

Abstract
A fundamental question concerning the ClC Cl−/H+ antiporters is the nature of their proton transport (PT) pathway. We addressed this issue by using a novel computational methodology capable of describing the explicit PT dynamics in the ClC-ec1 protein. The main result is that the Glu203 residue delivers a proton from the intracellular solution to the core of ClC-ec1 via a rotation of its side chain and subsequent acid dissociation. After reorientation of the Glu203 side chain, a transient water-mediated PT pathway between Glu203 and Glu148 is established that is able to receive and translocate the proton via Grotthuss shuttling after deprotonation of Glu203. A molecular-dynamics simulation of an explicit hydrated excess proton in this pathway suggests that a negatively charged Glu148 and the central Cl− ion act together to drive H+ to the extracellular side of the membrane. This finding is consistent with the experimental result that Cl− binding to the central site facilitates the proton movement. A calculation of the PT free-energy barrier for the ClC-ec1 E203V mutant also supports the proposal that a dissociable residue is required at this position for efficient delivery of H+ to the protein interior, in agreement with recent experimental results.
Introduction
The ClC family of chloride transport membrane proteins includes both Cl−-selective ion channels and H+-coupled secondary active Cl− transporters or “antiporters” (1–4). Channels mediate passive C1− diffusion/conduction, whereas transporters or exchangers transport ions against their electrochemical gradient by coupling the uphill movement of H+ to the downhill movement of Cl− ions, or vice versa.
ClC-ec1 is a ClC homolog from Escherichia coli. It belongs to the transporter subclass and stoichiometrically exchanges two Cl− ions for one H+ (1). This protein participates in the extreme acid-resistance response that enables the bacterium to survive in the acidic environment of the stomach (1,5). Its fundamental and practical importance as a potential target for novel antibiotics has attracted great interest over the last decade. The determination of a high-resolution crystal structure of ClC-ec1 in 2002 (6) not only prompted many functional studies of eukaryotic chloride channels (7–9), it also provided opportunities to investigate the coupling mechanism of the ClC Cl−/H+ antiporters.
Crystallographic, electrophysiological, and site-directed mutagenesis studies have yielded considerable insights into the structure-function relations of ClC-ec1 (10–15). One key mechanistic insight is that two proton transfer residues on each side of the membrane (Glu148 and Glu203) are required for proton transport (PT). Substitution of a neutral residue for either Glu148 or Glu203 leads to complete loss of the H+ translocation (10,11). On the extracellular side of the membrane, Glu148 mediates PT between the extracellular solution and the protein interior, and a movement of the Glu148 side chain is believed to link H+ and Cl− binding at some point in the transport cycle. On the opposite side of the membrane, the intracellular-facing Glu203 is thought to shuttle protons between the intracellular solution and the core of ClC-ec1. Glu203 is partially buried in the intracellular vestibule and resides away from the Cl− binding region. These structural features suggest that ClC-ec1 contains separate Cl− and H+ pathways that overlap at the location of Glu148 and then diverge toward the intracellular side (11,12).
The two proton-binding sites, Glu148 and Glu203, are separated by ∼15 Å. The way in which protons translocate between them, however, remains elusive. Certain experimental investigations (12,15) have focused on a highly conserved tyrosine residue, Tyr445, which lies approximately halfway between Glu148 and Glu203 and coordinates the central Cl− ion. Mutating Tyr445 to phenylalanine or tryptophan demonstrates no change in the protein's exchange transport function, suggesting that the hydroxyl group of Tyr445 is not essential to the PT of ClC-ec1. An examination of Tyr445 variants, however, revealed a correlation between the proton coupling efficiency and the central Cl− site occupancy, indicating that Cl− binding somehow facilitates the proton movement (12).
Several computational methods, ranging from continuum electrostatic calculations to kinetic Monte Carlo and classical molecular-dynamics (MD) simulations, have been applied to investigate anion conduction and gating mechanisms in ClC channels (16–21). However, systematic studies of the PT pathway and the mechanism of the H+ coupling are still lacking. A pore-searching algorithm, TransPath, was applied to the crystal structure of ClC-ec1 to explore the proton access pathway from the extracellular solution to Glu148 (22), as well as a possible proton pathway linking Glu148 and Glu203 (23). However, since PT pathways are often dynamic in nature, analysis of static structures alone is insufficient.
In this study, therefore, we aimed to identify the dynamical PT pathway that connects Glu203 to Glu148, and to reveal the nature of the proton pathway by virtue of novel atomistic MD computer simulations. We propose that H+ are carried from the intracellular solution to the core of ClC-ec1 by a reorientation of the Glu203 side chain. After this reorientation, a transient (metastable) water-mediated PT pathway is established, through which a hydrated proton can translocate along a hydrogen-bonded water chain from Glu203 to Glu148 after deprotonation of Glu203. The multistate empirical valence bond (MS-EVB) MD method (24–28) is utilized here to simulate the explicit translocation of an excess proton along this water chain. Additional simulations of the PT in the mutant E203V demonstrate that a dissociable proton-binding residue is required at this position for efficient delivery of H+ from the intracellular solution to the core of ClC-ec1 (29). The results presented in this work provide, for the first time to our knowledge, a detailed picture of the transient water distribution in the PT pathway, the side-chain motion of Glu203, and the resulting H+ translocation pathway.
Materials and Methods
System preparation
The ClC system was set up and equilibrated for 7 ns with the use of standard, classical MD as described in the Supporting Material. Various crevices and cavities were identified within the equilibrated structure of ClC-ec1. As many as seven water molecules could be placed in these crevices and cavities in long-lived metastable configurations, as discussed further below. As also discussed in detail below, protons were shown to then move along the hydrogen-bonded network defined by a reoriented Glu203 side chain, transiently bound water molecules, and Glu148. The free-energy profile, i.e., the potential of mean force (PMF), for the reorientation of the Glu203 side chain was calculated as a function of the χ2 angle using umbrella sampling. Twelve windows at an interval of 30° were sampled, and a force constant of 40 kcal/mol/rad2 was used for the umbrella restraint potential. The structure of the E203V mutant was obtained by replacing Glu203 with valine in the equilibrated structure of the wild-type protein and then equilibrating the system with standard MD as described in the Supporting Material.
MS-EVB simulations of water mediated PT
The MS-EVB model (24–28) can explicitly treat Grotthuss hopping of hydrated excess protons between water molecules within the framework of MD simulations, and, in turn, it can characterize the detailed solvation and transport of these excess protons. The low computational cost of this model relative to ab initio MD simulations has permitted the study of PT in a variety of aqueous and biomolecular systems (24,25). The second-generation MS-EVB2 model (28) with the third-generation state selection algorithm (30) was used to explicitly simulate the PT within ClC-ec1. To make the MS-EVB simulations more computationally tractable, a reduced system was simulated as described in the Supporting Material.
