-F loop moves away from the C-D loop. The analysis of the BR, N′, and N states thus indicates that the amplitude of conformational changes is larger in the N state than in the N′ state. However, it should be noted that changes in the distance distributions may be induced by the shift of an equilibrium between several possible conformational states of either bacteriorhodopsin or the spin label.
Figure 4.
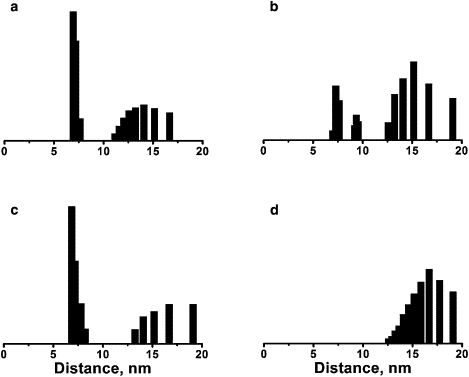
Distance distribution of V49A/A103R1/M163R1 calculated from the EPR spectra in Fig. 3, a and b. Panels a and b are distributions in the BR and N′ states, respectively, at pH 6; c and d are distributions in the BR and N states, respectively, at pH 9. The distance distribution was obtained by deconvoluting the spectra attributed to N or N′ after subtracting a scaled amount of the nonilluminated spectrum from the illuminated spectrum, according to the occupancy of N or N′ in Table 1. The vertical (normalized population) axes of the distributions are arbitrary and selected for convenience of display.
Distance change between helices C and F (V101R1/A168R1 and L100R1/T170R1)
It was previously reported that in V101R1/A168R1 a distance increase of ∼1 Å occurs between helices C and F in the M state, suggesting an outward tilt of helix F (25). A further counterclockwise rotation was suggested to take place in the succeeding intermediates (19). We used the same locations to determine whether the changes in the N and N′ states are different. In Fig. 3, c and d, the spin-interacting spectra of nonilluminated and illuminated samples show a medium extent of lineshape broadening and decrease of central peak amplitude when compared with the spectra expected for noninteracting spins. The lineshapes of illuminated and nonilluminated V101R1/A168R1 samples at pH 6 (Fig. 3 c) and pH 9 (Fig. 3 d) are so similar that the distance distribution analysis does not yield obviously changed populations or even significantly changed averaged distances. As shown in Table 2, the nonilluminated state at pH 6 contains a dominant population at ∼9 Å and a broad tail with a mean of ∼18 Å, and there is no detectable change in the illuminated state. At pH 9, the distance distribution is slightly narrower, but if there is a change between the illuminated and nonilluminated states, it is not statistically significant.
Table 2.
Distances between spin labels labeled at cysteine residues, with the V49A mutation as background
| Doubly labeledmutants | Distances at nonilluminated state (Å) | Distances at illuminated state (Å) | |||
|---|---|---|---|---|---|
| V101R1/A168R1 | pH 6 | 8.6 ± 1.9 | 17.8 ± 1.2 | 8.6 ± 1.9 | 17.8 ± 1.2 |
| pH 9 | 8.4 ± 1.5 | 17.3 ± 1.5 | 8.0 ± 1.0 | 17.1 ± 1.8 | |
| L100R1/T170R1 | pH 6 | 8.2 ± 0.9 | 15.8 ± 2.6 | 8.0 ± 0.8 | 15.6 ± 2.9 |
| pH 9 | 8.1 ± 0.8 | 15.7 ± 2.5 | 8.5 ± 0.9 | 15.8 ± 2.6 | |
| S169R1/S226R1 | pH 6 | 9.0 ± 3.1 | — | 9.2 ± 3.0 | — |
| pH 9 | 9.5 ± 3.5 | — | 9.8 ± 3.1 | — | |
| G106R1/G155R1 | pH 6 | 8.5 ± 1.0 | 14.7 ± 1.9 | 8.5 ± 1.1 | 14.7 ± 1.9 |
| pH 9 | 8.1 ± 1.0 | 15.7 ± 2.2 | 8.2 ± 1.9 | 15.7 ± 2.1 | |
The distances of nonilluminated and illuminated states under pH 6 and pH 9 were calculated from data in Fig. 3 by Fourier deconvolution (see Materials and Methods).
