

Abstract
The process of genetic recombination involves the formation of branched four-stranded DNA structures known as Holliday junctions. The Holliday junction is known to have an antiparallel orientation of its helices, i.e., the crossover occurs between strands of opposite polarity. Some intermediates in this process are known to involve two crossover sites, and these may involve crossovers between strands of identical polarity. Surprisingly, if a crossover occurs at every possible juxtaposition of backbones between parallel DNA double helices, the molecules form a paranemic structure with two helical domains, known as PX-DNA. Model PX-DNA molecules can be constructed from a variety of DNA molecules with five nucleotide pairs in the minor groove and six, seven or eight nucleotide pairs in the major groove. A topoisomer of the PX motif is the juxtaposed JX1 molecule, wherein one crossover is missing between the two helical domains. The JX1 molecule offers an outstanding baseline molecule with which to compare the PX molecule, so as to measure the thermodynamic cost of forming a crossover in a parallel molecule. We have made these measurements using calorimetric and ultraviolet hypochromicity methods, as well as denaturing gradient gel electrophoretic methods. The results suggest that in relaxed conditions, a system that meets the pairing requirements for PX-DNA would prefer to form the PX motif relative to juxtaposed molecules, particularly for the 6:5 structure.
Introduction
Homologous recombination is the mechanism by which genetic information can be exchanged in vivo (1–4). Fundamental to understanding the mechanism of recombination is the suggestion by Holliday (5) that heteroduplexes can form via basepairing in homologous regions, and lead to a junction in which strands cross over from one duplex to the other. The structure of the Holliday junction has been established by a variety of techniques, including hydroxyl radical autofootprinting (6), gel mobility (7,8), Förster resonance energy transfer (9), atomic force microscopy (10,11) and x-ray crystallography (12). A unified picture emerged from all of these studies: in the presence of divalent cations, the four double helical arms that flank the junction assort themselves into two stacking domains; the strands that do not participate in the crossover are oriented 40–60 from antiparallel. Despite earlier suggestions that the helical domains are parallel to each other (13), a single molecule Förster resonance energy transfer study by Ha et al. (14) has found that the parallel conformation is not a significant contributor to the single-crossover Holliday structure.
Nevertheless, homologous interactions and duplex parallelism present a very satisfying picture. For example, Kleckner and Weiner have suggested that chromosome pairing entails duplex-duplex recognition (15,16). Recently, we have found that head-to-head homology can lead to the relaxation of supercoiling in plasmids (X. Wang, X. Zhang, C. Mao, and N. C. Seeman, unpublished observations). The data suggest that the structure resembles PX-DNA, which is a four-stranded complex consisting of two parallel double helices (18). In contrast to the Holliday junction, which has only a single crossover between double helices, the PX structure can be derived from parallel double helices by forming a crossover at every possible juxtaposition between them (19). Thus, it becomes key to understand the structural and thermodynamic features of the parallel crossover, in addition to the twisted antiparallel single crossover. In this study, we present thermodynamic data on the parallel crossover to complement the thermodynamic data that have been gathered for the twisted antiparallel single crossover (20,21).
We use the PX structure and one of its topoisomers to facilitate our measurements. In the same way that previous experiments measuring the thermodynamics of chain branching in the Holliday junction use analogs of the Holliday junction lacking sequence symmetry, we use PX structures that lack sequence symmetry and therefore are stable in relaxed molecules. Several features are important to emphasize about the PX motif, a representation of which is shown in Fig. 1 a. The molecule is capable of containing a dyad symmetry axis along the backbone of the structure, and the midline through the vertical of the dyad axis alternates between the minor groove and major groove, indicated by N and W in Fig. 1. In this projection, a crossing of strands within a double helix is called a unit tangle (22), and two successive unit tangles make an approximate full helical turn with one minor groove spacing and one major groove spacing per helical turn. The repeat of the PX structure contains ∼2 helical turns of DNA in each of its domains. It is important to recognize that were a PX molecule to be produced by two double helices, such as the red and blue molecules of Fig. 1 a, there is an alternation along each domain of “intramolecular” pairs (red-red or blue-blue) and “intermolecular” pairs (red-blue or blue-red). It has been found in four-stranded molecules that stable structures can be formed at micromolar concentrations if the minor groove spacing is five nucleotide pairs, whereas the major groove spacing can be six, seven, or eight basepairs (18). These molecules are termed PX 6:5, PX 7:5, and PX 8:5 structures. Other values yield stable molecules if the concentration is lower (18) or fewer strands are involved, for example in the PX hairpins (23) used in paranemic cohesion (24). Other important features of these PX structures are that the circular dichroism spectra suggest B-form DNA and they are designed to contain only Watson-Crick basepairing.
Figure 1.
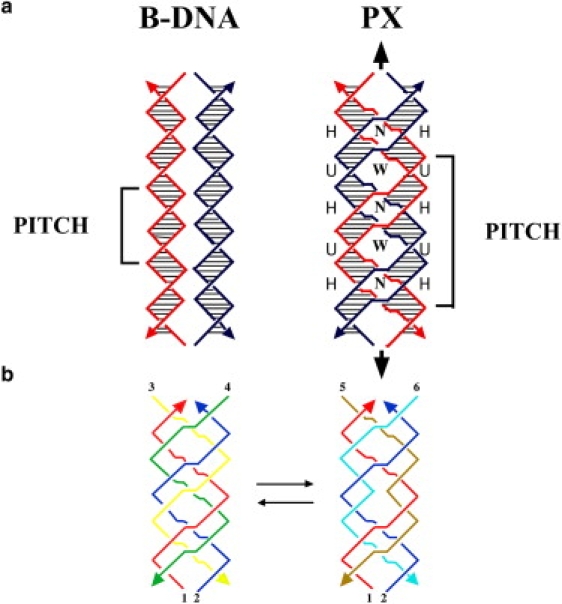
(a) Representation of B-DNA and PX-DNA, showing helical repeat units and crossovers in the PX forms. N and W refer to narrow and wide grooves in the crossover structure. (b) Depiction of PX and JX structures, identifying the strands required to form crossovers and juxtapositions in PX and JX forms. (See text for explanation.)
To study the energetics of a crossover, it is necessary to have a reference structure with which to compare properties. This requires a molecular species with an equivalent number of basepairs of the same sequence and a structure similar to the PX motif but with a single missing crossover compared with the PX form. Such structures are possible by choosing the four strands such as to provide the same five- or six-member building blocks (for the PX 6:5 motif), but placing them in a different order such that one crossover will be eliminated. In this case the appropriate strands will be juxtaposed across from each other without crossing over and are called JXi motifs, where the subscript refers to the number of missing crossovers. JX2 molecules are a key state in robust sequence-dependent nanomechanical devices (25–27). Thus, a 38-mer of alternating five and six nucleotides in the narrow and wide grooves will produce six crossovers in the PX 6:5 structure, whereas the corresponding JX1 6:5 structure will contain only five crossovers. The structures of PX molecules and JXn molecules have been simulated in molecular dynamics calculations by Maiti et al. (28,29). Their calculations indicate that motifs with large numbers of nucleotide pairs in the major groove appear to distort the structure by inducing a writhe in the double helix at those sites. It is worth noting that a JX-type molecule with only two remaining crossovers is a DX molecule (30), the parallel versions of which have been shown to be a meiotic intermediate (31).