The charge defect (net unit positive charge) arising from the excess proton propagates along the hydrogen-bonded network via Grotthuss hopping. Due to the delocalized nature of the charge defect associated with the excess hydrated proton, its position was described by the center of excess charge (CEC) (31) (see also Supporting Material). The free-energy profile for this CEC migration (PT) process through the water-mediated pathway was calculated using umbrella sampling with the weighted histogram analysis method (WHAM) (32,33). The free-energy profile for the PT in the E203V mutant was calculated in the same way as for the wild-type protein.
Results
Functional water molecule configurations
According to the Grotthuss shuttling mechanism, excess protons (H+) translocate within proteins via the rearrangement of hydrogen and covalent bonds through protonatable residues and water molecules (24,25,34–37). Because the two proton-binding sites, Glu148 and Glu203, are ∼15 Å apart, proton shuttling by water molecules or other groups between them is required.
Even though water is ubiquitous in biomolecular systems, crystal structures of proteins often will not reveal the presence of certain water molecules because these molecules can be quite mobile. Furthermore, certain configurations of water relevant to PT phenomena in proteins involve rare events (fluctuations) to form transient water “chains” or “wires”. The latter are characteristic of activated (Arrhenius law) rate processes that have an exponentially small probability of being observed in their transition state (or, in this case, their transition “pathway”) configurations. Such configurations are not likely to be seen in a crystal structure unless the protein somehow becomes trapped in such a state during crystallization. The fact that the experimentally observed proton flux is about one proton per millisecond, whereas the natural proton hopping time through water molecules in the absence of additional energetic barriers is 8–9 orders of magnitude faster, clearly supports the assertion that the PT process in the ClC-ec1 antiporter system is a rare (i.e., activated) event. Correspondingly, many (or most) of the participating proton shuttling water molecules from these activated events are not likely to be seen in crystal structures, even ones of very high resolution.
Despite the small likelihood of seeing functionally relevant water molecules for the PT pathway in crystal structures, to help identify possible pathways between Glu148 and Glu203 we first scrutinized the crystal structure of ClC-ec1. Several structural features of the protein are noted below. One feature is that on the intracellular side of the membrane, Glu203 forms a salt bridge with Arg28 from the other subunit. At the same time, it is within a hydrogen-bond distance to Glu113 from the same subunit. Two crystal water molecules are found near the side chain of Glu203. One of them is hydrogen bonded to the amide nitrogen atom of Tyr445. A second feature of the ClC-ec1 protein structure is that the side chain of Glu148 on the extracellular side of the membrane sits ∼4 Å above the central Cl− ion, which is coordinated by the hydroxyl group of Tyr445. Crystal water molecules are not seen in the anion conduction pore. A third feature is that Thr407 is hydrogen bonded to Glu202, which is located near the dimer interface.
Overall, ClC-ec1 is hydrophobic in the region between Glu148 and Glu203 (not including the above-mentioned locations), and thus Tyr445, Thr407, and Glu202 are the only residues that could mediate PT between Glu148 and Glu203. However, the hydroxyl group of Tyr445 was eliminated as a proton transfer site by a recent experiment (12). Moreover, the proton movement was found to be only partially uncoupled in the mutant E202Q (11), indicating that Glu202 apparently is not important for the PT. These experimental findings lead us to propose that transient water configurations play a significant role in the PT behavior of ClC-ec1. It is now well established (38,39) that water can transiently fill cavities in proteins (even hydrophobic ones), so this proposal is completely in line with the current understanding of such phenomena. Furthermore, a similar behavior has also been suggested (40–42) for cytochrome c oxidase; in that case, a glutamic acid deprotonates to a transient chain of water molecules, filling a hydrophobic cavity of the protein, and the excess proton then translocates along that chain to a proton loading site (43).
The DOWSER program (44) is often used to estimate the internal hydration of proteins, and in this work was used for that purpose in ClC-ec1 before the classical MD simulations were conducted (see Supporting Material). DOWSER searches for internal cavities and assesses the hydrophilicity of these cavities by calculating the interaction energy between a water molecule in the cavity and the surrounding protein atoms. The main assumption utilized in DOWSER is that the water-protein interaction energy represents the key factor involved in protein cavity hydration; however, the program does not include the water-water hydrogen-bonding energy for the waters that fill such cavities. The latter feature has been found to be critical for the water-filling behavior of carbon nanotubes (45) and for cavities in proteins (38,39), and was also accounted for in this work as described below.
Based on the established concept that waters in protein cavities and crevices can utilize hydrogen bonds among themselves to energetically stabilize transient water configurations, we were able to stabilize as many as seven water molecules in the crevices and cavities identified within the core of ClC-ec1 using a systematic procedure. In particular, after each water addition described below, 5000 steps of energy minimization were performed in NAMD with all atoms free to move. At the end of each energy minimization, the sum of water-protein and water-water interaction energies was calculated for all water molecules added in the crevices to gauge their stabilization energy. The total interaction energy, averaged by the number of water molecules, was then compared with the threshold value of −12 kcal/mol that is used by DOWSER to distinguish hydrated from empty cavities. The water configurations at the end of each energy minimization (Fig. S1) and their stabilization energy (Table S1) are provided in the Supporting Material. These configurations are all plausible transient structures at finite temperature.