Although deconvolution does not reveal any distance changes between the BR, N, and N′ states, they are clearly seen in difference EPR spectra between illuminated and nonilluminated samples (Fig. 5), which reveal the existence and direction, if not the exact amplitude, of the changes. For V101R1/A168R1, illumination causes an increase in the interspin distance at pH 6 (Fig. 5 c), but a decrease at pH 9 (Fig. 5 d). Because of the lesser spin interaction, the positive peak at the center and the two minima at the edges of the magnetic field sweep suggest that the distance increases in the N′ state, at pH 6. This characteristic pattern of distance increase is present also in A103R1/M163R1 (Fig. 5, a and b) as a reference with an authenticated and greater change. On the other hand, stronger spin interaction, indicated by a reversed pattern at pH 9, suggests that the distance decreases in the N state (Fig. 5 d).
Figure 5.
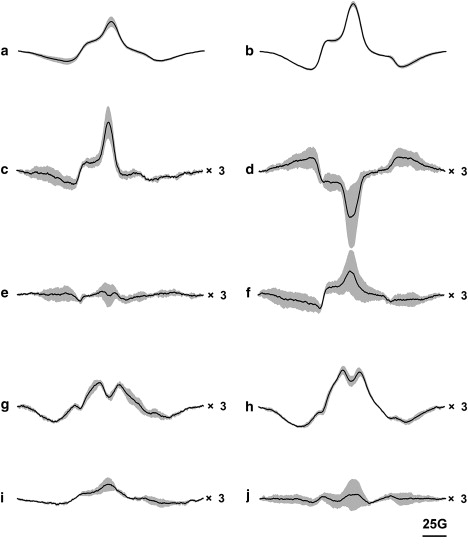
EPR difference spectra at 120 K, calculated by subtracting the nonilluminated EPR spectra from the illuminated spectra in Fig. 3. Panels a and b are A103R1/M163R1 at pH 6 and pH 9; c and d are V101R1/A168R1 at pH 6 and pH 9; e and f are L100R1/T170R1 at pH 6 and pH 9; g and h are S169R1/S226R1 at pH 6 and pH 9; and i and j are G106R1/G155R1 at pH 6 and pH 9, respectively. Black lines are the difference spectra, and gray shadows are the standard errors. Statistics were calculated from three sets of individual experiments for each nonilluminated and illuminated state. Spectra c–j were scaled up to a and b by multiplying by 3. The vertical axes are arbitrary.
The mutant L100R1/T170R1 was designed to investigate the possible rotation of helix F because this pair of labels is on the other side of the helix and rotation would produce a distance change opposite to what is observed with V101R1/A168R1. Similar to findings in V101R1/A168R1, the observed difference between nonilluminated and illuminated spectra of L100R1/T170R1 was small (Fig. 3, e and f). Deconvolution did not reveal distance changes between the BR, N, and N′ states (Table 2). Difference spectra (Fig. 5 f) revealed a pattern of small distance increases in the illuminated state at pH 9, and even smaller distance changes in the illuminated state at pH 6 (Fig. 5 e). Therefore, the distance should increase between L100R1 and T170R1 in the N state, whereas this change would be virtually completely reversed in the succeeding N′ state. The different behaviors of the V101R1/A168R1 and L100R1/T170R1 pairs suggest that in addition to an outward tilt, there may be a rotation of helix F.
Distance change between helices F and G (S169R1/S226R1)
If helix G is considered as another reference point, a more comprehensive understanding of the motion of helix F can be obtained by measuring the distance change between S169R1 and S226R1. In Fig. 3, g and h, the largest lineshape broadening and decrease of central peak amplitude reveals the strongest spin interaction in all five spin pairs examined, in both nonilluminated and illuminated samples. Deconvolution reveals one major population at ∼9 Å (Table 2) and little distance change in the illuminated state at pH 6. At pH 9 as well, little change is present between the illuminated and nonilluminated states. The difference spectra in Fig. 5, g and h, show an overall pattern of distance increases. The amplitudes are very small—about threefold less than A103R1/M163R1 (Fig. 5 a) at pH 6 and fourfold less than A103R1/M163R1 (Fig. 5 b) at pH 9. We note that both EPR difference spectra contain a small population of distance decreases, indicated by a negative peak at the center. Apparently, this suggests that a second conformer, which can arise whenever a spin label is attached to the protein, may undergo opposite movement. Nevertheless, Fig. 5, g and h, indicate an overall distance increase between S169R1 and S226R1 in the N state, and somewhat less in the N′ state.