To make both these PX and JX structures, a total of six strands is required, For example, in Fig. 1 b, the PX structure (left) requires that red strand 1 be complementary successively to a half-turn of green (strand 4), a half-turn of yellow (strand 3), a half-turn of green, a half-turn of yellow, another half-turn of green and another half-turn of yellow; likewise, blue strand 2 is complementary to successive half-turns of yellow and green in the opposite order. The JX1 molecule (right) is formed in a similar fashion, substituting ochre (strand 5) for green and aqua (strand 6) for yellow; however, there is a key difference: red strand 1 is paired to ochre strand 5 for two successive half-turns, and similarly blue strand 2 is paired with aqua strand 6 for two successive half-turns. This arrangement leads to juxtapositions in the middle of the structure, because one crossover in this region is eliminated. This method enables us to prepare PX 6:5 and JX1 6:5 motifs, PX 7:5 and JX1 7:5 motifs, and PX 8:5 and JX1 8:5 motifs. (See sequences in the Supporting Material.)
The purpose of this study is to determine the thermodynamic properties of melting of the six stable PX and JX1 structures so as to compare the energetics of formation of the PX structures that contain all possible crossovers with the energetics of forming the corresponding JX1 structures that lack one crossover in each case. The differences in properties of the two forms provide a measure of the thermodynamic stability of a crossover relative to juxtaposed strands. The effect of having six, seven, or eight basepair spacings in the major groove can also be evaluated. Melting is studied by differential scanning calorimetry (DSC), so that model-free thermodynamic properties can be determined. The results are compared with thermal melting measurements studied by ultraviolet (UV) absorption hyperchromicity changes. The effect of magnesium ion concentration on the stability of the two motifs is also studied for each case. The thermal transitions seem to have underlying sequential melting domains, so attempts are made to analyze the melting profile into a set of sequential steps. The calorimetric data are also compared with data obtained from denaturing gradient gel electrophoresis (32) (DGGE) experiments on cyclic molecules; these data provide an independent estimate of the relative melting properties of the various PX and JX1 molecules.
The results show clearly that the formation of both PX and JX1 molecules from relaxed DNA duplex molecules are clearly not favored. Nevertheless, they also show that the formation of PX DNA from JX1 DNA is approximately isoenergetic. Thus, once the price has been paid to bring two double helices together in a parallel arrangement, the formation of a further internal crossover does not entail an unacceptable thermodynamic cost.
Materials and Methods
These are described in the Supporting Material.
Results
Scanning calorimetry data
Fig. S1 in the Supporting Material shows an example of the raw data for a DSC scan, with the derived baseline shown in the figure. The baseline was calculated from a 4th order polynomial fit to the scans between 35 and 80° in the pre- and posttransitional regions. Scans were run on at least two different samples of each motif, to insure repeatability of the measured peak temperatures and areas. Fig. 2 presents the resulting thermal scans for both the PX and JX structures in high magnesium ion buffer (125 mM Mg2+) after subtracting the appropriate baselines, and after scaling the ordinate to cal/deg-mol by dividing by the scan rate in deg/s, and by the number of moles in the cell. Similar results were obtained in low magnesium ion buffer (12.5 mM Mg2+), and are presented in Fig. S2. Several features are evident from the data in Fig. 2 and Fig. S2. The first obvious characteristic of the scans is that none of the transitions is a simple two-state melt, but rather shows evidence of underlying complexity in the melting transitions. Virtually all curves show multiple melting domains with one or two intermediates in the melting profile. The symbols in the figures are the experimental data, and the solid lines are calculated assuming sequential transitions through intermediates (discussed below).
Figure 2.
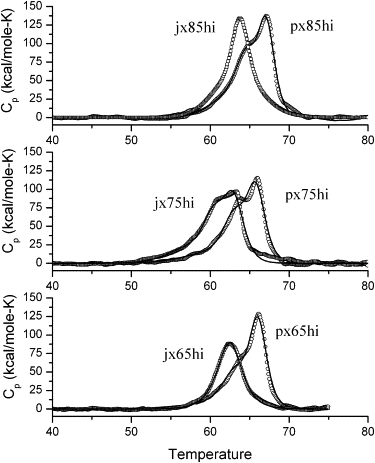
DSC scans of 1.5 micromolar solutions in cacodylate buffer containing 125 mM Mg2+ ion for various PX and JX1 structures with differing numbers of nucleotide bases in the major groove. Circles are experimental data, and the solid curves are theoretical curves assuming a sequential transition model, as described in the text. Polynomial baselines were subtracted from the raw data and the resulting values normalized to per mole of solute for 1°/min scan rate.
To examine the DSC data more quantitatively, Fig. 3 (high magnesium) and Fig. S3 (low magnesium concentration) display the values of the enthalpies, entropies and free energies of melting of the 12 different species. (A table of the data is presented in the Supporting Material.) Enthalpies are determined directly from the total areas of the thermal transitions, and ΔSm is calculated from the integral of a plot of Cp(ex)/T versus temperature, ΔSm = ∫Cp(ex)/T dT. Also shown in Fig. 3 and Fig. S3 (top panels) are the corresponding energetics per basepair, which factors out that there are differences in the number of basepairs for the 6:5 (76 bp), 7:5 (86 bp), and 8:5 (92 bp) motifs. Note that the free energy and entropy (T ΔS) contributions are calculated at 37°C. Values of TΔS are calculated from the experimental melting entropies, ΔSm, and the free energy determined from, ΔG37 = ΔHm − T37ΔSm, assuming that the enthalpy and entropy of melting are temperature independent. The issue of the constancy of ΔHm and ΔSm as temperature is changed is discussed in the supplementary material. Errors in the total enthalpies and entropies range from 25 to 40 kcal/mol based on averages obtained for measurements on three freshly prepared samples of the PX 6:5 motif and three samples of JX1 6:5, both in low magnesium ion buffer. The greatest contribution to error is related to the evaluation of the baseline, which in itself can cause variations of 5–10% in the areas.
Figure 3.