Although crystal water was not resolved in the anion conduction pores, previous computer simulations of anion conduction and channel gating mechanism seem to suggest that the permeating Cl− ions are partially hydrated (19–21). This leads us to propose a hydration site in the pore between Glu148 and the central Cl− ion. In a 20 ns MD simulation, a water molecule was observed diffusing into the pore between Glu148 and the central Cl− ion from the intracellular side of the protein. The open state of the transporter may be favorable for direct observation of water in the pore; however, this open conformation was never seen in the crystal structure of wild-type protein. Two water molecules can be inserted into the anion conduction pore, where they make hydrogen bonds with Glu148, the central Cl− ion, and each other. The energy required to transfer these two water molecules from vacuum into the pore is −41 kcal/mol. The average interaction energy is thus −20.5 kcal/mol, well below the threshold value of DOWSER. Two more water molecules were added in the immediate proximity of the pore, as depicted in Fig. 1 a, or one was added on the outer side of the central Cl− ion and the other next to the Thr407 hydroxyl group, as depicted in Fig. 1 b. The stabilization energy for both configurations was −18.8 kcal/mol. This procedure was repeated until as many as seven water molecules were placed in the cavities and crevices spanning the core of the protein, and a hydrogen-bonded wire formed between Glu148 and Thr407. It is noted that addition of these water molecules produced no significant perturbations to the protein structure. At finite temperature, the combined effects of kinetic energy and entropy then make these water configurations transient in nature during MD simulations, as they are likely to be in reality given their absence from the observed crystal structure of the protein. A detailed analysis of the lifetimes of these water configurations is provided in the Supporting Material. The four-water configuration is estimated to have a lifetime of 3.7 ns, and the seven-water configuration has a lifetime of 1.4 ns. The seven-water configuration also often turned into three- or four-water configurations during the simulations, as water molecules on the side of Thr407 and Glu202 escaped to the intracellular side of the protein. Hundreds of thousands of such water wire configurations can assemble and disassemble over the millisecond timescale it takes for a single PT event to be observed in the ClC-ec1 system. It is also noted that water in the anion conduction pore between Glu148 and the central Cl− ion never escaped during the 4.5 ns MD simulation.
Figure 1.
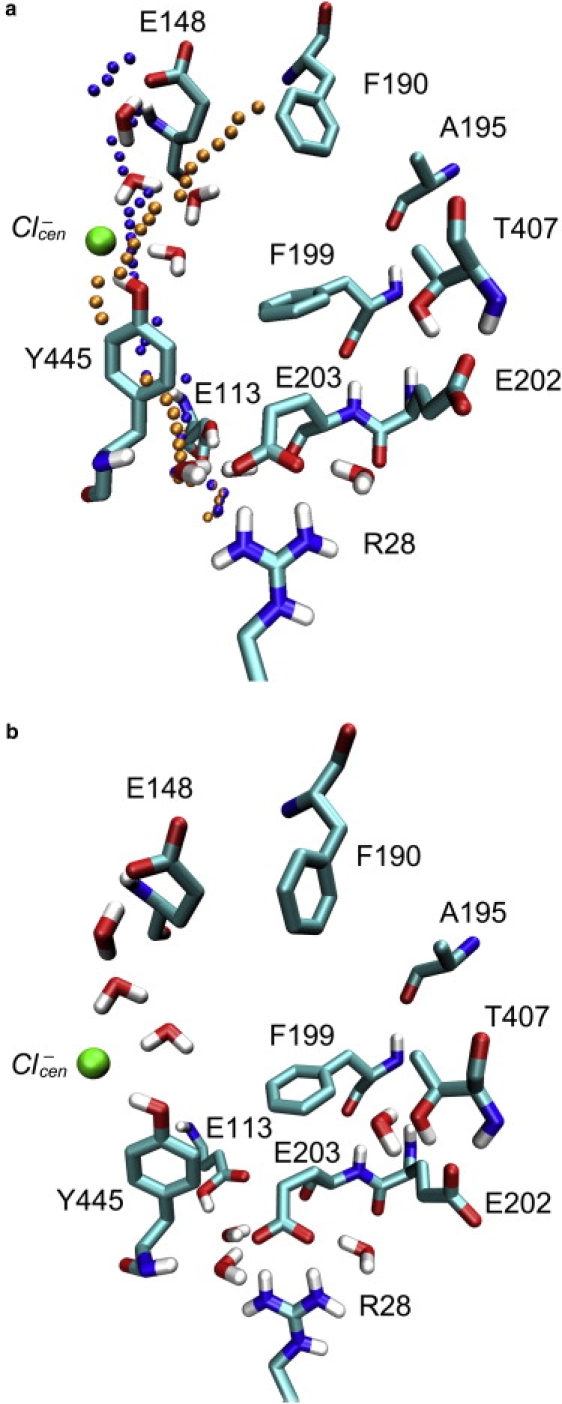
Hydration of the protein crevices and cavities identified between the two proton transfer sites. (a) With four water molecules near the Cl− permeation pathway. Two proton pathways suggested previously (23) based on the crystal structure of ClC-ec1 are depicted. Their P1 path is represented with orange spheres, and their P2 path is shown by blue spheres. P1 ends some distance from Glu148. The upper portion of P2 coincides with the hydration sites identified in this work. (b) With three water molecules in the anion conduction pore and one near Thr407 and Glu202. The protein is largely hydrophobic in the region between Glu148 and Glu203 except for the location of the central Cl−-binding site, Tyr445, Thr407, and Glu202. Two crystal water molecules near the Glu203 side chain are also shown. The pictures are generated with the VMD visualization software (52).
A pore-searching algorithm, TransPath, was previously applied to the crystal structure of ClC-ec1 to suggest a PT pathway between Glu148 and Glu203 (23). Two proton pathways (labeled P1 and P2) were proposed. As displayed in Fig. 1 a, the P2 path proceeds from Glu203 directly along the aromatic ring of Tyr445, past the central chloride-binding site, and then heads directly toward the Glu148 carboxyl along the Cl− permeation pathway. It is noted that the upper portion of P2 coincides with hydration of the anion conduction pore described above. The other path (P1) has a radius large enough to accommodate up to three water molecules near Glu203. However, except for one crystal water hydrogen bonded to the amide nitrogen atom of Tyr445, water molecules added in the predicted sites of P1 were not found to be stable in our classical MD simulations during the initial system setup phase. Moreover, since the P1 path ends some distance from Glu148, it seems unlikely to be involved in the PT.
As discussed above, the PT pathway of ClC-ec1 is in fact likely to be dynamic in nature, so analysis of crystal structure alone is insufficient. In particular, water molecules following the permeating anions into the pore likely play an important role in the H+ conduction. Although as many as seven water molecules can transiently occupy the crevices and cavities spanning the core of ClC-ec1, a completely static water-mediated pathway directly connecting Glu203 to Glu148 is not found or expected. Instead, it is suggested here that movement of the Glu203 side chain and its subsequent deprotonation to one of the transient water wires are required to initiate the transfer of H+ from the intracellular solution to the core of the protein, as described in the next section.
Reorientation of the Glu203 side chain
Glu203 was previously identified as a proton transfer site by a mutagenesis scan of inward-facing, carboxyl-bearing residues. This residue was therefore proposed to mediate PT between the intracellular solution and the protein interior (11). The way in which this intracellular-facing glutamate acts to shuttle H+, however, is not clear. In the crystal structure of wild-type protein, Glu203 forms a salt bridge with Arg28 from the other subunit of the homodimer, and a hydrogen bond with Glu113 from the same subunit. During the exchange transport, once Glu203 accepts a proton from the intracellular side of the membrane, the salt bridge and hydrogen-bond interactions will be disrupted.