Distance change between helices D and E (G106R1/G155R1)
The G106R1/G155R1 pair was chosen because little conformational change near the helices D and E region was reported in the M state. If the distance between these locations does not change, the largest conformational changes between the C-D and E-F loops, uncovered by A103R1/M163R1, should reflect the movement of helix F alone.
Fig. 3, i and j, show a medium extent of spin-spin interaction in both nonilluminated and illuminated samples. Deconvolution did not reveal any distance changes between the illuminated and nonilluminated states at pH 6 or pH 9 (Table 2). The more sensitive difference spectra also show hardly any changes. The amplitude is so small (∼10-fold less than A103R1/M163R1) that we believe no distance changes occur between G106R1 and G155R1, and further suggest that no conformational change takes place in nearby regions either.
Discussion
Structural changes in the M and N states
The structural changes of bacteriorhodopsin in the M and N states have been extensively investigated. Initial low-resolution diffraction maps (12,14,15,30) indicated that large conformational changes take place in helices F, G, and B in the cytoplasmic region. When investigators subsequently sought to assign these changes to specific intermediates, discrepancies arose among different groups. Kamikubo et al. (13,31) reported that in the N or late M (MN) state the most prominent structural changes occurred near helices F and G, but in the M state smaller changes occurred (at helices G and B, and much less so at helix F). The idea that different molecular events take place in the M and N states was further supported in subsequent reports (32–35). However, in studies using time-resolved electron diffraction, Subramaniam et al. (36,37) did not observe any significant difference changes between M and N, and concluded that these states are variations of one fundamental kind of conformational change. It should be noted that the interpretations of time-resolved and trapped-intermediates data rely on the accuracy of the photocycle kinetics and the yield and species of the intermediates trapped, respectively. The discrepancies may be explained by the fact that in some reports the kinetics were measured in purple membrane suspensions rather than in the crystals used for diffraction, and in other studies the identity and occupancy of the trapped intermediate were not measured.
On the other hand, although high-resolution diffraction data from 3D crystals clearly showed local structural changes in residues, and retinal and water molecules in the M1, M2, and N′ states, large movements of helices G, F, and B were absent (10,38,39). Of interest, the nonilluminated F219L mutant showed significant structural differences from wild-type bacteriorhodopsin in a low-resolution projection map (36), but not in a high-resolution x-ray diffraction map (10). Does this suggest that the crystal contacts in 3D crystals enforce the same internal structure as in the wild-type, despite the differences that exist in 2D crystals, or that small, nearly undetectable (but real) local structural differences result in the large density features observed in low-resolution maps? It is important to note that all arguments as to where large-scale conformational changes occur in the projection maps include such density features. Therefore, it is still not clear whether all of the large helical movements in the low-resolution data are real but inhibited by lattice forces in the 3D crystals, or some of the movements are smaller than assumed.
In time-resolved mobility measurements, the spin probes at some sites show mobility changes that begin before the decay of M, and vary little during the lifetime of M and N (16), whereas at other sites the changes arise and decay together with N (17,40). In previous studies it was concluded that structural changes had a larger amplitude in the N state than in the M state, as evidenced by distance changes during the lifetime of N or late M (18,19). If we assume that the mobility change reflects the structural change at the site where the spin label is located, and that information about mobility changes can be combined with conclusions drawn from distance changes, it appears that in the M state, structural changes are clustered at the E-F interhelical loop while in the N state, they spread over helices F, G, and B. Therefore, the movements of structural elements detected by EPR support the idea that different molecular events occur in the M and N states.
Structural changes in the N and N′ states detected by EPR
E-F loop
The average distance change between A103R1 and M163R1 increases ∼6 Å in the N state, and ∼3 Å in the N′ state (Fig. 4). Because both low-resolution diffraction and spin-labeling data suggest that there is only a minor movement of helices C and D, and the C-D loop, we assign this largest distance change reported so far to the conformational change of the E-F loop, where Met163 is located. Its side chain is downward in the BR state and points to the center of the putative proton channel (9). In agreement with this, its EPR spectrum contains a motionally restricted lineshape (18), and M163C is poorly accessible to the water-soluble reagent DTNB (41) even though the residue is on the otherwise mobile and exposed E-F loop. Time-resolved EPR previously revealed that its mobility increases during M formation, and this change decays with decay of the M and N states (16). Its access to DTNB increases in M (41). Therefore, it seems from these results that Met163 begins to leave the channel center in M and recovers as N decays via N′ (Fig. 1). On the same E-F loop, mobility changes with similar kinetics occur also at S158R1, A160R1, E161R1, and R164R1 (16). Taken together, these changes suggest that the extensive structural changes of the E-F loop begin with M and decay with N and then N′. Time-resolved EPR of A103R1/M163R1 further showed that the distance between helix C and the E-F loop increased during the lifetime of the N intermediate (18). We suggest that this distance begins to increase in late M, reaches a maximum of ∼6 Å in the N state, decreases to 3 Å in the state that follows (i.e., N′), and recovers during N′ decay to the O (24) and then BR states, and so do the amplitudes of other possible structural changes of the E-F loop.