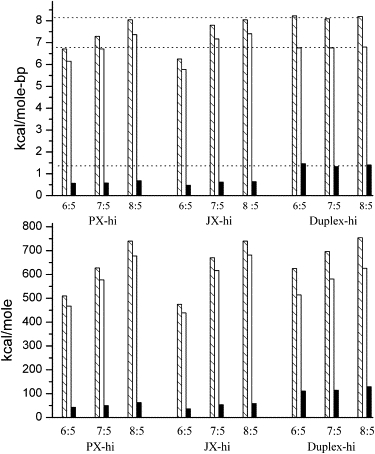
Thermodynamic values of melting for the various PX and JX motifs studied, along with corresponding melting data for B-DNA duplexes containing the same base-pairs, which were calculated from the nearest neighbor model of SantaLucia (33). Bottom panel is the energetics of melting for each structure. The top panel is the melting per basepair in each case. Enthalpies are the hatched bars, entropies (as T37ΔS) are the open bars, and free energies at 37°C are the solid bars. For comparison with the duplex melting data dashed lines are extrapolated across the figure from the ΔH (top), TΔS, and ΔG for the melting of the corresponding two duplex chains in each case. These data are with the buffer containing 125 mM Mg2+ ion.
For purposes of comparison also shown in Fig. 3 and Fig. S3 are estimates of the thermodynamics of melting as calculated from the nearest neighbor treatment of Santa Lucia (33), in which a set of parameters for enthalpy, entropy, and free energy of unfolding for nearest neighbor basepairs is summed over the sequence, assuming a normal B-form duplex DNA structure. A correction for the ion concentrations in solution is made to the total free energy and the total calculated enthalpy change is assumed to be independent of salt concentration. This salt correction has the effect of changing the entropy contribution slightly. The sequences for the 6:5, 7:5, and 8:5 structures were determined for the appropriate chains in the PX crossovers, and the nearest neighbor contributions added up. The corresponding JX1 sequences lead to essentially the same total values of the thermodynamic parameters because the basepairs are equivalent for each individual motif. (There are slight variations in the order of the sequences, which have little effect on the energetic totals.) These nearest neighbor totals are presented in Fig. 3 and Fig. S3 as the bars labeled “Duplex-hi” or “Duplex-lo” for the two salt concentration, and give a basis for comparison of the thermodynamics of melting (or forming) of normal duplex DNA of the same sequence as the PX or JX1 structures. We will discuss the trends in these data below, considering the formation of PX and JX1 derivatives from both single strands and from duplex DNA.
UV-melting transitions
In addition to the calorimetric data, melting transitions were monitored by UV-absorption hyperchromicity effects at 260 nm for purposes of comparison with the DSC melting. These studies also provide a means of determining the repeatability of the melting profiles in both heating and cooling modes. Fig. 4 shows derivative UV-heating and cooling scans for the PX and JX1 structures in 125 mM magnesium ion buffer. The data show heating scans of samples that had been prepared as described in Materials and Methods, followed by a cooling scan, and then a repeat heating scan, all done at a scan rate of 0.15°/min. Several notable features emerge from these studies. First, the repeat heating scans are virtually superimposible with the originals for all of the scans. Although the derivatives are a bit noisy, the positions of peak maxima and the general shapes of the curves are essentially the same in the heating scans. This suggests that on cooling back to room temperature the original structure of each motif is regenerated. The cooling scans, presented as black dots in Fig. 4, show some evidence of kinetic control of the reformation of the structures on lowering the temperature. For example, the PX 6:5 sample in Fig. 4 a on cooling shows complex shifts in the peak maxima of over 10° with the reformation transitions occurring at ∼54°C and 51°C, clearly quite different from the melting at ∼65°C. The corresponding JX1 6:5 structure, on the other hand, shows virtually no shift in the temperature of melting relative to reformation, suggesting no kinetic limitation to reforming the original structure from the single strands. The PX 7:5 and PX 8:5 structures show downward shifts in the temperature of reformation, but not quite so dramatically as for the PX 6:5 structure (Fig. 4, b and c) The JX1 7:5 and 8:5 motifs again show shifts of several degrees on reformation, but significantly less change than for the corresponding PX structures. The data broadly suggest that the JX1 melting or formation is basically reversible with little shift in the Tm whether melting or formation of the JX1 motif. On the other hand, the formation of PX with the additional crossover causes some kinetic limitation, but does indeed form the original structure on cooling 5–10° below the melting temperature.
Figure 4.
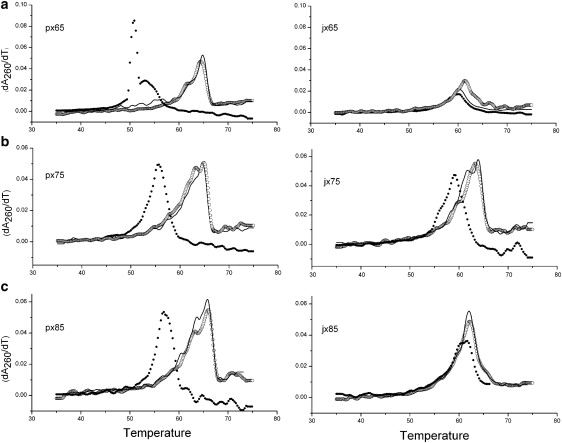
(a) Thermal melting of PX 6:5 (left) and JX1 6:5 (right) monitored by absorbance at 260 nm. Curves are temperature derivatives of absorbance, and the data are for the original upscan (open circles), followed by a downscan (black dots), and then a subsequent upscan (solid curve). All scans are at 0.15°/min, and are in buffer containing 125 mM magnesium ion. (b) Thermal melting of PX 7:5 (left) and JX1 7:5 (right) monitored by absorbance at 260 nm. Conditions and identification of scans are the same as in a. (c) Thermal melting of PX 8:5 (left) and JX1 8:5 (right) monitored by absorbance at 260 nm. Conditions and identification of scans are the same as in a.
One further point regarding UV-melting and DSC transitions is whether the shapes and values of Tm are the same by the two techniques. We find that the Tm values for the DSC scans are a degree or so higher than those of the UV peaks, a perhaps not surprising result, because the scan rates for the DSC runs were at 1°/min, whereas the UV-melts were at 0.15°/min. The general shapes of the melting curves in both cases are basically equivalent (data not shown), providing evidence of overlapping sequential melting through intermediate states.
Evidence of intermediate states in melting of PX and JX1 DNA
The UV-melting and DSC behavior of all of the PX and JX1 structures suggest that one cannot treat the melting transitions as simple reversible, two-state processes, but as the DSC data suggest, it is more likely that the transitions are complex, being made up of smaller domains that melt sequentially. In addition, evaluation of van 't Hoff enthalpies from the DSC transitions leads to values that are much less than the observed calorimetric enthalpies, the value of ΔHvH/ΔHcal being close to 0.5 for most scans. This behavior generally means that the transitions are not two-state, and are a result of sequential melting of intermediates. The UV-melting curves thus cannot be used to determine enthalpies or entropies in an unambiguous way. For this reason we attempt to analyze the thermal transitions in the DSC scans as melting of a set of components that transform in a sequential manner. The software provided by MicroCal (Amherst, MA) provides a method for deconvolution of sequential melting domains that was developed by Freire and Biltonen (34,35). The deconvolution generates Ti and ΔHi values for the conversion from the PX or JX1 motif through intermediates to the final state of melted strands. The solid curves in Fig. 3 and Fig. S3 show calculated curves that come from this sequential analysis of the DSC scans. The deconvolution procedure assumes a model for the sequential melting consisting of either one or two intermediate states:
| (1) |
All of the melting profiles, except that for the PX 6:5, yielded two intermediate states, whereas the PX 6:5 required only one intermediate to obtain the deconvolution of the curve. As can be seen from the calculated curves in Fig. 2 and Fig. S2, there is quite good agreement of the experimental data with the sequential melting model (36). The deconvoluted transitions show relatively small differences between the PX and JX1 forms for formation of I1 and I2, with Ti and enthalpies of the resolved transitions numerically quite close. The largest differences occur for the transformation from I2 to the single strands, for which the JX1 enthalpy values are significantly smaller than the corresponding PX transitions. Because the data provide no way of knowing the exact structural character of the intermediate states, we just conclude that to explain the calorimetric transition data, melting must occur through intermediate states; that is, these transitions are definitely not simple two-state conversions.