The classical MD simulations presented here show that Glu203, upon protonation, carries H+ from the intracellular surface to the core of the protein via a conformational change of the side chain. With seven water molecules solvating the crevices, reorientation of the Glu203 side chain was observed in several of the standard MD trajectories, each of 500 ps duration. After rotation of the Glu203 side chain, a water-mediated PT pathway is established between Glu203 and Glu148. We estimate that at least four water molecules are required to establish a water-mediated connection between the reoriented Glu203 and Glu148, as described earlier. The conformational change of the Glu203 side chain, as depicted in Fig. 2, involves a change of χ2 from 60° to 180°. Water molecules in the crevices rearrange to accommodate this conformation change and form a transient water “wire”. The aromatic ring of Phe199, which blocks the proton pathway as observed in the crystal structure of ClC-ec1, is flipped once the side chain of Glu203 is protonated and reorients during the transport cycle.
Figure 2.

The proposed PT pathway between Glu203 and Glu148. In this pathway, Glu203 is suggested to carry H+ from the intracellular solution to the core of ClC-ec1 via a rotation of its side chain. After reorientation of the Glu203 side chain, a transient water-mediated PT pathway is established. After deprotonation of Glu203, hydrated protons shuttle through this pathway along a hydrogen-bonded network defined by Glu203, intervening water molecules, and Glu148. Separate pathways for Cl− and H+ are depicted that overlap at the location of Glu148 and diverge toward the intracellular side.
To evaluate the thermodynamic stability of various conformers and the kinetic barrier height of isomerization, we calculated the free-energy curve (i.e., PMF) for the reorientation of the Glu203 side chain as a function of χ2. While Glu203 is in a deprotonated form, the conformer at 60° is favored because of the formation of a salt bridge with Arg28 and a hydrogen bond with Glu113 as observed in the crystal structure of ClC-ec1 (Fig. 3 a). However, it appears that the conformer at 180° is not in a thermodynamically stable state. While Glu203 is protonated in the transport cycle, all three conformers are within 1−2 kcal/mol of each other, and thus are likely equally populated (Fig. 3 b). More importantly, the energy barrier separating the minima at 60° and 180° is lowered from ∼15 kcal/mol to 5−6 kcal/mol upon Glu203 protonation, indicating that the reorientation of the Glu203 side chain is kinetically feasible once Glu203 takes up a proton from the intracellular side of the membrane. In the above calculations, Glu113 was protonated according to a previous study in which the pKa shifts of titratable residues were determined with the Poisson-Boltzmann method (16). However, in the transport cycle, Glu113 may become deprotonated when Glu203 is protonated, and thus it is useful to also calculate PMF for the side-chain rotation of Glu203 for comparison (Fig. 3 c). It is noted that deprotonation of Glu113 destabilizes the rotamer at 180° but has little influence on the barrier height separating the rotamers at 60° and 180°.
Figure 3.
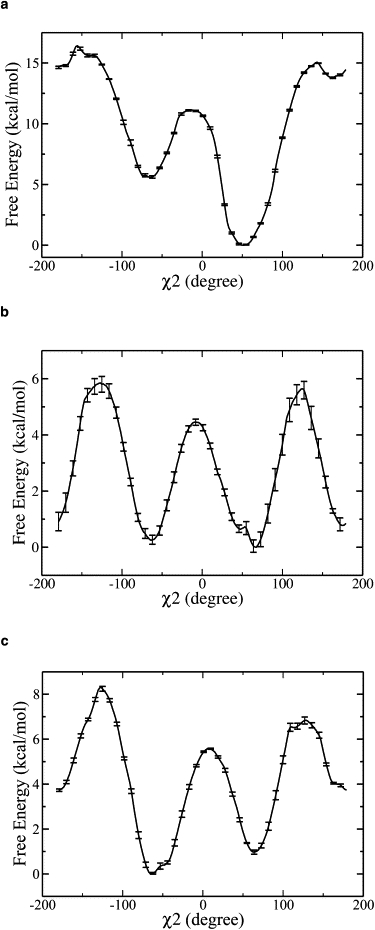
Free-energy profiles (PMFs) for rotation of the Glu203 side chain calculated as a function of the χ2 angle. (a) With negatively charged (deprotonated) Glu203 and neutral (protonated) Glu113. The conformer at 180° is not thermodynamically stable. The conformer at 60° is favored and observed in the crystal structure. (b) With both Glu203 and Glu113 neutral. All three conformers are within 1 kcal/mol of each other and thus likely are approximately equally populated. The energy barrier between the conformers at 60° and 180° is lowered from ∼15 kcal/mol to 5–6 kcal/mol upon protonation of Glu203. (c) With neutral Glu203 and negatively charged Glu113. The energy barrier between the conformers at 60° and 180° is ∼6 kcal/mol. The latter conformer is 3 kcal/mol less stable than the former. The error bars are shown in the plots.
The PMF curves shown in Fig. 3 were calculated with four water molecules solvating the protein crevice. In fact, more than four water molecules may be involved in transfering H+ from the reoriented Glu203 to Glu148 as described above. To test the robustness of this model under other circumstances, we also calculated the PMF for the rotation of Glu203 side chain in the case of seven water molecules transiently solvating the protein crevices. With seven transiently “bound” water molcules and protonated Glu203, the PMF profile again exhibits an energy barrier of 5–6 kcal/mol between the rotamers at 60° and 180° (Fig. S2), very similarly to the case of four water molecules. These results show that the movement of the Glu203 side chain is not sensitive to the number of transient water molecules in the crevices.
PT mediated by water molecules
Glu203 was proposed in the previous section to deliver H+ from the intracellular solution to the core of ClC-ec1 through a reorientation of its side chain. After the rotation of the Glu203 side chain, a metastable water-mediated PT pathway is established. After the deprotonation of Glu203, the hydrated proton is then proposed to move through this pathway along the hydrogen-bonded network via a Grotthuss shuttling mechanism. To quantify the latter process, explicit water mediated PT was simulated using the MS-EVB model. The deprotonation event of Glu203 was not studied, because the MS-EVB model used in this work does not include dissociable residues.