Helix F
In principle, the observed movement of the E-F loop could be driven by motion of helix E, helix F, or both. The G106R1/G155R1 pair showed little distance changes in either N or N′, which may suggest that helices D and E do not move (Fig. 1). Because this mutant produces L or M in the photostationary state, together with N′ or N (Table 1), the possible distance changes in the N or N′ states may be canceled out by unknown opposite trends in M or L. However, the absence of detectable EPR signal changes at D104R1, Q105R1, G106R1, and T107R1 (16), which are located at the cytoplasmic end of helix D, exclude movements of helix D during the photocycle. The absence of large density changes around helices E and D (13,36) in diffraction maps suggests this as well. Hence, the driving force of the structural changes in the E-F loop can be assigned only to helix F.
Helix F has been suggested to undergo complex movements that include a tilt away from the center of the protein and counterclockwise rotation. The latter was deduced by the relative position change of helix F against either helix B or helix G as reference. Because both helices B and G can move during the photocycle, but helix C apparently does not move (13,36), helix C is a better point of reference. We chose the pairs L100R1/T170R1 and V101R1/A168R1 to follow the motions of helix F. The opposite trend of distance changes at these two mutants in the N state, i.e., an increase for L100R1/T170R1 and a decrease for V101R1/A168R1, provides additional supporting evidence for the rotation model (Fig. 1). However, both T170R1 and V101R1 exhibit mobility changes during the photocycle, and possible side-chain repacking of these spin probes may affect the measured distance changes. A rotation of helix F seems to be contradicted by the observed immobilization at T170R1 and E166R1 after photoexcitation (16). If counterclockwise rotation happens in N, a mobility increase should be observed at both sites (Fig. 1). Again, however, repacking of the residues could well obviate this argument, because access to DTNB by both E166C and T170C increases rather than decreases in the M state, a measurement that is not subject to artifact from the reorientation of the spin label (41).
The interspin distance in V101R1/A168R1 begins to increase before the rise of N, and decays after the N or N′ state (25). The mobility signals of V170R1 and F171R1 rise and decay during the lifetime of N or N′ (16). These results, together with the distance analysis of the L100R1/T170R1 and V101R1/A168R1 pairs, suggest that the cytoplasmic end of helix F undergoes complex structural changes—an outward tilt and/or a possible rotation—in the second half of the photocycle. It should be noted that the mobility changes of V170R1 and F171R1 at the end of helix F lag behind those of S158R1, A160R1, E161R1, and M163R1 at the E-F loop (16). This appears to contradict the idea that the outward tilt of helix F triggers the structural changes of the E-F loop. A possible explanation is that the mobility of spin labels on helices is less sensitive to structural change than that on interhelical loops, because the motion of spins is generally more restricted inside the membrane than at the solvent-exposed loops. Alternatively, the rotation of helix F may unwind the twisted E-F loop without the kind of outward tilt that results in a change in mobility. We suggest that significant motion of helix F begins later than the changes in the E-F loop that occur in the late M state, reaches its full extent in the N state, and finally recovers during the decay of N′.
