

Abstract
The cd1 nitrite reductases, which catalyze the reduction of nitrite to nitric oxide, are homodimers of 60 kDa subunits, each containing one heme-c and one heme-d1. Heme-c is the electron entry site, whereas heme-d1 constitutes the catalytic center. The 3D structure of Pseudomonas aeruginosa nitrite reductase has been determined in both fully oxidized and reduced states. Intramolecular electron transfer (ET), between c and d1 hemes is an essential step in the catalytic cycle. In earlier studies of the Pseudomonas stutzeri enzyme, we observed that a marked negative cooperativity is controlling this internal ET step. In this study we have investigated the internal ET in the wild-type and His369Ala mutant of P. aeruginosa nitrite reductases and have observed similar cooperativity to that of the Pseudomonas stutzeri enzyme. Heme-c was initially reduced, in an essentially diffusion-controlled bimolecular process, followed by unimolecular electron equilibration between the c and d1 hemes (kET = 4.3 s−1 and K = 1.4 at 298K, pH 7.0). In the case of the mutant, the latter ET rate was faster by almost one order of magnitude. Moreover, the internal ET rate dropped (by ∼30-fold) as the level of reduction increased in both the WT and the His mutant. Equilibrium standard enthalpy and entropy changes and activation parameters of this ET process were determined. We concluded that negative cooperativity is a common feature among the cd1 nitrite reductases, and we discuss this control based on the available 3D structure of the wild-type and the H369A mutant, in the reduced and oxidized states.
Introduction
The cd1 nitrite reductases (NiRs) (EC 1.9.3.2) constitute a family of enzymes catalyzing the single-electron reduction of nitrite to nitric oxide in bacterial energy conversion denitrification processes. Three-dimensional structures have been determined so far for Paracoccus pantotrophus (1) and Pseudomonas aeruginosa (2) NiRs. Both are homodimers of ∼60 kDa subunits, each of which contains one covalently bound heme-c and one heme-d1, each residing in distinct domains of the monomer. Heme-c is the electron entry site, whereas heme-d1 constitutes the catalytic center where the single electron is transferred to the substrate, nitrite ion. Intramolecular electron transfer (ET), between c and d1 hemes is an essential step in the catalytic cycle and has been studied by several groups (3,4). Sequence comparison of cd1 NiRs shows a striking difference among the P. aeruginosa, P. pantotrophus, and Pseudomonas stutzeri (P. stutzeri) enzymes. We have previously investigated the internal ET in the P. stutzeri enzyme using the pulse radiolysis method and have measured the kinetics of ET equilibration between the c and d1 heme sites (5,6) as part of a more comprehensive study of intramolecular ET in proteins initiated by pulse radiolysis (7). Significantly, the internal heme-c to heme-d1 ET rate constant was found to decrease by close to three orders of magnitude upon increasing the extent of P. stutzeri NiR reduction from less than one electron-equivalent per mole enzyme to full reduction, resolving an allosteric control mechanism of this intramolecular process. The internal distribution of reduction equivalents between the heme sites was also shown to depend on the number of electrons taken up by the P. stutzeri enzyme. Static redox titrations of the cd1 NiR enzymes isolated from both P. aeruginosa and P. pantotrophus have also demonstrated substantial cooperativity between the hemes c and d1 (8,9). Reductive titrations of the P. aeruginosa enzyme suggested the presence of thermodynamic and probably electronic interactions between the hemes. The model used for fitting the data implied positive cooperativity between the two d1-type hemes and negative cooperativity of similar magnitude between the two c-type hemes (7). However, no knowledge of the 3D structure of the P. aeruginosa-NiR was available at that time. Hence, the proposed model required assumptions that now need reexamination. Similarly, the 3D structure of the P. stutzeri enzyme has still not been determined. Hence, structural implications of our studies of this enzyme are still pending. Therefore, a detailed kinetic study by pulse radiolysis of the wild-type (WT) P. aeruginosa-NiR and the single-site mutant H369A (in which one of the invariant histidines on the distal side of heme-d1 has been substituted by an alanine) has been carried out, and the results are interpreted using the available 3D structures.
Material and Methods
Mutagenesis and protein purification
The NiR was purified from anaerobically grown P. aeruginosa cells (10). Mutagenesis of His 369 to Ala, protein purification, and reconstitution with d1 heme has been described previously (11).