The measurements above are subject to the potential criticism that we do not know exactly which nucleotide pairs are melting at any time. As noted above, PX molecules derived from a pair of double helices (perhaps under superhelical stress) containing homology or “PX homology” (18) contain nucleotide pairs derived both from the original double helices, “intraduplex pairs”, and from interactions between the two double helices, “interduplex pairs”. A remarkable and important consequence of the PX structure is that the strands of the two interwrapped helices are completely unlinked, so that PX molecules can be built from DNA dumbbells (18). When this notion is combined with DGGE, it is possible to monitor the melting only of the interduplex pairs. DGGE is a method for separating DNA fragments according to their mobility under increasingly denaturing conditions, in particular increasing concentrations of a urea-formamide mixture (32). If a 7 M urea/40% (vol/vol) formamide gel is regarded as a 100% denaturing condition, each 1% of denaturant corresponds to ∼0.3°C (37). A denaturing gradient perpendicular to the electrophoresis direction enables the visualization of the melting profile. We visualize the melting of PX molecules built from two dumbbells into their constituent units. This approach allows us to know that it is the dissociation of the PX structure that we are monitoring, rather than the dissociation of helices within the strands that do not provide information about the stability of the intermolecular PX unit.
Fig. 5 illustrates the melting of PX 6:5, 7:5, and 8:5 molecules in this context. The chamber was held at 50°C, and the gradient extends from 0% to 50% across the gels. It is evident that the PX molecules are stable at low denaturant concentrations, because only a single band is seen. However, proceeding across the gel from left to right, two other bands of higher mobility emerge, and eventually the original band disappears. The range where both the original band and the higher mobility bands are seen is the melting region. These same species have been melted in traditional UV melting experiments, and the results are qualitatively similar. As with the other experiments, the PX 6:5 molecule contains fewer cohesive nucleotide pairs than the PX 7:5 molecules, which contain fewer than the 8:5 molecules. Table 1 summarizes the melting data, and again it is evident that PX 7:5 molecules are the least stable species, particularly when normalized for the number of interactions. The agreement between these data and the calorimetric and UV melting data confirm that the all experiments are examining features of PX molecules that are involved in their interhelix stability.
Figure 5.
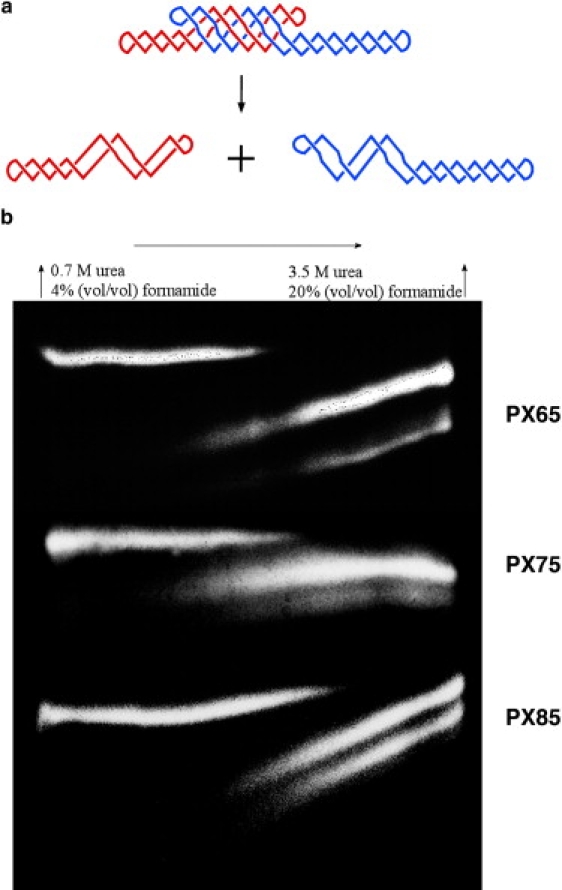
DGGE analysis of PX melting. (a) The PX melting transition is shown, as the complex melts into red and blue dumbbell components. (b) The melting of the three different species of PX is shown. The melting transition is occurring when the initial species (left) and the final species (right) are both present. Differing quantities of the product species are the result of differential labeling.
Table 1.
Comparison of DGGE and thermal melting for PX DNA
| PX65 | PX75 | PX85 | |
|---|---|---|---|
| DGGE | |||
| Melting range (%) | 22–33 | 21–34 | 28–40 |
| Midpoint (%) | 27.5 | 27.5 | 34 |
| Conversion to thermal Tm (°C) | 58.3 | 58.3 | 60.2 |
| Thermal denaturing | |||
| Tm (°C) | 57.6 | 55.1 | 60.8 |
Discussion
Comparisons of data for formation of PX, JX, and duplex DNA structures from single strands
The data in the Fig. 3 and Fig. S3 for the thermodynamics of melting show several expected trends. First, because the number of basepairs in the 6:5, 7:5m and 8:5 motifs increases, the nonnormalized energetics of melting shown in the bottom panels of Fig. 3 and Fig. S3 increase with number of basepairs in the structure. Keep in mind that the data in the figures are for the melting of the various PX and JX1 structures. The enthalpies are endothermic on melting, and the entropy increases on unfolding of DNA, so because the enthalpy is larger than TΔS, the free energy of melting at 37° is also positive. (ΔG37 = ΔHm − T37ΔSm) For the reverse process of forming the folded structures from single strands the thermodynamic properties would be the negative of values shown in Fig. 3 and Fig. S3, indicating a favorable enthalpy of folding that dominates the entropy decrease to yield small negative free energies of folding from the single strands. The data show clearly the enthalpy-entropy compensation effects that most proteins and nucleic acids reveal on folding (38–40), and show that despite significant nonbonding interactions that produce large enthalpic stabilization, the entropic penalties required to fold into highly ordered structures offsets the favorable energies, leading to relatively small stabilizing free energies.