Helix G
Of interest, the EPR spectrum of L100R1 shows a considerably motionally restricted lineshape, which suggests that its side chain is immobilized by the local environment (16). Structure analysis reveals that Leu100 is tightly surrounded by Val167, Thr170, and Leu174 on helix F, and Leu223, Ala228, and Ile229 on helix G (PDB code: 1C3W). Therefore, it is a sensitive site for detecting local structural changes in helices F and G (Fig. 1). However time-resolved EPR did not reveal any motional changes at this site during any part of the photocycle (16). This suggests two simple possibilities: 1), neither helix F nor helix G moves; or 2), helix F moves concurrently with helix G (Fig. 1). The latter might be understood if helix G moved ∼2 Å toward the center and helix F tilted outward. This would explain why the motions of L100R1, tightly restricted by the proximity of helix G, do not increase as helix F moves. In this case, if helix G moves inward concurrently with the outward tilt of helix F (Fig. 1), we will not see significant distance changes between helices G and F. Indeed, S169R1/S226R1 reveals only a small interspin distance increase in the N and N′ states. This is consistent with the observed small distance increase of the V173R1/I222R1 pair (19), which represents the same structural elements. The fact that there is any distance increase (Fig. 5, g and h) suggests that the motion of helix F is somewhat larger than that of helix G. The inward motion of helix G is supported by the distance decrease of the L100R1/S226R1 pair (25), as well as by mercury labeling (34). Therefore, we suggest that helix G begins to move toward the center concurrently with helix F, reaches its full extent in the N state, and recovers as the N′ (or O) state decays.
To summarize, we observed larger structural changes in the N state than in the N′ state. The E-F loop undergoes more structural changes, with greater distance changes, than any of the helices. In the N (but not N′) state, there may be a counterclockwise rotation of helix F in addition to its tilt. The motions of the structural elements in the photocycle can be described as follows: 1), In the M or late M (MN or M2) state, structural changes in the E-F loop occur first, and then helices F and G begin to move together. 2), In the N state, the inward movement of helix G reaches its full extent, and so does the tilt out of helix F with a possible counterclockwise rotation as it unwinds the turn in the E-F loop. The structural changes in the E-F loop also reach their maximum extent. 3), In the N′ state, all motions in the preceding N state have reversed their directions. The structural changes at the cytoplasmic surface should recover as N′ decays and new helical tilts appear in the extracellular region in the O state (24,42).
Implications for the mechanism of proton translocation
The nature and extent of structural changes that occur in different states of the BR photocycle have been of central interest during the past few decades because they are relevant to proton transport. Among the M- or N-like structures (PDB code 1C8S, 1FBK, 1CWQ, and 1IW9), only the D96G/F171C/F219L mutant exhibits an open conformation with significant movements of helices F and G, as revealed by EPR data. We consider the details of the crystal structures of the M state of E204Q (38), the D96G/F171C/F219L mutant (43), and the N′ state of V49A (10) in the order of M2→N→N′ in the photocycle to construct a model for the proton translocation mechanism.
Proton translocation in the cytoplasmic channel requires the release of a proton from Asp96 and a complete proton-conduction pathway that extends from Asp96 to the Schiff base. To facilitate the proton translocation, the energy stored in the photoisomerized retinal begins to spread to the protein in the M state as conformational changes. The 13-cis retinal has an upward-moving 13-methyl group that displaces Trp182, and there is also a significantly changed conformation at Lys216 that may induce displacements of nearby residues and movements of helices F and G (38). This results in an ∼1 Å separation of Thr46 in helix B from Asp96 on helix C, which will break its hydrogen bond to Asp96 and decrease the proton affinity of the carboxyl group. Two additional water molecules assemble along the putative proton pathway, which is not yet long enough to connect Asp96 to the Schiff base. Together, the results suggest that the structure begins to change in the M2 or late M state, and to prepare the release of the proton from Asp96 to the Schiff base.
In the D96G/F171C/F219L mutant, which assumes an M- or N-like state without illumination, the inward and outward movements of helices G and F are clearly revealed (43). Thr46 is separated from Asp96/Gly96 even further, consistent with deprotonation of Asp96 in the N state. The formation of this aspartate anion, which is buried inside the hydrophobic channel with no counterion, is energetically unfavored. As the hydration increases during the M to N transition, the open structure induced by movements of helices F, G, and B should accommodate sufficient water inside the channel to form a pathway connecting Asp96 and the Schiff base, and also to stabilize the aspartate anion. Unfortunately, because of the limitations of the 3.2 Å crystallographic resolution, we cannot see in this model where the water molecules are positioned, or how the 13-cis configuration of retinal affects Trp182.
In the N′ state that follows, the movements of helices F and G are as small as they were in M2, which suggests that the open structure partly closes as the N state decays to the N′ state (10). Two distinct structural features are observed at this stage: 1), recovery of the connection of Thr46 to Asp96; and 2), a complete proton pathway between Asp96 and the retinal Schiff base, formed by four water molecules. The latter is likely to be the pathway created in the preceding N state, whereas the former facilitates increase of the pKa of Asp96 and its reprotonation in the N→N′ reaction that ensures that once the Schiff base is reprotonated it will lose its connection to the bulk in the N′ state, making the proton transport across the cytoplasmic channel unidirectional (44).