The initial configuration for the MS-EVB simulation of the PT between Glu203 and Glu148 was obtained as follows: With four transiently bound water molecules, the configuration taken from the umbrella sampling of the rotation of Glu203 side chain was first relaxed with the MD simulation. During relaxation, an additional water molecule also diffused into the protein crevices. Fig. 4 a shows the resulting configuration, in which the aromatic ring of Phe199 is flipped, opening a water-mediated pathway through which H+ can move from Glu203 to Glu148. Glu203 is then considered to deprotonate to a water molecule in the pathway, i.e., the one hydrogen bonded to Glu203 in Fig. 4 a. The configuration thus generated is taken as the initial configuration for the MS-EVB simulation of the explicit PT through the water chain. In a single 200 ps trajectory, the excess proton was seen to propagate along the hydrogen-bonded water network toward Glu148. A snapshot of the proton propagation in the pathway is shown in Fig. 4 b. In comparison with the hydrogen-bonded network shown in Fig. 4 a, the presence of an excess proton in the pathway was found to affect the water-water hydrogen-bonding interactions, and to reverse orientations of the water molecules in the pathway. In the trajectory, the hydrated excess proton first spends ∼100 ps on the side of Glu203, during which it forms an ion pair with Glu203. It then travels quickly toward Glu148, hopping between consecutive water molecules, and driven at least in part by its electrostatic interaction with the Cl− anion.
Figure 4.

Snapshot of the proposed proton propagation pathway along the transient water chain. (a) Before the deprotonation of Glu203. The configuration taken from the umbrella sampling of the rotation of Glu203 side chain with bound four water molecules is first relaxed with the MD simulation. During relaxation, an additional water molecule diffuses into the cavities, after which there are five water molecules in the proton pathway. (b) After the deprotonation of Glu203. A hydrated excess proton (colored in yellow) shuttles between consecutive water molecules via the Grotthuss mechanism. As H+ propagates toward Glu148, the side chain of Glu203 reverts to its original position and the hydrogen bond with Glu113 is restored.
The full free-energy profile (PMF) for the PT through the water-mediated pathway was then calculated with the use of umbrella sampling. As the proton propagated through the pathway, the position of the CEC as defined in Materials and Methods was recorded and projected along the vector connecting the carboxyl carbon atom of Glu203 to the carboxyl carbon atom of Glu148. It should be noted that as the excess proton moved toward Glu148, the Glu203 side chain reverted to its original position seen in the crystal structure. To facilitate the convergence of the umbrella sampling of the proton CEC movement through the water chain, the carboxyl carbon atom of Glu203 was therefore tethered to its reoriented position with a harmonic potential having a force constant of 5 kcal/mol/Å2. Tethering of the Glu203 side chain in this fashion will hinder the proton hopping through the water chain toward Glu148, and thus lead to a modest overestimation of the barrier height that the excess protons encounter while moving from Glu203 to Glu148 (cf. Fig. 5 a).
Figure 5.

Free-energy profiles (PMFs) for the PT through the proposed transient water-mediated pathways. (a) In wild-type protein. Glu203 first carries H+ from the intracellular surface to the core of the protein, and then deprotonates to the transient water wire. This PMF thus characterizes the PT free energy along the water wire between the reoriented and deprotonated Glu203 and Glu148. The minimum on the left represents structures in which an ion pair is formed between deprotonated Glu203 and the hydrated excess proton; the minimum on the right represents structures in which the excess proton is associated with Glu148 and the central Cl− ion. The intrinsic proton wire barrier height is thus only 2 kcal/mol for the proton to move from near Glu203 to near Glu148. (b) In the mutant E203V. In the absence of the shuttling Glu203 residue, hydrated excess protons must travel all the way from the intracellular surface to Glu148. They therefore cross a high barrier while moving through a large hydrophobic region. The minimum on the left represents configurations in which the excess proton is in the vicinity of Glu113; the minimum on the right represents configurations in which the excess proton is located near Glu148 and the central Cl− ion. The error bars are shown in the plots. Note the differing scales on the vertical axes when comparing panels a and b.
The PMF profile for the hydrated excess proton through the transient water-mediated PT pathway (Fig. 5 a) shows two minima, with the one on the right being ∼2 kcal/mol lower in energy. The minimum on the left represents structures in which an ion pair is formed between deprotonated Glu203 and the hydrated excess proton. The minimum on the right represents structures in which the excess proton is located near Glu148 and the central Cl− ion. To move from the side of Glu203 to the side of Glu148, protons have to cross an energy barrier of only ∼2 kcal/mol.
It should be noted that the internal (intracellular side) Cl− ion, which diffused out of its binding site during the MD equilibration, is located 10 Å away from the proposed PT pathway (cf. Fig. 2). The PMF for PT along the water chain with the internal Cl− ion instead restrained to its binding site was also calculated (see Fig. S3) and shows a similar barrier height of ∼2 kcal/mol for the excess proton to shuttle from Glu203 to Glu148 through the water-mediated pathway. Therefore, it seems likely that the internal chloride anion primarily affects the pKa of Glu203 and not the subsequent PT through the transient water chain to the region of the central Cl− and Glu148.
E203V mutant
In this work, it is proposed that Glu203 shuttles protons between the intracellular solution and the core of ClC-ec1. Glutamate at this position is strictly conserved in all known ClC transporters, whereas valine is always found at this position in ClC channels. Therefore, PT in the E203V mutant was also examined using the MS-EVB methodology. Experimental results for this and other mutants were given in a recent work by Lim and Miller (29).
After valine was substituted for glutamic acid at position 203 in the equilibrated structure of the wild-type protein, the structure of the mutant E203V was obtained and equilibrated with a classical MD simulation. Since the goal of these particular calculations was to probe the role of a nonionizable residue substitution at the position of Glu203, propagation of the hydrated excess proton in the mutant was started in the vicinity of Glu113, as in the wild-type protein. However, in a mutant without a proton transfer residue near the intracellular surface, hydrated protons have to travel through a large hydrophobic region to arrive at Glu148, even with as many as seven water molecules solvating the crevices spanning the protein core. The PMF profile for the PT in the mutant E203V was thus calculated for the latter case. The reaction coordinate was defined as the excess proton CEC positon projected along the vector connecting the carboxyl oxygen atom of Glu113 to the carboxyl oxygen atom of Glu148. Of importance, the resulting PMF profile (Fig. 5 b) shows a high barrier of ∼12 kcal/mol that hydrated protons taken up at the intracellular surface must overcome to reach their destination on the extracellular side. This result clearly supports the concept that a proton shuttle is required at the position of Glu203 for efficient transfer of H+ from the intracellular solution to the core of the protein, in agreement with the experimental results presented by Lim and Miller (29).