It is informative to examine the data in Fig. 3 and Fig. S3 in the top panels, which show the thermodynamic parameters for melting per basepair. These numbers expose small but significant differences in the PX and JX motifs. For example, in the PX structures there is over a kilocalorie difference in both enthalpy and entropy between the 6:5 and 8:5 configurations per basepair, suggesting greater enthalpic stability for the 8:5, but again compensated by larger entropy effects. The free energies per basepair are rather small (∼500 cal/mol-bp) for all three PX structures. The JX molecules show similar increases per basepair proceeding from 6:5 to 8:5 configurations, although in the low magnesium solutions the 8:5 structure values of enthalpy and entropy actually decrease slightly from the 7:5 motif. Increased salt content (Mg2+ ion) in the buffer causes a trend toward increased enthalpy and entropy of melting when the Mg2+ ion concentration is increased from 12.5 to 125 mM. The free energies per basepair are also somewhat larger in the higher salt concentration (∼500 cal/mole-bp vs. 400 cal/mol base-bp). These results can be interpreted as a result of lowering the electrostatic repulsion between phosphates on individual DNA chains from magnesium ion condensation along the duplex (41), and from the observation from 25Mg NMR experiments that Mg2+ can bind specifically at phosphate sites in DNA (42). Both of these effects would favor formation of hydrogen bonding and stacking interactions in higher magnesium ion concentration. Again the free energy for formation of the PX and JX1 structures from single strands, does not change much with ion concentration because of the compensation between a stabilizing enthalpy and the destabilizing entropy penalty for forming complex organized structures, including the condensation of ions associated with phosphate charges.
Per basepair there is another comparison that is noticeable. The thermodynamic values of the PX 6:5 and JX1 6:5 are clearly both significantly lower than the 7:5 or 8:5 derivatives, and further, the JX1 6:5 has enthalpy, entropy, and free energy values below those of the PX 6:5. In the high Mg ion solutions the PX 8:5 and JX1 8:5 have the same values within the experimental error, and for the PX 7:5 the enthalpies and entropies are a bit lower than the JX1 7:5. Similar results are obtained in the lower salt solutions; that is, the PX 6:5 derivative is more stable than the JX1 6:5 per basepair. This result suggests that the PX 6:5 crossover structure would be the preferred form relative to the JX1, juxtaposed strand structure. However, there is little thermodynamic advantage for formation of the crossover PX derivative relative to JX1 for the 7:5 or 8:5 structures.
More details of the effects of number of basepairs per twist are shown in the comparison of PX and JX1 derivatives with the calculated properties per basepair for normal duplex DNA, which are also shown in the top panels of Fig. 3 and Fig. S3. The enthalpy values for duplex melting are quite typical (∼8 kcal/mol-bp) for sequences of 50–60% GC basepairs (33), which is what these structures contain. The typical stabilities (free energies) of ∼1.5 kcal/mol-bp are a result of significantly lower entropies of formation of duplex basepairs, relative to the entropies for the PX and JX1 derivatives. It is interesting to compare the PX 8:5 and JX1 8:5 values with duplex parameters. All three structures have comparable enthalpies of ∼8 kcal/mol-bp (see top dotted line in Figs. 3 and S3), but notice that the entropies for the PX and JX1 forms are significantly larger than for duplex, so that the greater stability of duplex DNA is a result of paying less of an entropy penalty to form the duplex relative to the crossover or juxtaposed structures. The JX1 7:5 shows similar trends, but the PX 7:5 enthalpy and entropies are a bit smaller than the corresponding duplex parameters. Again the outliers in these data are the PX 6:5 and JX1 6:5, both of which show significantly lower values for all three thermodynamic properties. The conclusion one is led to is that for the 8:5 and perhaps the 7:5 structures, the enthalpies are comparable to those of the normal duplex DNA, but the entropies of forming crossovers and juxtaposed strands, which are more compact and complicated than normal duplex, causes the entropy decrease to be larger, and thus the structures are less stable than the simple duplex. This penalty results in about a kcal/mol-bp greater stability for the duplex relative to either JX1 or PX forms. The data also suggest that the PX 6:5 and JX1 6:5 structures, most likely through differences in hydration effects, have significantly lower enthalpies of formation relative to duplex DNA of comparable sequence, and again the entropy decrease on forming such structures is a greater fraction of the enthalpy, leading to lower free energy to form the crossover structure from single strands. Further, the juxtaposition of a section of the strand in the JX1 form is even less similar to normal duplex paired bases and is more destabilized by the entropy-enthalpy compensation.
Formation of crossover structures from duplex DNA
There have been several studies regarding the thermodynamics of forming junctions between two duplex DNA chains (20,21). The formation of a single immobile Holliday-type four-arm junction from separate duplexes is slightly unfavorable (free energy difference of +1.1 kcal/mol), with a rather large unfavorable enthalpy of formation (+27.1 kcal/mol) (21). This branched junction is not exactly equivalent to a parallel crossover in our study, but both structures require bringing two duplex chains in close proximity to form basepairs on opposite chains. We can compare these effects by looking at the thermodynamic parameters for the process of converting two duplex chains to a PX or JX1 structure:
| (2) |
This process is not the same as forming the crossover structures from single strands, as discussed above. Fig. S4 shows pictorially the results of subtracting the data in Fig. 3 and Fig. S3 for the duplex formation from single strands, obtained from the data of Santa Lucia (33), from the corresponding values for PX (or JX1) formation from single strands, which should correspond to the conversion illustrated in Eq. 2. The data in Fig. S4 show that it definitely is unfavorable to form polycrossover structures from intact relaxed duplexes, the positive, unfavorable free energy being between 60–75 kcal/mol in high Mg2+ concentration, and 35–50 kcal/mol in low Mg2+. These numbers, compared with the negative free energies of formation of the crossover structures from single strands, suggest that it is the favorable contributions from basepairing and stacking when the single strands of the right sequences react with their complementary bases along the chains, that provide the necessary stabilization for PX or JX1 structures. If the equivalent basepairing sequences are rearranged such as to favor duplex formation, there is no doubt that the duplex forms would be preferred over the crossover structures by a rather significant amount. The numerical differences presented in Fig. S4 factor out the basepair hydrogen bonding and stacking formation energetics, so that the remaining numbers should show the thermodynamic effects of bringing two isolated duplexes together to form the paranemic crossover structures. Any excess solvation or ion condensation or conformational effects that have energetic and entropic consequences should be included in these effects.