Acknowledgments
We thank M. P. Krebs at the University of Florida for providing the expression system MPK409, and C. Altenbach at the University of California, Los Angeles, and R. Langen at the University of Southern California for the EPR software package and instructions. We are grateful for the critical reading of the manuscript by S. P. Balashov, and instructive discussions with J. M. Wang, E.S. Imasheva, A. K. Dioumaev, V. Krishnamani, and M. Goldfeld at the University of California, Irvine.
This work was supported by grants from the National Institutes of Health (GM29498 to J.K.L.) and the Department of Energy (DEFG03-86ER13525).
References
- 1.Lozier R.H., Bogomolni R.A., Stoeckenius W. Bacteriorhodopsin: a light-driven proton pump in Halobacterium halobium. Biophys. J. 1975;15:955–962. doi: 10.1016/S0006-3495(75)85875-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heyn M.P., Borucki B., Otto H. Chromophore reorientation during the photocycle of bacteriorhodopsin: experimental methods and functional significance. Biochim. Biophys. Acta. 2000;1460:60–74. doi: 10.1016/s0005-2728(00)00130-4. [DOI] [PubMed] [Google Scholar]
- 3.Lanyi J.K. Proton transfers in the bacteriorhodopsin photocycle. Biochim. Biophys. Acta. 2006;1757:1012–1018. doi: 10.1016/j.bbabio.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Varo G., Lanyi J.K. Thermodynamics and energy coupling in the bacteriorhodopsin photocycle. Biochemistry. 1991;30:5016–5022. doi: 10.1021/bi00234a025. [DOI] [PubMed] [Google Scholar]
- 5.Zscherp C., Schlesinger R., Tittor J., Oesterhelt D., Heberle J. In situ determination of transient pK(a) changes of internal amino acids of bacteriorhodopsin by using time-resolved attenuated total reflection Fourier-transform infrared spectroscopy. Proc. Natl. Acad. Sci. USA. 1999;96:5498–5503. doi: 10.1073/pnas.96.10.5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haupts U., Tittor J., Oesterhelt D. Closing in on bacteriorhodopsin: progress in understanding the molecule. Annu. Rev. Biophys. Biomol. Struct. 1999;28:367–399. doi: 10.1146/annurev.biophys.28.1.367. [DOI] [PubMed] [Google Scholar]
- 7.Kandori H. Hydration switch model for the proton transfer in the Schiff base region of bacteriorhodopsin. Biochim. Biophys. Acta. 2004;1658:72–79. doi: 10.1016/j.bbabio.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Lanyi J.K. Bacteriorhodopsin. Annu. Rev. Physiol. 2004;66:665–688. doi: 10.1146/annurev.physiol.66.032102.150049. [DOI] [PubMed] [Google Scholar]
- 9.Luecke H., Schobert B., Richter H.T., Cartailler J.P., Lanyi J.K. Structure of bacteriorhodopsin at 1.55 A resolution. J. Mol. Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 10.Schobert B., Brown L.S., Lanyi J.K. Crystallographic structures of the M and N intermediates of bacteriorhodopsin: assembly of a hydrogen-bonded chain of water molecules between Asp-96 and the retinal Schiff base. J. Mol. Biol. 2003;330:553–570. doi: 10.1016/s0022-2836(03)00576-x. [DOI] [PubMed] [Google Scholar]
- 11.Luecke H., Schobert B., Richter H.T., Cartailler J.P., Lanyi J.K. Structural changes in bacteriorhodopsin during ion transport at 2 angstrom resolution. Science. 1999;286:255–261. doi: 10.1126/science.286.5438.255. [DOI] [PubMed] [Google Scholar]