Of interest, the experiments reported by Lim and Miller (29) show that replacement of Glu203 with histidine will not alter the protein's PT function. Substitution by aspartate at this position does not undermine the transport mechanism either, as disclosed in an earlier mutagenesis study (11). Preliminary MD simulations by our group showed that the positively charged histidine may form a salt bridge with Glu113 in the mutant E203H. It can also be presumed that the histidine at this position acts as a proton shuttle in the mutant in a similar fashion to the glutamate in the wild-type protein. Explicit PT behavior in the E203H mutant will be examined in a future study.
Conclusions
The PT pathway of the ClC Cl−/H+ antiporters is a topic of great interest concerning these and related proteins. Although a high-resolution structure of ClC-ec1 was determined in 2002, details regarding the proton pathway have remained an open question. Except for two proton transfer residues residing near the cellular surfaces, little was known about the nature of this pathway. The novel computer simulations presented here therefore provide new insight into the PT behavior of ClC-ec1. Protein crevices and cavities spanning the core of ClC-ec1 were identified at the location of the Glu148 carboxyl, the central Cl− ion, and the Tyr445 and Thr407 hydroxyl groups. The crevices can accommodate as many as seven water molecules, albeit in a transient fashion. These hydrogen-bonded water molecules, however, seem insufficient by themselves to establish a proton pathway between the two proton-binding sites, Glu148 and Glu203. Therefore, a key proposal here is that Glu203, upon protonation, carries H+ from the intracellular surface to the protein interior via a side-chain rotation. After reorientation of the Glu203 side chain, a metastable hydrogen-bonded network defined by Glu203, water molecules lining the crevices, and Glu148 is established. After deprotonation of the Glu203, the proton then translocates along this hydrogen-bonded network via the Grotthuss shuttling mechanism from the intracellular side of the membrane to the extracellular side. We note that a similar mechanism for translocating protons beyond a glutamic acid through a hydrophobic region has been proposed for cytochrome c oxidase (40–42). In contrast to the alternating-site mechanism in which a single protonatable locus alternates its exposure to the two sides of the membrane through a cycle of conformational changes (46–49), the conformational change of the Glu203 side chain in ClC-ec1 is rather local.
Such a Glu203 side-chain orientation has not been observed crystallographically. In the crystal structure of wild-type protein, Glu203 forms a salt bridge with Arg28 from the other subunit of the homodimer, and a hydrogen bond with Glu113 in the same subunit. An analog to the protein with Glu203 protonated is the mutant of E203Q. In that structure of E203Q, the Gln203 side chain occupies the same position as Glu203 in the wild-type protein (11). This result does not contradict our proposal. First of all, the free-energy profile for the isomerization of the Glu203 side chain shows that all three conformers are likely to be equally populated while Glu203 is in a protonated state. Therefore, it is perhaps not surprising to see the Q203 side chain in E203Q occupy roughly the same position as Glu203 in the wild-type protein. Second, the crystal structure does not represent all possible conformational states visited by the protein, so the absence of a crystallographically observed conformational change does not mean that such a change will not take place. Lastly, the population of the three rotamers may well be different under the crystallographic conditions.
In support of this proposal, our free-energy calculations show that the rotation of the Glu203 side chain is plausible in terms of both the energetics and kinetics. Indeed, a side-chain movement of protonatable residues is sometimes invoked in other protein proton conduction mechanisms, such as the Glu242 residue of (bovine) cytochrome c oxidase, which is proposed to deliver a proton from the top of the D-channel to the binuclear center via such a side-chain rotation (40–42,50). In the transport cycle of ClC-ec1, Glu203 takes up a proton from the intracellular solution. A change in the protonation state of Glu203 makes the side-chain movement kinetically feasible. Once Glu203 releases a proton to the core of ClC-ec1, it will revert to its original position, and the disrupted interactions with Arg28 and Glu113 will be restored.
Lim and Miller (29) also found experimentally that replacing Glu203 with protonatable residues such as aspartate and histidine does not abolish H+ conduction. In fact, the E203D and E203H mutants behave like wild-type protein. Preliminary simulation results obtained by our group show that in the mutants of E203D and E203H the Arg28 side chain is most likely disordered. This is confirmed by the crystal structure of E203H (29). The results of these mutagenesis studies on Glu203 and previous studies on R28L (11) support the notion that Arg28 is not a necessary factor in H+ conduction.
The importance of Glu203 as a proton shuttle was further demonstrated in this work by an examination of the PT barrier in the mutant E203V. Substitution of Glu203 by valine reveals that in the absence of a proton-binding site at this position, H+ have to travel through a large hydrophobic area to arrive at Glu148. The energy barrier that these protons must cross is as high as 12 kcal/mol. This result indicates that a proton shuttle is required in part near the intracellular surface to deliver H+ from the intracellular solution to the protein interior. The fact that E203D and E203H mutants have also been found to transport protons implies that the aspartate and histidine residues may serve as proton shuttles in a fashion similar to that of glutamate in wild-type protein. The important role of an intracellular titratable residue in the coupled transport of mammalian ClC-4 and ClC-5 was also recently identified (51).
The proton movement from Glu203 to Glu148 is suggested here to consist of a series of steps, including binding of H+ to Glu203, reorientation of the Glu203 side chain, release of H+ by Glu203 to the core of ClC-ec1, and H+ shuttling transfer to Glu148 mediated by transient chains of water molecules. In this work, the free-energy curves for the rotation of the Glu203 side chain and for the water-mediated PT were calculated. In the reorientation of Glu203, a barrier of 5−6 kcal/mol was found, whereas for the proton propagation along a hydrogen-bonded water chain toward Glu148, a barrier of ∼2 kcal/mol was obtained. Therefore, it is likely that the rate-limiting step in the transfer of H+ from Glu203 to Glu148 is the reorientation of the Glu203 side chain combined with the deprotonation event of the Glu203. It should be noted that the overall proton flux of ClC-ec1 has been measured to be ∼2000 ion/s (15). This level of flux is in line with that expected for a barrier of ∼6 kcal/mol given the experimental pH conditions.
Hydrated proton movement along a transient water chain via the Grotthuss mechanism is not naturally downhill without an additional environmental electrostatic influence. On the intracellular side, an ion pair between Glu203 and the hydrated excess proton is formed. On the opposite side, the excess proton is located between Glu148 and the central Cl− ion. The minimum on the Glu148 side is ∼2 kcal/mol lower in energy, with a free-energy barrier of ∼2 kcal/mol for PT along the water chain from Glu203 to Glu148. These results indicate that binding of a Cl− ion to the central site promotes proton movement through the water chain, and that electrostatic interactions between the excess proton and the negatively charged residues are the major driving force for that movement. A synergism between the proton coupling ratio and the central site occupancy of Cl− ion has been experimentally observed in the mutants of Tyr445 (12), suggesting that Cl− binding to the central site facilitates the proton movement. This finding is consistent with our conclusions.