Kinetic measurements
Time-resolved measurements were performed on the pulse radiolysis system based on the Varian (Palo Alto, CA) V-7715 linear accelerator at the Hebrew University in Jerusalem, Israel. Five MeV accelerated electrons were employed using pulse lengths in the range 0.1–1.5 μs. To produce 1-methylnicotinamide radicals (1-MNA∗), argon-saturated solutions containing 5 mM 1-MNA∗ and 10 mM potassium phosphate at pH 5.5–7.5 were employed. For production of CO2− radicals, N2O-saturated solutions containing 100 mM sodium formate and 10 mM potassium phosphate at pH 5.5 or 7.5 were used. A 10.00 mm Spectrosil (Hellma, Müllheim, Germany) cuvette was employed, using either one or three light passes, which resulted in an overall optical path length of either 1 or 3 cm. A 150 W xenon lamp produced the analyzing light beam, together with a Bausch & Lomb (Rochester, NY) double grating monochromator. Appropriate optical filters with cutoff at 285 or 385 nm were used to reduce photochemical and light-scattering effects. For both WT and H369A P. aeruginosa-NiR the redox state of heme-c could be monitored at 551 nm, where heme-d1 has an isosbestic point. Reduction of heme-d1 was followed at 640 nm in the WT enzyme, whereas, in the H369A mutant, the examining wavelength was 460 nm, exploiting the different isosbestic points of heme-c here. The data acquisition system consisted of a Tektronix 390 A/D transient recorder (Sony-Tektronix, Tokyo, Japan) attached to a personal computer. The temperature of the reaction solutions in the cuvette (15 different temperatures in the range 2–42°C) was controlled by a thermostating system and continuously monitored by a thermistor attached to the cuvette. Reactions were generally performed under pseudo-first-order conditions, with typically a 20-fold excess of oxidized protein over reducing radicals. In each experiment, 2000 data points were collected and divided equally between two different time scales, each comprising at least three half-lives. Each kinetic run was repeated at least four times. The data were analyzed by fitting to a sum of exponentials using a nonlinear least-squares program written in MATLAB (The MathWorks, Natick, MA) to give rate constants, kET, and amplitudes. The equilibrium constants were evaluated based on the amplitude of the fast initial reduction of heme-c and the amplitude of the subsequent reoxidation in the following way: In each experiment the amplitude of the fast heme-c(III) reduction was a measure of the number of reduction equivalents added to the enzyme in this particular pulse (Atot). Part of heme-c(II) was then being reoxidized by internal ET to heme-d1(III). This was Areox. The remaining part of the reduced heme-c(II) was Ared. Thus, for each pulse Atot = Areox + Ared, and R551 = Areox/Ared. When R551 was plotted against the total number of reduction equivalents added to the enzyme during the pulse radiolytic reduction titration, a bell-shaped figure was obtained. A best fit was then calculated from the model using the intrinsic equilibrium constants as variables. The forward and reverse rate constants were calculated based on their relationship to the observed kET values and equilibrium constants. For example, in the initial pulses, kET = k23 + k32 and K23 = k23/k32 (for numbering, see Scheme 1). Simulation of the observed rate constants was accomplished by implementing the mechanism displayed in Scheme 1 using the Kintecus (V. 3.2) program package, converting the species concentrations into absorbance values, and fitting absorbance versus time data, after 1 ms, to two exponentials. The simulation started with a 20 μM solution of cd1 enzyme homodimers, then introduced a 3 μM pulse of 1-MNA∗ radicals. Fifty seconds were allowed for the system to evolve, and then another pulse of radicals was introduced. This was repeated for 27 pulses to produce 27 absorbance versus time data sets and to achieve full reduction.
Scheme 1.
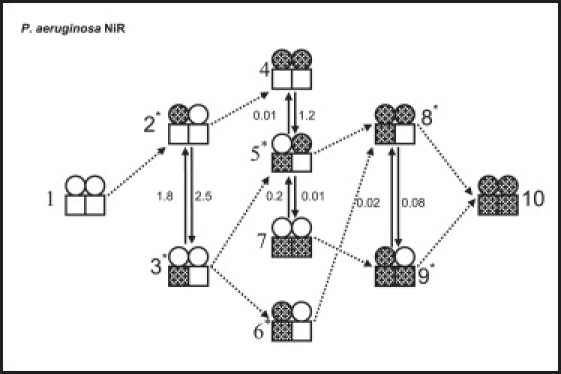
Proposed mechanism for the stepwise reduction of P. aeruginosa-NiR. Circles represent heme-c and squares heme-d1. Open symbols represent an oxidized site; solid ones, a reduced site. Each protein subunit includes one heme-c and the heme-d1. The single, dotted arrows represent the bimolecular reduction of a heme-c by the external reductant. These reactions are all assumed to be irreversible and to occur with the same, near diffusion controlled rate. The double, extended arrows represent reversible intramolecular electron transfer within a subunit. It is assumed that there is no intramolecular electron transfer between subunits, and that there is no intermolecular protein-protein electron transfer during the examined time domain. The asterisk indicates that there are two forms of the species. They differ by the subunit on which the electron resides. For example,  and
and  are the two forms of species 5. Rate constants are experimental values determined at 25°C.
are the two forms of species 5. Rate constants are experimental values determined at 25°C.
Results
The reduction of P. aeruginosa-NiR was investigated in several sets of experiments using either the CO2− or, mainly, 1-MNA∗ radicals, at pH 7.5, over a temperature range of 2–42°C. Almost identical results were obtained with each one of the two radicals. The reduction states of both heme types were monitored independently by measuring time-resolved absorption changes at 551 nm (for heme-c) and 640 nm (for heme-d1). The initial, fast process observed at 551 nm was assigned to a bimolecular reaction proceeding at rates close to the diffusion-controlled limit where the radicals reduce heme-c. Interestingly, this reaction was biphasic, showing two pseudo-first-order rate constants that led to two second-order rate constants, 2.4 × 109 M−1s−1 and 4.0 × 108 M−1s−1 at 298 K (Fig. 1). Significantly, no absorption changes at 640 nm were observed in this submillisecond time range, demonstrating that the radicals do not directly reduce heme-d1. Subsequently, a much slower decline in absorption at 551 nm and 640 nm was observed in a seconds time range. These processes were always well separated from the initial bimolecular step (Fig. 2). Because these absorption changes were found to be synchronous and concentration independent, they were assigned to an internal ET from heme-c to heme-d1, analogous to the reduction pattern observed for the P. stutzeri-NiR. Sequential introduction of pulses into the solution resulted in an increasing degree of NiR reduction. Rate and equilibrium constants for the internal ET process were determined together (Tables 1A and 1B). The extent of enzyme reduction was calculated by adding up the observed reduction amplitudes of the initial electron uptake by heme-c as monitored at 551 nm. Again, analogous to the results obtained with P. stutzeri-NiR, increasing the enzyme's degree of reduction beyond one electron equivalent per monomer caused the internal heme-c to heme-d1 ET rate constant to decrease by more than one order of magnitude (e.g., from ∼2 s−1 to <0.1 s−1 at 11°C), as illustrated in Fig. 3. The internal electron distribution between the c and d1 hemes in each monomer was similarly found to depend on the number of reduction equivalents taken up by the enzyme (Fig. 4). The same pattern of a decline in the internal electron distribution and transfer rates has been observed over the entire temperature range examined (2–40°C). From the temperature dependence, activation parameters as well as standard enthalpy and entropy changes for the intraprotein ET are presented in Tables 1A and 1B.