It is interesting in comparing the 6:5, 7:5, and 8:5 derivatives that whereas the free energies are numerically similar, there are rather major differences in the enthalpies and entropies among the isomers. For the PX 6:5 and JX1 6:5 homologs the enthalpies are very endothermic with a counterbalancing entropy increase, which leads to relatively large positive free energy. It is intriguing to attribute these effects to changes in solvation (hydration) as the two helices are brought together to form crossovers. The large positive enthalpy could be a result of removal of water from the surfaces of duplex DNA as formation of the tightly bound crossovers occurs. The relatively large entropy increases would correlate with release of the hydration water to bulk solvent. The enthalpy dominates so that there is insufficient energy to yield a net conversion. That de-hydration effects may be important in determining the magnitudes of the thermodynamic values for forming PX and JX1 DNA is supported by the work of Rau et al. (43) and Leikin et al. (44) on the condensation of DNA duplexes under osmotic stress. The interhelical spacing for the condensed hexagonal phase for DNA duplex strands is dependent on water activity, and these authors find that in the presence of a variety of ions release of structured water in the vicinity of the duplex to bulk solvent is responsible for the large entropic increases associated with the formation of the condensed state of the DNA. As the duplex chains approach each other there is an exponential increase in repulsive energy that is associated with the “hydration force”. For example, to bring duplex chains to an interhelical distance of ∼7 Å in a hexagonal configuration requires ∼12 kcal for 40 basepairs (44). Obviously, to form PX and JX1 crossovers, the helices would have to be in near contact, so that dehydration free energies would be significantly higher. These results add support to the idea that the formation of crossover structures, particularly for the PX 6:5 and JX1 6:5, have such positive enthalpies and entropies because bringing the duplexes into the closely packed structure of these motifs requires significant dehydration of the duplexes to form compact PX and JX1 contacts.
Proceeding to the 7:5 and 8:5 structures the positive enthalpies decrease and the entropy terms actually become negative, but again the numbers lead to large destabilizing positive free energies. In these cases if we again assume solvation changes are responsible for the magnitudes and signs of the thermodynamic values, the results suggest that there is less water displaced for the 7:5 and 8:5 motifs and the resulting enthalpy and entropy changes reflect more basic losses in degrees of freedom in forming the crossovers relative to duplex strands. In addition, ion condensation on or near the phosphate sites would contribute negative entropy, which may be more important in the 7:5 and 8:5 homologs. The data in Fig. S4 suggest that the 7:5 and 8:5 derivatives are more open and solvent accessible, perhaps a consequence of forcing 7 and 8 basepairs in the major groove per helical twist in these structures. A pair of theoretical studies by Maiti et al. (28,29) on PX and JX1 crossover structures supports these ideas. Based on molecular dynamics simulations on PX-5:5, 6:5, 7:5, 8:5, and 9:5 crossover structures, several features become apparent (28). First, the simulations show that all of the PX motifs are more rigid than normal B-DNA, but there are variations in the overall flexibility in the series of compounds. Looking at dynamic deviations from the average structures obtained through the simulations, the PX 6:5 and 8:5 seem to show less flexibility whereas the PX 7:5 exhibits large fluctuations from the average structure. Calculation of structural strain energies shows that the PX 6:5 is the least structurally strained, whereas all of the others have significantly higher instability from structural strain. In examining the helical properties of the various forms, PX 6:5, as expected, has properties such as twist, roll, tilt, and rise that are quite similar to normal B-DNA. The other forms deviate more from the canonical B-DNA properties somewhat. All of these simulations are consistent with the idea that the PX 6:5 structurally is the most stable configuration in the PX series. In another theoretical study similar calculations were carried out on JX motifs containing varying numbers of missing crossovers (29). For example, JX1 is the same structure as the PX 6:5 derivative except that one of the crossovers is missing. JX2, JX3, and JX4 have 2, 3, and 4 missing crossovers, respectively. As expected, as the number of missing crossovers increases, the flexibility of the molecule increases, and the structural stability decreases. The JX1 is of interest to us because it would correspond to the JX1 6:5 derivative in our study. The simulations show that both PX 6:5 and JX1 (JX1 6:5) are more structurally stable than the others, and that there is a small increase in strain energy for JX1 relative to PX 6:5. The simulation results are basically consistent with the thermodynamic arguments we presented above. First, the 6:5 compounds are the easiest to form, but also sacrifice the most solvation on formation of the structure. The net thermodynamic stability is thus lowered because the gains in inherent stability are offset by changes in hydration. It seems consistent that the 7:5 and 8:5 are more flexible and open, more degrees of freedom, and thus perhaps more likely to have a larger surface area exposed to solvent, leading to lower enthalpy changes and more negative entropy changes for conversion from duplex to crossover structures, as shown in Fig. S4. Thus, although the six crossover structures all have very positive values of the free energy for formation from duplex chains, the reasons for the instability are actually quite different in each case. The conclusions from these thermodynamic analyses of the PX and JX1 data are that formation of crossover structures from single strands is favorable, whereas formation from relaxed duplex chains is not. The reason that formation of crossovers from the single strands has a negative free energy is that the enthalpy and entropy of basepairing and stacking is great enough to overcome the unfavorable energetics of de-hydration as the total surface area decreases in bringing the strands together. It thus seems that for parallel duplexes to form crossovers, other sources of energy, such as from supercoiling or from protein complexes that open the duplexes to single strands, are required to compensate for the unfavorable free energy of formation from duplex chains.
Another point from these data is illustrated in Table 2, showing the data for the conversion of JX ⇒ PX for the three derivatives. Although all show very small free energies, essentially identical within the experimental error, the 6:5 and 8:5 compounds seem to have a slight free energy advantage for forming PX from JX1 crossover structures. It is notable that for the 6:5 conversion, the enthalpies and entropies are negative, so the small negative free energy is a result of excess negative energy of formation. The 7:5 motif shows positive values for both enthalpy and entropy and for the 8:5 there is virtually no difference between the PX and JX1 thermodynamic values. The DGGE experiments are in agreement with this finding. These results point out that the subtle differences in structural features can influence the thermodynamics rather drastically.
Table 2.
Thermodynamic values for the conversion JX1 ⇒ PX
| ΔH | T37ΔS | ΔG37 | |
|---|---|---|---|
| 6:5hi | −35 | −28 | −7 |
| 7:5hi | +43 | +40 | +3 |
| 8:5hi | 0 | +5 | −5 |
| 6:5lo | −55 | −53 | −2 |
| 7:5lo | +6 | +10 | −4 |
| 8:5lo | −31 | −25 | −6 |
All values are in kcal/mol.
Recognition of homology by formation of PX DNA
Wilson (45) and McGavin (46) proposed early models for four-stranded recognition of homology. Other data are consistent with the formation of four-stranded structures between homologous duplex molecules (47,48). The recent work of Wang et al. (17) provides evidence that double helices can indeed recognize double helices in a superhelical context. An inherent feature suggested by the experiments of Wang et al. (17), is the formation of a structure resembling PX-DNA, which requires a series of parallel crossovers to be formed, but in a paranemic context. In the comparison of PX and JX1 we have examined the formation of a parallel crossover in the context of a structure (JX1) that has already paid the price of forming previous parallel crossovers. According to the results of this study, the conclusion seems that certainly in the 6:5 and 8:5 motifs there is sufficient free energy available so that a small preference would be given to the parallel crossover structure relative to juxtaposed chains in high magnesium ion concentration. Even in the 7:5 structure the free energy difference between PX and JX is only a few kcal/mol. Thus, for the transformation,
in high magnesium ion buffer there is sufficient free energy to yield a parallel crossover and although there is no dramatic energetic advantage of one form over the other, it seems that both could be present in a simple equilibrium mixture. Finally, even though the PX 6:5 motif, overall is less thermodynamically stable than the other structures with more nucleotides in the major groove, it does show the greatest difference between the PX and JX forms. The PX 6:5 structure of the three possibilities is closest to the nucleotide count per helical turn of normal B-form DNA. (11 vs. 10.5) The other two motifs with totals of 12 or 13 would have to accommodate the extra nucleotide pairs by either twisting or writhing in such a way that could be responsible for the destabilization observed. The insight into these twisting or writhing adjustments provided by molecular dynamics simulations of the PX structures actually shows that the 7:5 motif is the most flexible of the three (28,29).