- 12.Dencher N.A., Dresselhaus D., Zaccai G., Buldt G. Structural changes in bacteriorhodopsin during proton translocation revealed by neutron diffraction. Proc. Natl. Acad. Sci. USA. 1989;86:7876–7879. doi: 10.1073/pnas.86.20.7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamikubo H., Kataoka M., Varo G., Oka T., Tokunaga F. Structure of the N intermediate of bacteriorhodopsin revealed by x-ray diffraction. Proc. Natl. Acad. Sci. USA. 1996;93:1386–1390. doi: 10.1073/pnas.93.4.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koch M.H., Dencher N.A., Oesterhelt D., Plohn H.J., Rapp G. Time-resolved X-ray diffraction study of structural changes associated with the photocycle of bacteriorhodopsin. EMBO J. 1991;10:521–526. doi: 10.1002/j.1460-2075.1991.tb07978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramaniam S., Gerstein M., Oesterhelt D., Henderson R. Electron diffraction analysis of structural changes in the photocycle of bacteriorhodopsin. EMBO J. 1993;12:1–8. doi: 10.1002/j.1460-2075.1993.tb05625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rink T., Pfeiffer M., Oesterhelt D., Gerwert K., Steinhoff H.J. Unraveling photoexcited conformational changes of bacteriorhodopsin by time resolved electron paramagnetic resonance spectroscopy. Biophys. J. 2000;78:1519–1530. doi: 10.1016/S0006-3495(00)76704-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinhoff H.J., Mollaaghababa R., Altenbach C., Hideg K., Krebs M. Time-resolved detection of structural changes during the photocycle of spin-labeled bacteriorhodopsin. Science. 1994;266:105–107. doi: 10.1126/science.7939627. [DOI] [PubMed] [Google Scholar]
- 18.Thorgeirsson T.E., Xiao W., Brown L.S., Needleman R., Lanyi J.K. Transient channel-opening in bacteriorhodopsin: an EPR study. J. Mol. Biol. 1997;273:951–957. doi: 10.1006/jmbi.1997.1362. [DOI] [PubMed] [Google Scholar]
- 19.Xiao W., Brown L.S., Needleman R., Lanyi J.K., Shin Y.K. Light-induced rotation of a transmembrane α-helix in bacteriorhodopsin. J. Mol. Biol. 2000;304:715–721. doi: 10.1006/jmbi.2000.4255. [DOI] [PubMed] [Google Scholar]
- 20.Eaton S.S., Eaton G.R. Distance measurements by CW and pulsed EPR. In: Berliner L.J., Eaton S.S., Eaton G.R., editors. Biological Magnetic Resonance: Distance Measurements in Biological Systems by EPR. Kluwer Academic/Plenum; New York: 2000. pp. 2–27. [Google Scholar]
- 21.Hustedt E.J., Beth A.H. Nitroxide spin-spin interactions: applications to protein structure and dynamics. Annu. Rev. Biophys. Biomol. Struct. 1999;28:129–153. doi: 10.1146/annurev.biophys.28.1.129. [DOI] [PubMed] [Google Scholar]
- 22.Krebs M.P., Mollaaghababa R., Khorana H.G. Gene replacement in Halobacterium halobium and expression of bacteriorhodopsin mutants. Proc. Natl. Acad. Sci. USA. 1993;90:1987–1991. doi: 10.1073/pnas.90.5.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dioumaev A.K., Brown L.S., Needleman R., Lanyi J.K. Coupling of the reisomerization of the retinal, proton uptake, and reprotonation of Asp-96 in the N photointermediate of bacteriorhodopsin. Biochemistry. 2001;40:11308–11317. doi: 10.1021/bi011027d. [DOI] [PubMed] [Google Scholar]
- 24.Chen D., Wang J.M., Lanyi J.K. Electron paramagnetic resonance study of structural changes in the O photointermediate of bacteriorhodopsin. J. Mol. Biol. 2007;366:790–805. doi: 10.1016/j.jmb.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Radzwill N., Gerwert K., Steinhoff H.J. Time-resolved detection of transient movement of helices F and G in doubly spin-labeled bacteriorhodopsin. Biophys. J. 2001;80:2856–2866. doi: 10.1016/S0006-3495(01)76252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gergely C., Zimanyi L., Varo G. Bacteriorhodopsin intermediate spectra determined over a wide pH range. J. Phys. Chem. B. 1997;101:9390–9395. [Google Scholar]
- 27.Rabenstein M.D., Shin Y.K. Determination of the distance between two spin labels attached to a macromolecule. Proc. Natl. Acad. Sci. USA. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altenbach C., Oh K.J., Trabanino R.J., Hideg K., Hubbell W.L. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- 29.Brown L.S., Gat Y., Sheves M., Yamazaki Y., Maeda A. The retinal Schiff base-counterion complex of bacteriorhodopsin: changed geometry during the photocycle is a cause of proton transfer to aspartate 85. Biochemistry. 1994;33:12001–12011. doi: 10.1021/bi00206a001. [DOI] [PubMed] [Google Scholar]