Figure 1.
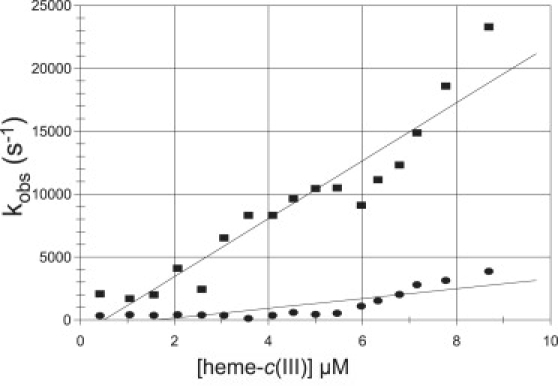
Observed rate constants of the heme-c fast reduction phases in WT P. aeruginosa-NiR plotted as function of [Fe(III)heme-c]. Concentrations: 9.7 μM enzyme; 5 mM 1-methyl nicotinamide; 7 mM phosphate; 0.1 M tert-BuOH; Anaerobic, Argon saturated. pH 7.0; optical path length 3.0 cm; pulse width 0.1 μs; temperature 25°C. Formation of Fe(II)heme-c was found to be bi-exponential, and the observed rate constants of the two phases are shown as ■ and ●, respectively, in the plot.
Figure 2.
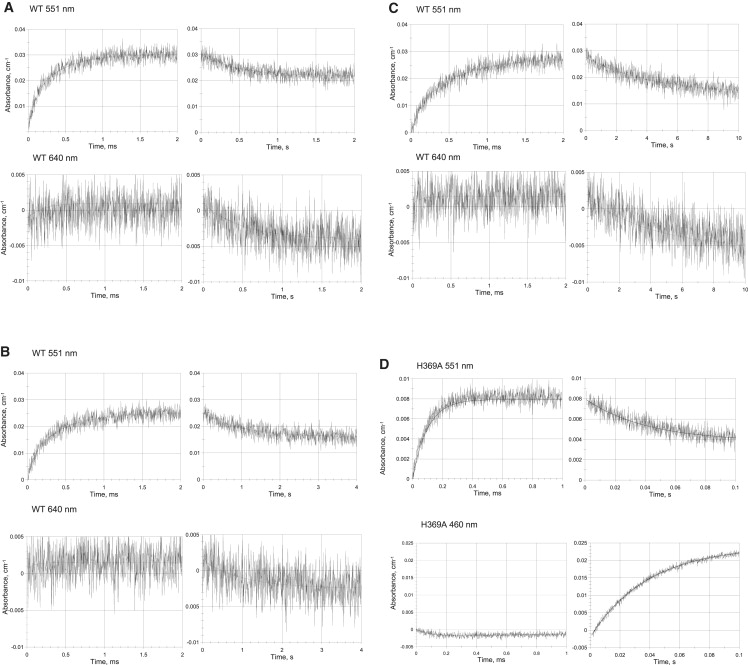
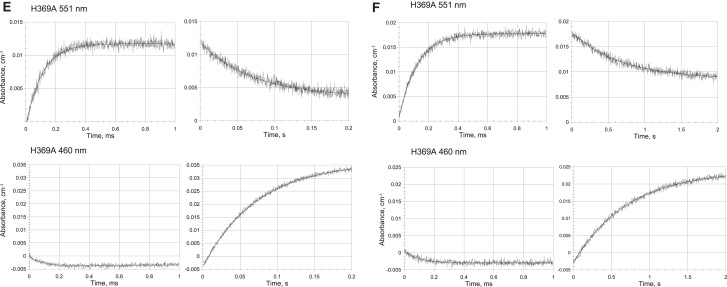
Time-resolved absorption changes upon reduction by 1-MNA∗ radicals. Concentrations: WT P. aeruginosa-NiR 9.6 μM, 5 mM 1-methyl nicotinamide, 5 mM phosphate, 0.1 M tert-BuOH, argon saturated, pH 7.0. Optical path length 3.0 cm, pulse width 0.1 μs, temperature 11.2°C. Measurements were performed at 551 nm, monitoring heme-c reduction (left panel: fast time scale) and heme-c reoxidation (right panel: slow time scale), and at 640 nm, monitoring heme-d1 reduction (left panel: fast time scale) and heme-d1 reoxidation (right panel: slow time scale). (A) WT initial pulses after <0.5 reduction equivalents were added. (B) WT after addition of ∼1.1 reduction equivalents were added. (C) WT after addition of ∼2.1 reduction equivalents were added. Concentrations: H369A P. aeruginosa-NiR mutant 48 μM, 2 mM 1-methyl nicotinamide, 10 mM phosphate, 0.1 M tert-BuOH, argon saturated. pH 7.0 Optical path length 3.0 cm, pulse width 1.0 μs; temperature 25.0°C. Measurements were performed at 551 nm, monitoring heme-c reduction (left panel: fast time scale) and heme-c reoxidation (right panel: slow time scale) and at 460 nm, monitoring heme-d1 reduction (left panel: fast time scale) and heme-d1 reoxidation (right panel: slow time scale). (D) H369A. Initial pulses after <0.5 reduction equivalents were added. (E) H369A, after addition of ∼1.1 reduction equivalents were added. (F) H369A, after addition of ∼2.1 reduction equivalents were added. Note that, in the case of the mutant, the rate of the faster phase at 551 nm is greater than that with the WT, consistent with the higher enzyme concentration employed.