Conclusions
We believe that the thermodynamic data presented in this study suggest that the PX DNA motif could be used as a model for homology recognition. The results suggest that in relaxed conditions, a system that meets the homology requirements for the PX form would prefer the PX motif relative to juxtaposed duplexes, particularly for the 6:5 structure. It is also important to recognize that we have not yet established the type of PX structure that is formed in superhelical DNA; indeed, the most dramatic results are seen with a 10:5 molecule that is only marginally stable in a four-stranded context (X. Wang, X. Zhang, C. Mao, and N. C. Seeman, unpublished observations). The PX motif is more stable in higher magnesium ion content and the 7:5 and 8:5 structures have a slightly lower tendency to form PX from JX1. However, PX 6:5 seems to represent a good model for the formation of fused heteroduplexes that could be a general intermediate in recombination events. Whether there would be kinetic limitations to forming these motifs is an open question. The presence of an apparent thermodynamic driving force for the formation of the paranemic crossover from juxtaposed duplexes indicates there is a potential for this conversion to occur.
Supporting Material
Materials and methods, two tables, references, and four figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00974-6.
Supporting Material
Acknowledgments
This work has been supported by the National Institute of General Medical Science (GM-29554), the National Science Foundation (DMI-0210844, EIA-0086015, CCF-0432009, CCF-0523290, CTS-0548774, CTS-0608889), Army Research Office (48681-EL, W911NF-07-1-0439), Department of Education (subcontract from the Research Foundation of the State University of New York; DE-FG02-06ER64281), the office of Naval Research (N000140910181), and the W. M. Keck Foundation (grant to N.C.S.).
References
- 1.Low K.B. Academic Press; Orlando, FL: 1988. The Recombination of Genetic Material. [Google Scholar]
- 2.Craig N.L. The mechanism of conservative site-specific recombination. Annu. Rev. Genet. 1988;22:77–105. doi: 10.1146/annurev.ge.22.120188.000453. [DOI] [PubMed] [Google Scholar]
- 3.Messelson M.S., Radding C.M. General model for genetic recombination. Proc. Natl. Acad. Sci. USA. 1975;72:358–361. doi: 10.1073/pnas.72.1.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dressler D., Potter H. Molecular mechanisms in genetic recombination. Annu. Rev. Biochem. 1982;51:727–761. doi: 10.1146/annurev.bi.51.070182.003455. [DOI] [PubMed] [Google Scholar]
- 5.Holliday R. Mechanism for gene conversion in fungi. Genet. Res. 1964;5:282–304. doi: 10.1017/S0016672308009476. [DOI] [PubMed] [Google Scholar]
- 6.Churchill M.E.A., Tullius T.D., Kallenbach N.R., Seeman N.C. A Holliday recombination intermediate is twofold symmetric. Proc. Natl. Acad. Sci. USA. 1988;85:4653–4656. doi: 10.1073/pnas.85.13.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooper J.P., Hagerman P.J. Gel electrophoretic analysis of the geometry of a DNA 4-way junction. J. Mol. Biol. 1987;198:711–719. doi: 10.1016/0022-2836(87)90212-9. [DOI] [PubMed] [Google Scholar]
- 8.Lilley D.M.J., Clegg R.M. The structure of the 4-way junction in DNA. Annu. Rev. Biophys. Biomol. Struct. 1993;22:299–328. doi: 10.1146/annurev.bb.22.060193.001503. [DOI] [PubMed] [Google Scholar]
- 9.Clegg R.M., Murchie A.I., Lilley D.M.J. The solution structure of the 4-way DNA junction at low salt conditions—a fluorescence resonance energy transfer analysis. Biophys. J. 1994;66:99–109. doi: 10.1016/S0006-3495(94)80765-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mao C., Sun W., Seeman N.C. Designed two-dimensional DNA Holliday junction arrays visualized by atomic force microscopy. J. Am. Chem. Soc. 1999;121:5437–5443. [Google Scholar]
- 11.Sha R., Liu F., Millar D.P., Seeman N.C. Atomic force microscopy of parallel DNA branched junction arrays. Chem. Biol. 2000;7:743–751. doi: 10.1016/s1074-5521(00)00024-7. [DOI] [PubMed] [Google Scholar]
- 12.Ho P.S., Eichman B.F. The crystal structures of DNA Holliday junctions. Curr. Opin. Struct. Biol. 2001;11:302–308. doi: 10.1016/s0959-440x(00)00219-0. [DOI] [PubMed] [Google Scholar]
- 13.Sigel N., Alberts B. Genetic recombination—nature of a crossed strand exchange between 2 homologous DNA molecules. J. Mol. Biol. 1972;71:789–793. doi: 10.1016/s0022-2836(72)80039-1. [DOI] [PubMed] [Google Scholar]
- 14.McKinney S.A., Freeman A.D.J., Lilley D.M.J., Ha T.J. Observing spontaneous branch migration of Holliday junctions one step at a time. Proc. Natl. Acad. Sci. USA. 2005;102:5715–5720. doi: 10.1073/pnas.0409328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiner B.M., Kleckner N. Chromosome pairing via multiple interstitial interactions before and during meiosis in yeast. Cell. 1994;77:977–991. doi: 10.1016/0092-8674(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 16.Kleckner N., Weiner B.M. Potential advantages of unstable interactions for pairing of chromosomes in meiotic, somatic and premeiotic cells. Cold Spring Harb. Symp. Quant. Biol. 1993;58:553–565. doi: 10.1101/sqb.1993.058.01.062. [DOI] [PubMed] [Google Scholar]
- 17.Reference deleted in proof.