- 30.Nakasako M., Kataoka M., Amemiya Y., Tokunaga F. Crystallographic characterization by X-ray diffraction of the M-intermediate from the photo-cycle of bacteriorhodopsin at room temperature. FEBS Lett. 1991;292:73–75. doi: 10.1016/0014-5793(91)80837-s. [DOI] [PubMed] [Google Scholar]
- 31.Kamikubo H., Oka T., Imamoto Y., Tokunaga F., Lanyi J.K. The last phase of the reprotonation switch in bacteriorhodopsin: the transition between the M-type and the N-type protein conformation depends on hydration. Biochemistry. 1997;36:12282–12287. doi: 10.1021/bi9712302. [DOI] [PubMed] [Google Scholar]
- 32.Vonck J. A three-dimensional difference map of the N intermediate in the bacteriorhodopsin photocycle: part of the F helix tilts in the M to N transition. Biochemistry. 1996;35:5870–5878. doi: 10.1021/bi952663c. [DOI] [PubMed] [Google Scholar]
- 33.Vonck J. Structure of the bacteriorhodopsin mutant F219L N intermediate revealed by electron crystallography. EMBO J. 2000;19:2152–2160. doi: 10.1093/emboj/19.10.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oka T., Kamikubo H., Tokunaga F., Lanyi J.K., Needleman R. Conformational change of helix G in the bacteriorhodopsin photocycle: Investigation with heavy atom labeling and X-ray diffraction. Biophys. J. 1999;76:1018–1023. doi: 10.1016/S0006-3495(99)77266-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oka T., Yagi N., Fujisawa T., Kamikubo H., Tokunaga F. Time-resolved x-ray diffraction reveals multiple conformations in the M-N transition of the bacteriorhodopsin photocycle. Proc. Natl. Acad. Sci. USA. 2000;97:14278–14282. doi: 10.1073/pnas.260504897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramaniam S., Lindahl M., Bullough P., Faruqi A.R., Tittor J. Protein conformational changes in the bacteriorhodopsin photocycle. J. Mol. Biol. 1999;287:145–161. doi: 10.1006/jmbi.1999.2589. [DOI] [PubMed] [Google Scholar]
- 37.Subramaniam S., Henderson R. Crystallographic analysis of protein conformational changes in the bacteriorhodopsin photocycle. Biochim. Biophys. Acta. 2000;1460:157–165. doi: 10.1016/s0005-2728(00)00136-5. [DOI] [PubMed] [Google Scholar]
- 38.Luecke H., Schobert B., Cartailler J.P., Richter H.T., Rosengarth A. Coupling photoisomerization of retinal to directional transport in bacteriorhodopsin. J. Mol. Biol. 2000;300:1237–1255. doi: 10.1006/jmbi.2000.3884. [DOI] [PubMed] [Google Scholar]
- 39.Lanyi J., Schobert B. Crystallographic structure of the retinal and the protein after deprotonation of the Schiff base: the switch in the bacteriorhodopsin photocycle. J. Mol. Biol. 2002;321:727–737. doi: 10.1016/s0022-2836(02)00682-4. [DOI] [PubMed] [Google Scholar]
- 40.Rink T., Riesle J., Oesterhelt D., Gerwert K., Steinhoff H.J. Spin-labeling studies of the conformational changes in the vicinity of D36, D38, T46, and E161 of bacteriorhodopsin during the photocycle. Biophys. J. 1997;73:983–993. doi: 10.1016/S0006-3495(97)78131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown L.S., Needleman R., Lanyi J.K. Conformational change of the E-F interhelical loop in the M photointermediate of bacteriorhodopsin. J. Mol. Biol. 2002;317:471–478. doi: 10.1006/jmbi.2002.5428. [DOI] [PubMed] [Google Scholar]
- 42.Okumura H., Murakami M., Kouyama T. Crystal structures of acid blue and alkaline purple forms of bacteriorhodopsin. J. Mol. Biol. 2005;351:481–495. doi: 10.1016/j.jmb.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 43.Subramaniam S., Henderson R. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 2000;406:653–657. doi: 10.1038/35020614. [DOI] [PubMed] [Google Scholar]
- 44.Balashov S.P. Protonation reactions and their coupling in bacteriorhodopsin. Biochim. Biophys. Acta. 2000;1460:75–94. doi: 10.1016/s0005-2728(00)00131-6. [DOI] [PubMed] [Google Scholar]