Table 1A.
Kinetics and thermodynamics of heme-c to heme-d1 intramolecular ET process in WT P. aeruginosa-NiR at 298 K
| Kinetic parameters | k/s−1 | ΔH‡/kJ mol−1 | ΔS‡/J K−1 mol−1 |
|---|---|---|---|
| forward, k23 | 2.5 ± 0.3 | 84.8 ± 7.2 | +47 ± 4 |
| reverse, k32 | 1.8 ± 0.2 | 44.5 ± 8.5 | −91 ± 17 |
| Equilibrium parameters | K23 | ΔH0/kJ mol−1 | ΔS0/J K−1 mol−1 |
| 1.4 ± 0.1 | 40.4 ± 4.7 | +139 ± 16 | |
Data are presented for the initial, first electron equilibration.
Table 1B.
Kinetics and thermodynamics of heme-c to heme-d1 intramolecular ET process in H369A P. aeruginosa-NiR at 298 K
| Kinetic parameters | k/s−1 | ΔH‡/kJ mol−1 | ΔS‡/J K−1 mol−1 |
|---|---|---|---|
| forward, k23 | 12.8 ± 1.6 | 53 ± 10 | −48 ± 8 |
| reverse, k32 | 11.7 ± 1.5 | 58 ± 10 | −32 ± 5 |
| Equilibrium parameters | K23 | ΔH0/kJ mol−1 | ΔS0/J K−1 mol−1 |
| ∼1.1 | Not determined | Not determined | |
Data are presented for the initial, first electron equilibration.
Because of the limited amount of the H369A mutant, the results are based on measurements at three different temperatures, only.
Figure 3.
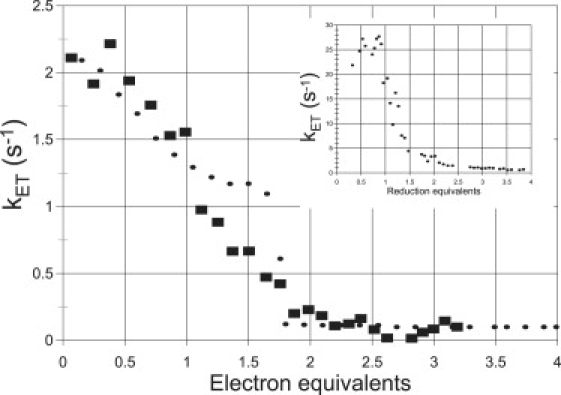
(A) The observed (■) and simulated (●) rate constants of intramolecular ET in P. aeruginosa-NiR at 11.2°C as a function of the state of reduction of the enzyme based on 551 nm absorption (reoxidation of reduced heme-c). The simulation represents the dominant process obtained by simulating the mechanism in Scheme 1 as described above for 27 pulses of 1-MNA∗ radicals, resulting in complete reduction of the four sites. The rate constants (s−1) used for this temperature were: k23, 0.60; k32, 1.5; k45, 0.35; k54, 0.0046; k57, 0.0010; k75, 0.049; k89, 0.082; and k98, 0.018. (B) Observed rate constants of intramolecular heme-c to heme-d1 ET in the H369A mutant of P. aeruginosa-NiR based on 551 and 460 nm data, plotted as a function of the reduction state of the enzyme at 25°C.
Figure 4.
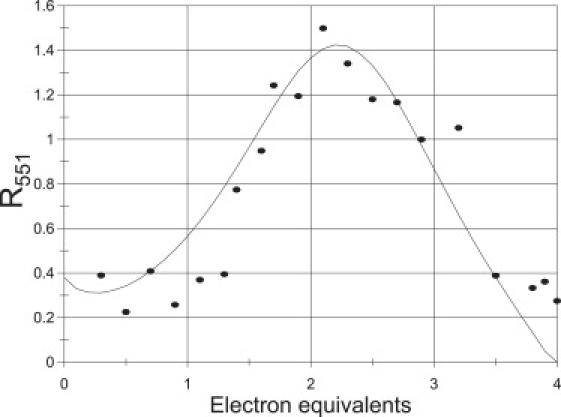
R551 is the ratio between reduced and reoxidized heme-c produced in each pulse radiolysis experiment. The curve represents the best fit to the experimental data at 11.2°C based on the reaction mechanism presented in Scheme 1.
The above results have been analyzed using the same model described previously (5). It assumes that electron uptake by heme-c is followed by equilibration between hemes c and d1 within the same subunit. Intersubunit ET equilibration has been ignored, because the heme-heme separation distances in the dimer are too large (4.2 nm) (12) to allow its occurrence during the examined time domain. Similarly, intermolecular ET between enzyme dimers was not observed in the time range employed. The model depicted in Scheme 1 required only four equilibration steps where intrasubunit ET can take place.