- 18.Shen Z., Yan H., Wang T., Seeman N.C. Paranemic crossover DNA: a generalized Holliday structure with applications in nanotechnology. J. Am. Chem. Soc. 2004;126:1666–1674. doi: 10.1021/ja038381e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seeman N.C. Nicks and nodes and nanotechnology. Nano Lett. 2001;1:22–26. [Google Scholar]
- 20.Lu M., Guo Q., Marky L.A., Seeman N.C., Kallenbach N.R. Thermodynamics of DNA chain branching. J. Mol. Biol. 1992;223:781–789. doi: 10.1016/0022-2836(92)90989-w. [DOI] [PubMed] [Google Scholar]
- 21.Marky L.A., Kallenbach N.R., McDonough K.A., Seeman N.C., Breslauer K.J. The melting behavior of a nucleic acid junction: a calorimetric and spectroscopic study. Biopolymers. 1987;26:1621–1634. doi: 10.1002/bip.360260912. [DOI] [PubMed] [Google Scholar]
- 22.Sumners D.W. Untangling DNA. Math. Intelligencer. 1990;12:71–80. [Google Scholar]
- 23.Zhang X., Yan H., Shen Z., Seeman N.C. Paranemic cohesion of topologically closed DNA molecules. J. Am. Chem. Soc. 2002;124:12940–12941. doi: 10.1021/ja026973b. [DOI] [PubMed] [Google Scholar]
- 24.Shih W.M., Quispe J.D., Joyce G.F. A 1.7-kilobase single-stranded DNA that folds into a nanoscale octahedron. Nature. 2004;427:618–621. doi: 10.1038/nature02307. [DOI] [PubMed] [Google Scholar]
- 25.Yan H., Zhang X., Shen Z., Seeman N.C. A robust DNA mechanical device controlled by hybridization topology. Nature. 2002;415:62–65. doi: 10.1038/415062a. [DOI] [PubMed] [Google Scholar]
- 26.Liao S., Seeman N.C. Translation of DNA signals into polymer assembly instructions. Science. 2004;306:2072–2074. doi: 10.1126/science.1104299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding B., Seeman N.C. Operation of a DNA robot arm inserted into a 2D DNA crystalline substrate. Science. 2006;314:1583–1585. doi: 10.1126/science.1131372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiti P.K., Pascal T.A., Vaidehi N., Heo J., Goddard W.A. Atomic-level simulations of Seeman DNA nanostructures: the paranemic crossover in salt solution. Biophys. J. 2006;90:1463–1479. doi: 10.1529/biophysj.105.064733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maiti P.K., Pascal T.A., Vaidehi N., Goddard W.A. The stability of Seeman JX DNA topoisomers of paranemic crossover (PX) molecules as a function of crossover number. Nucleic Acids Res. 2004;32:6047–6056. doi: 10.1093/nar/gkh931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu T.-J., Seeman N.C. DNA double crossover molecules. Biochemistry. 1993;32:3211–3220. doi: 10.1021/bi00064a003. [DOI] [PubMed] [Google Scholar]
- 31.Schwacha A., Kleckner N. Identification of double Holliday junctions as intermediates in meiotic recombination. Cell. 1995;83:783–791. doi: 10.1016/0092-8674(95)90191-4. [DOI] [PubMed] [Google Scholar]
- 32.Fischer S.G., Lerman L.S. 2-Dimensional fractionation of DNA restriction fragments in polyacrylamide gels. Cell. 1979;16:191–200. doi: 10.1016/0092-8674(79)90200-9. [DOI] [PubMed] [Google Scholar]
- 33.SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freire E., Biltonen R.L. Statistical mechanical deconvolution of thermal transitions in macromolecules 1. Theory and application to homogeneous systems. Biopolymers. 1978;17:463–479. [Google Scholar]
- 35.Freire E., Biltonen R.L. Statistical mechanical deconvolution of thermal transitions in macromolecules 3. Application to double-stranded to single-stranded transitions of nucleic acids. Biopolymers. 1978;17:497–510. [Google Scholar]
- 36.Spink C.H. Differential scanning calorimetry. Methods Cell Biol. 2008;84:115–141. doi: 10.1016/S0091-679X(07)84005-2. [DOI] [PubMed] [Google Scholar]
- 37.Abrams E.S., Stanton V.P. Use of denaturing gradient gel electrophoresis to study conformational transitions in nucleic acids. Methods Enzymol. 1992;212:71–104. doi: 10.1016/0076-6879(92)12006-c. [DOI] [PubMed] [Google Scholar]
- 38.Liu L., Yang C., Guo Q.X. A study on the enthalpy-entropy compensation in protein unfolding. Biophys. Chem. 2000;84:239–251. doi: 10.1016/s0301-4622(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 39.Petruska J., Goodman M.F. Enthalpy-entropy compensation in DNA melting thermodynamics. J. Biol. Chem. 1995;270:746–750. doi: 10.1074/jbc.270.2.746. [DOI] [PubMed] [Google Scholar]
- 40.Rentzeperis D., Kupke D.W., Marky L.A. Volume changes correlate with entropies and enthalpies in the formation of nucleic acid homoduplexes—differential hydration of A-conformation and B-conformation. Biopolymers. 1993;33:117–125. doi: 10.1002/bip.360330111. [DOI] [PubMed] [Google Scholar]
- 41.Record M.T., Anderson C.F., Lohman T.M. Thermodynamic analysis of ion effects on binding and conformational equilibria of proteins and nucleic acids—roles of ion association or release, screening and ion effects on water activity. Q. Rev. Biophys. 1978;11:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- 42.Berggren E., Nordenskold L., Braulin W.H. Interpretation of 25Mg spin relaxation in Mg-DNA solutions—temperature variation and chemical exchange effects. Biopolymers. 1992;32:1339–1350. doi: 10.1002/bip.360321008. [DOI] [PubMed] [Google Scholar]
- 43.Rau D.C., Lee B., Parsegian V.A. Measurement of the repulsive force between poly-electrolyte molecules in ionic solution—hydration forces between parallel double helices. Proc. Natl. Acad. Sci. USA. 1984;81:2621–2625. doi: 10.1073/pnas.81.9.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leikin S., Rau D.A., Parsegian V.A. Measured entropy and enthalpy of hydration as a function of distance between DNA double helices. Phys. Rev. A. 1991;44:5272–5278. doi: 10.1103/physreva.44.5272. [DOI] [PubMed] [Google Scholar]
- 45.Wilson J.H. Nick-free formation of reciprocal heteroduplexes—simple solution to the topological problem. Proc. Natl. Acad. Sci. USA. 1979;76:3641–3645. doi: 10.1073/pnas.76.8.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGavin S. Models of specifically paired like (homologous) nucleic acid structures. J. Mol. Biol. 1971;55:293–298. doi: 10.1016/0022-2836(71)90201-4. [DOI] [PubMed] [Google Scholar]
- 47.Inoue S., Sugiyama S., Travers A.A., Oyama T. Self-assembly of double-stranded DNA molecules at nanomolar concentrations. Biochemistry. 2007;46:164–171. doi: 10.1021/bi061539y. [DOI] [PubMed] [Google Scholar]
- 48.Baldwin G.S., Brooks N.J., Robson R.E., Wynveen A., Goldar A. DNA double helices recognize mutual sequence homology in a protein free environment. J. Phys. Chem. B. 2008;112:1060–1064. doi: 10.1021/jp7112297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.