Finally, this work shows that the PT pathway in ClC-ec1 is likely to be dynamical in nature, which makes the analysis of static crystal structures alone insufficient, and simulations or other experimental measures of the explicit PT valuable. Our classical MD and MS-EVB simulations of the PT within ClC-ec1 therefore complement crystallographic, site-directed mutagenesis and electrophysiological experiments. Insights gained from such simulations are invaluable for revealing the possible mechanism of coupled transport in the Cl−/H− antiporter, providing new predictions for experimentalists to test, and helping to build a complete picture of the structure-function relationship for these interesting and important proteins.
Supporting Material
Further information on the MD simulations, PT free energy calculations, and internal hydration of the CIC-ec1 protein is available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00904-7.
Supporting Material
Acknowledgments
We are grateful to our collaborator Professor Chris Miller of Brandeis University for many stimulating discussions, and to Professor Tom Beck of the University of Cincinnati for sending us coordinate files of their proposed proton pathways located with the TransPath program. We thank Dr. Hanning Chen for assistance in setting up the MS-EVB simulations, Dr. Jiancong Xu for helpful discussions, and Dr. Jessica Swanson for a critical reading of the manuscript. The WHAM code written by Dr. Alan Grossfield was used for data analysis. Computational resources were provided by the National Science Foundation through TeraGrid computing resources administered by the Texas Advanced Computing Center and the Pittsburgh Supercomputing Center.
This work was supported by the National Institutes of Health (R01-GM053148).
Footnotes
Dong Wang's present address is Department of Chemistry, Beijing Normal University, Beijing, China.
References
- 1.Accardi A., Miller C. Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature. 2004;427:803–807. doi: 10.1038/nature02314. [DOI] [PubMed] [Google Scholar]
- 2.Jentsch T.J., Neagoe L., Scheel O. ClC chloride channels and transporters. Curr. Opin. Neurobiol. 2005;15:319–325. doi: 10.1016/j.conb.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Picollo A., Pusch M. Chloride/proton antiporter activity of mammalian ClC proteins ClC-4 and ClC-5. Nature. 2005;436:420–423. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- 4.Scheel O., Zdebik A.A., Lourdel S., Jentsch T.J. Voltage-dependent electrogenic chloride/proton exchange by endosomal ClC proteins. Nature. 2005;436:424–427. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]
- 5.Iyer R., Iverson T.M., Accardi A., Miller C. A biological role for prokaryotic ClC chloride channels. Nature. 2002;419:715–718. doi: 10.1038/nature01000. [DOI] [PubMed] [Google Scholar]
- 6.Dutzler R., Campbell E.B., Cadene M., Chait B.T., MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 Angstrom reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 7.Chen M.F., Chen T.Y. Side-chain charge effects and conductance determinants in the pore of ClC-0 chloride channels. J. Gen. Physiol. 2003;122:133–145. doi: 10.1085/jgp.200308844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Traverso S., Elia L., Pusch M. Gating competence of constitutively open ClC-0 mutants revealed by the interaction with a small organic inhibitor. J. Gen. Physiol. 2003;122:295–306. doi: 10.1085/jgp.200308784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estevez R., Schroeder B.C., Accardi A., Jentsch T.J., Pusch M. Conservation of chloride channel structure revealed by an inhibitor binding site in ClC-1. Neuron. 2003;38:47–59. doi: 10.1016/s0896-6273(03)00168-5. [DOI] [PubMed] [Google Scholar]
- 10.Dutzler R., Campbell E.B., MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- 11.Accardi A., Walden M., Nguitragool W., Jayaram H., Williams C. Separate ion pathways in a Cl+/H+ exchanger. J. Gen. Physiol. 2005;126:563–570. doi: 10.1085/jgp.200509417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Accardi A., Lobet S., Williams C., Miller C., Dutzler R. Synergism between Halide binding and proton transport in a ClC-type exchanger. J. Mol. Biol. 2006;362:691–699. doi: 10.1016/j.jmb.2006.07.081. [DOI] [PubMed] [Google Scholar]
- 13.Nguitragool W., Miller C. Uncoupling of a ClC Cl−/H+ exchange transporter by polyatomic anions. J. Mol. Biol. 2006;362:682–690. doi: 10.1016/j.jmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 14.Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006;440:484–489. doi: 10.1038/nature04713. [DOI] [PubMed] [Google Scholar]
- 15.Walden M., Accardi A., Wu F., Xu C., Williams C. Uncoupling and turnover in a Cl−/H+ exchange transporter. J. Gen. Physiol. 2007;129:317–329. doi: 10.1085/jgp.200709756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faraldo-Gomez J.D., Roux B. Electrostatics of ion stabilization in a ClC chloride channel homologue from Escherichia coli. J. Mol. Biol. 2004;339:981–1000. doi: 10.1016/j.jmb.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 17.Miloshevsky G.V., Jordan P.C. Anion pathway and potential energy profiles along curvilinear bacterial ClC Cl− pores: electrostatic effects of charged residues. Biophys. J. 2004;86:825–835. doi: 10.1016/S0006-3495(04)74158-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miloshevsky G.V., Jordan P.C. Permeation and gating in proteins: kinetic Monte Carlo reaction path following. J. Chem. Phys. 2005;122:214901. doi: 10.1063/1.1924501. [DOI] [PubMed] [Google Scholar]
- 19.Cohen J., Schulten K. Mechanism of anionic conduction across ClC. Biophys. J. 2004;86:836–845. doi: 10.1016/S0006-3495(04)74159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gervasio F.L., Parrinello M., Ceccarelli M., Klein M.L. Exploring the gating mechanism in the ClC chloride channel via metadynamics. J. Mol. Biol. 2006;361:390–398. doi: 10.1016/j.jmb.2006.06.034. [DOI] [PubMed] [Google Scholar]
- 21.Bostick D.L., Berkowitz M.L. Exterior site occupancy infers chloride-induced proton gating in a prokaryotic homolog of the ClC chloride channel. Biophys. J. 2004;87:1686–1696. doi: 10.1529/biophysj.104.042465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jian Y., Kuang Z.F., Mahankali U., Beck T.L. Ion transit pathways and gating in ClC chloride channels. Proteins. 2004;57:414–421. doi: 10.1002/prot.20208. [DOI] [PubMed] [Google Scholar]