The rate constants determined at 298 K are included in Scheme 1. The calculated, detailed internal electron distribution depends strongly on the enzyme's reduction state: In contrast to an equal partition of the first reducing equivalent between hemes c and d1 observed for P. stutzeri-NIR, in P. aeruginosa-NiR, a slight preference for the latter (heme d1) is observed (K23 = 1.4). Adding a second equivalent to P. aeruginosa-NiR favors species 5 (K45 = 120 and K75 = 20); i.e., the enzyme molecules that prevail have one heme site (c or d1) in distinct subunits reduced. As far as the distribution of the third equivalent is concerned, preference is found for species 9, namely having both hemes d1 reduced (K89 = 4.0). The marked decrease in observed internal ET rate constant with the extent of reduction of the enzyme is similar to that measured for P. stutzeri, although somewhat less extreme; a factor of 40 is measured in P. aeruginosa-NiR, whereas the factor is 200 in P. stutzeri, both at 25°C. The observed intramolecular rate constant reflects the ET process of the predominant species present at each pulse.
We have also studied the reduction pattern and internal ET rates in the single-site H369A mutant of the P. aeruginosa enzyme (cf. Figs. 2 and 3). It is noteworthy that the observed internal ET rates were significantly faster: at 25°C, a rate constant of 24.5 s−1 was determined—i.e., almost one order of magnitude larger than that measured for the WT enzyme (cf. Tables 1A and 1B). Similarly, a decrease in the internal ET rates upon increased reduction was also seen in the mutant with the rate constant leveling off at 0.8 s−1.
Discussion
We have investigated the reduction process of P. aeruginosa-NiR using the pulse radiolysis method by employing 1-MNA∗ and CO2− radicals as reductants. These pulse radiolytically produced radicals were found to directly reduce only the heme-c, as in P. stutzeri-NiR (6) as well as in earlier pulse radiolysis studies of the P. pantotrophus and P. aeruginosa-NiRs (Table 2); however, the current observation of two distinct phases of heme-c reduction is a feature not observed in the previous pulse-radiolysis studies of any NiR. Still, in studies of P. aeruginosa-NiR reduction by excess reduced azurin or chromium(II) ions, two concentration-dependent phases were also observed. This was explained by hypothesizing the existence of two different enzyme populations (14). In that study (14), the reducing agent was in large excess, whereas, in the presented study, the protein sites in excess; nevertheless, heterogeneity of the heme-c reduction rate was still observed. The currently observed two reduction phases cannot represent reduction of two distinct enzyme populations (e.g., two forms of the heme-c in the dimer), because this situation would lead to a pseudo-first-order process in which the observed rate constant would be the sum of the two individual rate constants. Instead, we suggest that the two phases could be the result of two consecutive processes in which heme-c Fe(III) first undergoes reduction, followed by a structural rearrangement causing spectral changes at 551 nm.
Table 2.
Rate constants for intramolecular ET in nitrite reductases from different sources
| Source | kET/s−1 | Conditions | Reference |
|---|---|---|---|
| Pseudomonas aeruginosa | 4.3∗ | pH 7.0; 298 K | The presented study |
| Pseudomonas aeruginosa H369A mutant | 24.5∗ | pH 7.0; 298 K | The presented study |
| Pseudomonas aeruginosa | 3.0∗ | pH 7.0; 298 K | 13 |
| Pseudomonas aeruginosa | 0.25–0.81† | pH 7.0; 298 K | 14,16,17 |
| Paracoccus pantotrophus | 1400∗ | pH 7.0 | 4 |
| Pseudomonas stutzeri | 23∗ | pH 7.0; 298 K | 5 |
Measured by pulse radiolysis.
Measured by stopped flow.
The subsequent, slower reaction observed in P. aeruginosa-NiR solutions involved the synchronous oxidation of heme-c and reduction of heme-d1, both independent of protein concentration, and the reaction is therefore attributed to the internal (heme-c to heme-d1) electron transfer process.
Both our earlier study and this pulse radiolysis study of NiRs isolated from P. stutzeri (5) and P. aeruginosa, respectively, took advantage of the possibility of gradual reduction of the enzymes by sequential pulses and resolved the marked dependence of the internal ET rate on the degree of the reduction. This procedure apparently was not feasible in other pulse radiolysis studies of NiRs (4,13). Early kinetic studies by pulse radiolysis of cd1 NiRs isolated from P. aeruginosa (13), P. pantotrophus (4), and P. stutzeri (5) yielded considerably different rate constants for this step: ∼3 s−1, 1400 s−1, and 23 s−1, respectively, at 298 K and pH 7.0 (Table 2). The rate constant for heme-c to heme-d1 ET measured for the fully oxidized NiR in this study (kET = 4.3 s−1) is in agreement with the rate measured by Kobayashi et al. (13) and is of the same order of magnitude as is the turnover number of P. aeruginosa-NiR at pH 7.0°C and 20°C (4–6 sec−1) (15). Previous kinetic studies carried out by stopped-flow on the P. aeruginosa enzyme yielded somewhat slower rates for the heme-c to heme-d1 ET (14,16,17) (Table 2), probably reflecting the different experimental conditions employed (e.g., rates obtained in this study at high degrees of NiR reduction). Because internal ET to heme-d1 is required for nitrite reduction, only steps 2→3 and 4→5 in Scheme 1 are candidates for participating in the catalytic cycle. However, the presence of nitrite and its preferential binding to heme-d1 are expected to change the reduction potential of this site and possibly the rates of formation of species 3 and beyond. Nitric oxide (NO) is the final product of nitrite reduction at the reduced heme-d1 site. The stability constant for heme-d1 Fe(II) complex formation with NO is in the range 107−108 M−1, markedly different from that of most reduced heme proteins, which display higher affinities, usually ∼1011 M−1. The lower affinity of reduced P. aeruginosa-NiR is mainly due to an unexpectedly high rate of dissociation (koff = 70 s−1) (15), excluding significant product inhibition by NO binding to reduced heme-d1. If NO also binds to the oxidized heme-d1, then the reduction potential of this site, as well as the heme-c to heme-d1 electron transfer rate constant, would be enhanced. It has indeed been observed in our earlier experiments (S. Wherland and I. Pecht, unpublished results) that adding NO to partially reduced P. stutzeri-NiR caused reoxidation of the heme-c, presumably by inducing intramolecular electron transfer to the oxidized heme-d1-NO complex.
For WT P. aeruginosa-NiR, the rate-limiting step of the catalytic cycle in the presence of nitrite could be the rate of the first and second reduction equivalents arriving at the ligand-free heme-d1 to form species 3 and 5. These species then bind nitrite, and the steps afterward (ET, protonation, water, and NO dissociation) could be fast, in agreement with recent results obtained for the P. pantotrophus-NiR, showing that pseudoazurin may act as an electron mediator between enzyme molecules having different reduction states, thereby accelerating the observed catalytic rate of nitrite reduction (18). Therefore, the rate of formation of species 3 and 5, which should be unaffected by nitrite, controls the overall catalytic reaction rate in WT P. aeruginosa-NiR.
We have also examined the internal ET in the single-site mutant of the P. aeruginosa-NiR, where the invariant His369 on the distal side of heme-d1 has been substituted with Ala and is catalytically inactive toward nitrite (11). The internal ET rate was found to be significantly faster (kET = 24.5 s−1 at 298 K) when <1 electron equivalent has been added, almost one order of magnitude larger than that observed for the WT enzyme.
First we wish to present a possible structural interpretation for the different internal ET rates observed for the NiR species. Examination of available structural data obtained by crystallography of WT P. aeruginosa-NiR (2) suggests that dissociation of the OH− bound on the distal side of the heme-d1 Fe(III) may be involved in rate control. The possible importance of the Fe(III) heme-d1 ligand OH− in controlling the internal ET rate was suggested by Kobayashi et al. (13), based on pulse radiolysis experiments on P. aeruginosa-NiR. The structure of the oxidized form shows the OH− to be stabilized by interactions with Tyr-10, protruding into the active site and originating from the partner monomer (2). This is a remarkable case of “domain swapping”, which is typical of P. aeruginosa-NiR (2) activity, but not observed in the oxidized P. pantotrophus-NiR (1). Moreover, it should be added that, upon reduction, Tyr-10 changes its conformation (19), and this slippage is associated with disappearance of the hydroxide ion from the active site pocket. It was also shown (11,20) that binding of anions to heme-d1 is stabilized by the two invariant distal His; therefore, it may be anticipated that stability of the Fe(III)-bound OH− would decrease if one or both His were removed. The observation that the heme-c to heme-d1 internal ET rate constant is increased by ∼10-fold in the His369A mutant seems consistent with this hypothesis.
The second and more intriguing observation is the decrease in the internal heme-c to heme-d1 ET rate constant (by more than one order of magnitude) as the number of electrons pumped in is increased, as shown in Fig. 3 for the WT enzyme. Modeling of the current results requires the assumption of site-site interaction within a dimer. After one reducing equivalent has been taken up by the enzyme, the rate constant declines to a value approximately one half of the maximum value of 2 s−1 (at 11°C in Fig. 3). However, a more dramatic drop in rate constant is observed upon addition of more reducing equivalents, as the second heme-d1 starts being reduced. In both the WT and the H369A mutant the heme-c to heme-d1 rate constant drops as the fractional reduction increases, by approximately the same amount (∼30-fold), leveling off at 0.08 s−1 at 25°C in WT. These results provide clear kinetic evidence for negative cooperativity between the two heme-d1 sites in the dimer. Using the 3D structures now available for oxidized and reduced P. aeruginosa-NiR (2,19), we present below a structural hypothesis for this remarkable effect.
The rate constants observed for the individual internal ET steps (Scheme 1) deserve attention in the context of the 3D structure. Using the separation distance between the iron centers of the two hemes in one monomer, 2.0 nm (2,19), we calculate the expected species 2→species 3 intramolecular ET rate constant in the activationless case to be kMAX ≈ 104 s−1. The experimentally observed rate constant is 2.5 s−1 and the driving force, −ΔG0, is 0.009 eV (K = 1.4 at 298 K). From these we may calculate a reorganization energy for heme-c to heme-d1 ET of 0.87 eV (84 kJ/mol−1). This is typical for c-type heme reorganization, 0.8 eV (21). The 30-fold drop in rate constant to 0.08 s−1 mentioned in the previous paragraph precludes changes in the driving force having a major influence, because this would require a K89 = 0.002 for the heme-c to heme-d1 electron equilibration, whereas the observed equilibrium constant is 4.0 (Scheme 1). Changes in reorganization energy alone would require an increase of no less than 0.35 eV, which appears quite unrealistic. Hence, this observed decrease in rate must involve changes taking place in the 3D structure of the enzyme. Several conformational differences between reduced and oxidized P. aeruginosa-NiR have been reported (13), which may provide a rationale for the steep decline in rates shown in Fig. 3. Structural changes causing a decrease in the electronic coupling between donor and acceptor may in principle lead to a decrease in ET rate constant (21). Thus, for instance, breaking one hydrogen bond, forcing a through-space jump, could account for a ∼50-fold drop in rate constant. Candidates for such hydrogen bonds in the P. aeruginosa-NiR structure have been identified as two water molecules bridging hemes c and d1 (2). An important redox-linked conformational change was observed by crystallography: In the reduced form, a loop (residues 56–62) belonging to the heme-c domain moves to form a hydrogen bond between Thr-59 and Gln-11 (coming from the N-terminal arm), as shown in Fig. 5.
Figure 5.