- 23.Kuang Z., Mahankali U., Beck T.L. Proton pathways and H+/Cl− stoichiometry in bacterial chloride transporters. Proteins. 2007;68:26–33. doi: 10.1002/prot.21441. [DOI] [PubMed] [Google Scholar]
- 24.Voth G.A. Computer simulation of proton solvation and transport in aqueous and biomolecular systems. Acc. Chem. Res. 2006;39:143–150. doi: 10.1021/ar0402098. [DOI] [PubMed] [Google Scholar]
- 25.Swanson J.M.J., Maupin C.M., Chen H.N., Petersen M.K., Xu J.C. Proton solvation and transport in aqueous and biomolecular systems: insights from computer simulations. J. Phys. Chem. B. 2007;111:4300–4314. doi: 10.1021/jp070104x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmitt U.W., Voth G.A. Multistate empirical valence bond model for proton transport in water. J. Phys. Chem. B. 1998;102:5547–5551. [Google Scholar]
- 27.Schmitt U.W., Voth G.A. The computer simulation of proton transport in water. J. Chem. Phys. 1999;111:9361–9381. [Google Scholar]
- 28.Day T.J.F., Soudackov A.V., Cuma M., Schmitt U.W., Voth G.A. A second generation multistate empirical valence bond model for proton transport in aqueous systems. J. Chem. Phys. 2002;117:5839–5849. [Google Scholar]
- 29.Lim H.-H., Miller C. Intracellular proton-transfer mutants in a ClC C1−/H+ exchanger. J. Gen. Physiol. 2009;133:131–138. doi: 10.1085/jgp.200810112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y., Chen H., Wang F., Paesani F., Voth G.A. An improved multistate empirical valence bond model for aqueous proton solvation and transport. J. Phys. Chem. B. 2008;112:467–482. doi: 10.1021/jp076658h. [DOI] [PubMed] [Google Scholar]
- 31.Cuma M., Schmitt U.W., Voth G.A. A multi-state empirical valence bond model for weak acid dissociation in aqueous solution. J. Phys. Chem. A. 2001;105:2814–2823. [Google Scholar]
- 32.Kumar S., Rosenberg J.M., Bouzida D., Swendsen R.H., Kollman P.A. Multidimensional free-energy calculations using the weighted histogram analysis method. J. Comput. Chem. 1995;16:1339–1350. [Google Scholar]
- 33.Roux B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 1995;91:275–282. [Google Scholar]
- 34.Agmon N. The Grotthuss mechanism. Chem. Phys. Lett. 1995;244:456–462. [Google Scholar]
- 35.Decoursey T.E. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- 36.Wraight C.A. Chance and design—proton transfer in water, channels and bioenergetic proteins. Biochim. Biophys. Acta. 2006;1757:886–912. doi: 10.1016/j.bbabio.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 37.Cukierman S. Et tu, Grotthuss! And other unfinished stories. Biochim. Biophys. Acta. 2006;1757:876–885. doi: 10.1016/j.bbabio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Yin H., Hummer G., Rasaiah J.C. Metastable water clusters in the nonpolar cavities of the thermostable protein tetrabrachion. J. Am. Chem. Soc. 2007;129:7369–7377. doi: 10.1021/ja070456h. [DOI] [PubMed] [Google Scholar]
- 39.Rasaiah J.C., Garde S., Hummer G. Water in nonpolar confinement: from nanotubes to proteins and beyond. Annu. Rev. Phys. Chem. 2008;59:713–740. doi: 10.1146/annurev.physchem.59.032607.093815. [DOI] [PubMed] [Google Scholar]
- 40.Kaila V.R.I., Verkhovsky M.I., Hummer G., Wikström M. Glutamic acid 242 is a valve in the proton pump of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 2008;105:6255–6259. doi: 10.1073/pnas.0800770105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tuukkanen A., Kaila V.R.I., Laakkonen L., Hummer G., Wikström M. Dynamics of the glutamic acid 242 side chain in cytochrome c oxidase. Biochim. Biophys. Acta. 2007;1767:1102–1106. doi: 10.1016/j.bbabio.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Wikström M., Verkhovsky M.I., Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta. 2003;1604:61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 43.Xu J., Voth G.A. Redox-coupled proton pumping in cytrochrome c oxidase: further insights from computer simulation. Biochim. Biophys. Acta. 2008;1777:196–201. doi: 10.1016/j.bbabio.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L., Hermans J. Hydrophilicity of cavities in proteins. Proteins. 1996;24:433–438. doi: 10.1002/(SICI)1097-0134(199604)24:4<433::AID-PROT3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 45.Hummer G., Rasaiah J.C., Noworyta J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 2001;414:188–190. doi: 10.1038/35102535. [DOI] [PubMed] [Google Scholar]
- 46.Abramson J., Iwata S., Kaback H.R. Lactose permease as a paradigm for membrane transport proteins (review) Mol. Membr. Biol. 2004;21:227–236. doi: 10.1080/09687680410001716862. [DOI] [PubMed] [Google Scholar]
- 47.Hunte C., Screpanti E., Venturi M., Rimon A., Padan E. Structure of Na+/H+ antiporter and insights into mechanism of action and regulation by pH. Nature. 2005;435:1197–1202. doi: 10.1038/nature03692. [DOI] [PubMed] [Google Scholar]
- 48.Knauf P.A., Law F.Y., Leung T.W.V., Gehret A.U., Perez M.L. Substrate-dependent reversal of anion transport site orientation in the human red blood cell anion-exchange protein, Ae1. Proc. Natl. Acad. Sci. USA. 2002;99:10861–10864. doi: 10.1073/pnas.162402399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lemieux M.J., Huang Y.F., Wang D.N. The structural basis of substrate translocation by the Escherichia coli glycerol-3-phosphate transporter: a member of the major facilitator superfamily. Curr. Opin. Struct. Biol. 2004;14:405–412. doi: 10.1016/j.sbi.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 50.Riistama S., Hummer G., Puustinen A., Dyer R.B., Woodruff W.H. Bound water in the proton translocation mechanism of the haem-copper oxidases. FEBS Lett. 1997;414:275–280. doi: 10.1016/s0014-5793(97)01003-x. [DOI] [PubMed] [Google Scholar]
- 51.Zdebik A.A., Zifarelli G., Bergsdorf E.Y., Soliani P., Scheel O. Determinants of anion-proton coupling in mammalian endosomal ClC proteins. J. Biol. Chem. 2008;283:4219–4227. doi: 10.1074/jbc.M708368200. [DOI] [PubMed] [Google Scholar]
- 52.Humphrey W., Dalke A., Schulten K. VMD:visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.