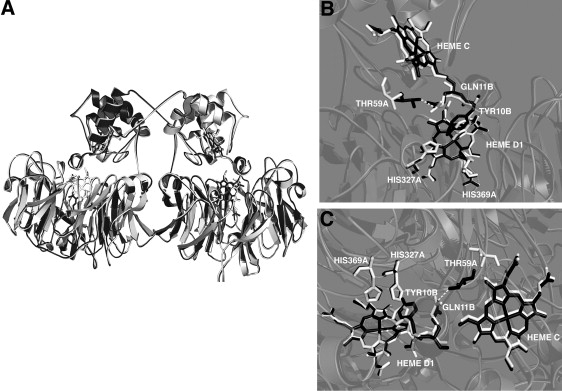
Conformational changes occurring upon reduction of P. aeruginosa-NiR. (A) Superposition of the 3D structure of the oxidized (white) and reduced (black) dimeric enzyme, highligthing the swapping of the N-termini. Two different views (B and C) of the interface between the heme-c and heme-d1 domains in the two oxidation states are presented. Three relevant residues in the heme-d1 pocket are depicted: His-369 and His-327 belonging to monomer A (HIS-327A and HIS-369A), and Tyr-10 coming from the other monomer B (TYR-10B). The reorganization of the 56-62 loop and the new H-bond (dashed line) between Thr-59 (from monomer A) and Gln-11 (from monomer B) formed upon reduction are shown (THR-59A and GLN-11B).
In summary, we propose that the observed allosteric control of the rate constants for intramolecular electron transfer between the hemes is a result of a complex interplay among several conformational changes occurring upon reduction and resolved by the 3D structural analysis—namely, the relocation of Tyr-10, the coupled dissociation of the OH− ligand, and the disruption of the hydrogen bonds at the interface between the two heme domains (see Fig. 5). This interpretation is consistent with the finding that intramolecular ET is faster in the H369A mutant, given the decrease of positive charge density on the distal side of heme-d1.
The mechanism of communication between monomers, underlying the decrease in ET rate with the extent of the enzyme's reduction (Fig. 3), is not immediately apparent but may be interpreted as follows: The structural analysis resolved a rather large sliding motion of the whole heme-c domain relative to the heme-d1 domain in the His mutants (11,12). This rather large conformational change was not observed upon reduction of the WT enzyme (19); however, we assume that this is due to the fact that the structure of the reduced WT was obtained by reduction of oxidized crystals rather than by crystallization of the reduced protein, possibly imposing a constraint that prevented this transition. In fact, in the case of P. pantotrophus-NiR, Hajdu and coworkers (23) observed a large heme-c domain rotation only when the enzyme was crystallized in the reduced state. Therefore, we propose that allosteric communication between identical monomers may depend on a perturbation of the domain-domain interface within each monomer and a coupled large relocation of the heme-c domain, upon reduction of the enzyme. These conformational changes perturb the above-mentioned hydrogen bonds, decreasing the electronic coupling between heme-c and heme-d1. Thus, control of the electron transfer rates involves contributions from local changes, affecting the driving force of the intramolecular ET, and more global changes (such as the relocation of the c-heme versus d1-heme domains), which may affect the electronic coupling between sites (within and between monomers), further increasing the energy barrier for ET as the enzyme undergoes reduction.
Acknowledgments
O.F. acknowledges the generous support extended by the Kimmelman Center for Biomolecular Structure and Assembly at the Weizmann Institute of Science. We thank Mr. Eran Gilad of the Hebrew University in Jerusalem for his excellent technical support in running the accelerator.
These studies were supported by grants from Ministero della Università e Ricerca of Italy (RBIN04PWNC_000 to M.B. and RBIN04PWNC_002 to F.C.) and from the Ministero degli Affari Esteri of Italy (L.401/90-2006) to M.B. for supporting the Italy-Israel research project.
References
- 1.Fülöp V., Moir J.W.B., Ferguson S.J., Hajdu J. The anatomy of a bifunctional enzyme: structural basis for reduction of oxygen to water and synthesis of nitric oxide by cytochrome cd1. Cell. 1995;81:369–377. doi: 10.1016/0092-8674(95)90390-9. [DOI] [PubMed] [Google Scholar]
- 2.Nurizzo D., Silvestrini M.-C., Mathieu M., Cutruzzolà F., Bourgeois D. N-terminal arm exchange is observed in the 2.15 Å crystal structure of oxidized nitrite reductase from Pseudomonas aeruginosa. Structure. 1997;5:1157–1171. doi: 10.1016/s0969-2126(97)00267-0. [DOI] [PubMed] [Google Scholar]
- 3.Silvestrini M.C., Tordi M.G., Musci G., Brunori M. The reaction of Pseudomonas nitrite reductase and nitrite. A stopped-flow and EPR study. J. Biol. Chem. 1990;265:11783–11787. [PubMed] [Google Scholar]
- 4.Kobayashi K., Koppenhoefer A., Ferguson S.J., Tagawa S. Pulse radiolysis studies on cytochrome cd1 nitrite reductase from Thiosphaera pantotropha: evidence for a fast intramolecular electron transfer from c-heme to d1-heme. Biochemistry. 1997;36:13611–13616. doi: 10.1021/bi971045o. [DOI] [PubMed] [Google Scholar]
- 5.Farver O., Kroneck P.M.H., Zumft W.G., Pecht I. Intramolecular electron transfer in cytochrome cd1 nitrite reductase from Pseudomonas stutzeri; kinetics and thermodynamics. Biophys. Chem. 2002;98:27–34. doi: 10.1016/s0301-4622(02)00082-0. [DOI] [PubMed] [Google Scholar]
- 6.Farver O., Kroneck P.M.H., Zumft W.G., Pecht I. Allosteric control of internal electron transfer in cytochrome cd1 nitrite reductase. Proc. Natl. Acad. Sci. USA. 2003;100:7622–7625. doi: 10.1073/pnas.0932693100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farver O., Pecht I. Elucidation of electron-transfer pathways in copper and iron proteins by pulse radiolysis experiments. Prog. Inorg. Chem. 2007;55:1–78. [Google Scholar]
- 8.Blatt Y., Pecht I. Allosteric cooperative interactions among redox sites of Pseudomonas cytochrome oxidase. Biochemistry. 1979;18:2917–2922. doi: 10.1021/bi00580a037. [DOI] [PubMed] [Google Scholar]
- 9.Koppenhofer A., Turner K.L., Allen J.W.A., Chapman S.K., Ferguson S.J. Cytochrome cd1 from Parococcus pantotrophus exhibits kinetically gated, conformationally dependent, highly cooperative two-electron redox behavior. Biochemistry. 2000;39:4243–4249. doi: 10.1021/bi000192a. [DOI] [PubMed] [Google Scholar]
- 10.Parr S.R., Barber D., Greenwood C.A. A purification procedure for the soluble cytochrome oxidase and some other respiratory proteins from Pseudomonas aeruginosa. Biochem. J. 1976;157:423–430. doi: 10.1042/bj1570423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cutruzzolà F., Brown K., Wilson E.K., Bellelli A., Arese M. The nitrite reductase from Pseudomonas aeruginosa: essential role of two active-site histidines in the catalytic and structural properties. Proc. Natl. Acad. Sci. USA. 2001;98:2232–2237. doi: 10.1073/pnas.041365298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown K., Roig-Zamboni V., Cutruzzolà F., Arese M., Sun W.L. Domain swing upon His to Ala mutation in nitrite reductase of Pseudomonas aeruginosa. J. Mol. Biol. 2001;312:541–554. doi: 10.1006/jmbi.2001.4986. [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi K., Koppenhoefer A., Ferguson S.J., Watmough N.J., Tagawa S. Intramolecular electron transfer from c heme to d1 heme in bacterial cytochrome cd1 nitrite reductase occurs over the same distances at very different rates depending on the source of the enzyme. Biochemistry. 2001;40:8542–8547. doi: 10.1021/bi002534i. [DOI] [PubMed] [Google Scholar]
- 14.Parr S.R., Barber D., Greenwood C., Brunori M. The electron-transfer reaction between azurin and the cytochrome c oxidase from Pseudomonas aeruginosa. Biochem. J. 1977;167:447–455. doi: 10.1042/bj1670447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rinaldo S., Arcovito A., Brunori M., Cutruzzolà F. Fast dissociation of nitric oxide from ferrous Pseudomonas aeruginosa cd1 nitrite reductase: a novel outlook on the catalytic mechanism. J. Biol. Chem. 2007;282:14761–14767. doi: 10.1074/jbc.M700933200. [DOI] [PubMed] [Google Scholar]
- 16.Schichman S.A., Gray H.B. Kinetics of the anaerobic reduction of ferricytochrome-cd1 by Fe(EDTA)2—evidence for bimolecular and intramolecular electron transfers to the d1 hemes. J. Am. Chem. Soc. 1981;103:7794–7795. [Google Scholar]
- 17.Cutruzzolà F., Arese M., Grasso S., Bellelli A., Brunori M. Mutagenesis of nitrite reductase from Pseudomonas aeruginosa: tyrosine-10 in the c heme domain is not involved in catalysis. FEBS Lett. 1997;412:365–369. doi: 10.1016/s0014-5793(97)00583-8. [DOI] [PubMed] [Google Scholar]
- 18.Sam K.A., Fairhurst S.A., Thorneley R.N.F., Allen J.W.A., Ferguson S.J. Pseudoazurin dramatically enhances the reaction profile of nitrite reduction by Paracoccus pantotrophus cytochrome cd1 and facilitates release of product nitric oxide. J. Biol. Chem. 2008;283:12555–12563. doi: 10.1074/jbc.M800954200. [DOI] [PubMed] [Google Scholar]
- 19.Nurizzo D., Cutruzzolà F., Arese M., Bourgeois D., Brunori M. Conformational changes occurring upon reduction and NO binding in nitrite reductase from Pseudomonas aeruginosa. Biochemistry. 1998;37:13987–13996. doi: 10.1021/bi981348y. [DOI] [PubMed] [Google Scholar]
- 20.Sun W., Arese M., Brunori M., Nurizzo D., Brown K. Cyanide binding to cd1 nitrite reductase from Pseudomonas aeruginosa: role of the active-site His369 in ligand stabilization. Biochem. Biophys. Res. Commun. 2002;291:1–7. doi: 10.1006/bbrc.2002.6391. [DOI] [PubMed] [Google Scholar]
- 21.Gray H.B., Winkler J.R. Electron tunneling through proteins. Q. Rev. Biophys. 2003;36:341–372. doi: 10.1017/s0033583503003913. [DOI] [PubMed] [Google Scholar]
- 22.Beratan D.N., Betts J.N., Onuchic J.N. Protein electron-transfer rates set by the bridging secondary and tertiary structure. Science. 1991;252:1285–1288. doi: 10.1126/science.1656523. [DOI] [PubMed] [Google Scholar]
- 23.Sjögren T., Hajdu J. The structure of an alternative form of Paracoccus pantotrophus cytochrome cd1 nitrite reductase. J. Biol. Chem. 2001;276:29450–29455. doi: 10.1074/jbc.M103657200. [DOI] [PubMed] [Google Scholar]