

Abstract
Aromatic amino acids play an important role in ultraviolet (UV)-induced photochemical reactions in proteins. In this work, we aim at gaining insight into the photochemical reactions induced by near-UV light excitation of aromatic residues that lead to breakage of disulfide bridges in our model enzyme, Fusarium solani pisi cutinase, a lipolytic enzyme. With this purpose, we acquired transient absorption data of cutinase, with supplemental experimental data on tryptophan (Trp) and lysozyme as reference molecules. We here report formation kinetics and lifetimes of transient chemical species created upon UV excitation of aromatic residues in proteins. Two proteins, lysozyme and cutinase, as well as the free amino acid Trp, were studied under acidic, neutral, and alkaline conditions. The shortest-lived species is assigned to solvated electrons (lifetimes of a few microseconds to nanoseconds), whereas the longer-lived species are assigned to aromatic neutral and ionic radicals, Trp triplet states, and radical ionic disulphide bridges. The pH-dependent lifetimes of each species are reported. Solvated electrons ejected from the side chain of free Trp residues and aromatic residues in proteins were observed 12 ns after excitation, reaching a maximum yield after ∼40 ns. It is interesting to note that the formation kinetics of solvated electrons is not pH-dependent and is similar in the different samples. On the other hand, a clear increase of the solvated electron lifetime is observed with increasing pH. This observation is correlated with H3O+ being an electron scavenger. Prolonged UV illumination of cutinase leads to a larger concentration of solvated electrons and to greater absorption at 410 nm (assigned to disulphide electron adduct RSSR •−), with concomitant faster decay kinetics and near disappearance of the Trp• radical peak at 330 nm, indicating possible additional formation of TyrO• formed upon reaction of Trp• with Tyr residues. Prolonged UV illumination of cutinase also leads to a larger concentration of free thiol groups, known to originate from the dissociation of RSSR •−. Additional mechanisms that may lead to the near disappearance of Trp• are discussed. Our study provides insight into one key UV-light-induced reaction in cutinase, i.e., light-induced disruption of disulphide bridges mediated by the excitation of aromatic residues. Knowledge about the nature of the formed species and their lifetimes is important for the understanding of UV-induced reactions in humans that lead to light-induced diseases, e.g., skin cancer and cataract formation.
Introduction
Atmospheric gases of our planet Earth block solar ultraviolet (UV) radiation fairly efficiently, but a nonnegligible amount of near-UV light still reaches the surface. Although none of the amino acids naturally found in proteins absorbs in the visible spectrum, residues like tryptophan, tyrosine, phenylalanine, and cystine are known to absorb in the near-UV region. Several reviews have been published on the photochemistry and photophysics of tryptophan (1,2), tyrosine (3,4), phenylalanine (5), and cystine (6). Tryptophan has the highest absorption in the near-UV region, followed by tyrosine (2). Flash photolysis studies have revealed two nonradiative relaxation channels from singlet excited tryptophan (1). The first is electron ejection to the solvent, yielding solvated electrons, e-aq, which have a broad absorption peak centered at ∼720 nm and a tryptophan radical cation, , which absorbs at ∼560 nm and deprotonates rapidly, yielding a neutral radical, , that absorbs at ∼510 nm. First results on the solvated electron optical properties indicate a broad absorption band centered at 720 nm (7). The same spectral feature is observed in water after one-photon or multiple-photon absorption in the UV region (8–12).
| (1) |
| (2) |
The neutral radical can then react with, e.g., molecular oxygen, tyrosine, or cystine, and can lead to peptide chain cleavage. The second of these relaxation channels is intersystem crossing, yielding the triplet-state 3Trp, which absorbs at ∼450 nm. Triplet lifetimes range from 11 to 16 μs independent of pH from ∼2.5 to 10.5 (1). Shorter-lived transients (T1) with λmax at 450 nm and lifetimes of ∼20–45 ns have also been observed and tentatively suggested to be triplet states (1). Triplet-state tryptophan can then react with molecular oxygen (Scheme 5, (27)) or transfer an electron to a nearby disulfide bridge (Scheme 6), with RSSR•− absorbing at ∼420 nm (1).
| (3) |
| (4) |
| (5) |
| (6) |
At low pH, it is possible to generate a tryptophan hydrogen adduct (Scheme 9) that absorbs at ∼400 nm (13). It can either be generated directly by capture of a hydrogen atom, or in a two-step process, where a solvated electron is caught by tryptophan, which then reacts with a proton from the solvent (13,14):
| (7) |
| (8) |
| (9) |
These species and their absorption maxima are listed in Table 1. At neutral pH, tyrosine has absorption maxima at 220 nm and 275 nm (4). At alkaline pH, tyrosine deprotonates, and the resulting tyrosinate ( ) has slightly red-shifted absorption compared to tyrosine, with maxima at 240 nm and 290 nm (4). Photoexcited tyrosine can fluoresce, decay nonradiatively, or undergo intersystem crossing to a triplet state. Alternatively, at neutral pH, tyrosine can be photoionized through a biphotonic process that involves absorption of a second photon from the triplet state, leading to a solvated electron and a radical cation that rapidly deprotonates (Schemes 11 and 12). Photoionization of tyrosinate at high pH results in a neutral radical and a solvated electron (Scheme 12).
| (10) |
| (11) |
| (12) |
Table 1.
Transient chemical species and wavelength at maximum absorption, with their extinction coefficients and lifetimes
| Chemical species | Wavelength at maximum absorption, extinction coefficient, and lifetime | Reference | |
|---|---|---|---|
| 220, 280, 288, (265) nm | (2) | ||
| 450 nm | (1) | ||
| 330, 560 nm | (1)(45) | ||
| 320, 510 nm | (2,13,17,45,46) | ||
| pH 7–12 | |||
| pH 7 | |||
| pH7 | |||
| 320, 360, 420 nm | (17) | ||
| 250, 295, 575 nm | (3) | ||
| 220, 275 nm | (4) | ||
| 240, 290 nm | (4) | ||
| 250, 295, 575 nm | (3)(3) | ||
| Lifetimes 3-10 μs in water | |||
| Phenoxyl-type tyrosyl radical. Also called Tyr· | 245, 290, 390, 400, 405, 407, 410 nm | (3,37,46–49) | |
| pH 7 | |||
| pH 7 | |||
| 720 nm (very broad) | (1)(50) | ||
| Disulphide electron adduct | 420 nm | (1)(15,48) | |
| Lifetime in the μs range | |||
| Cystine pH 7.7 | |||
| perthiyl radical | 380 nm, in deaerated samples | (38) | |
| 380 nm | (38) | ||
| 245 nm | (3) | ||
Neutral radical absorption partly overlaps triplet-triplet absorption for tyrosine (3) (Table 1). However, the triplet-state tyrosine is rapidly quenched by molecular oxygen, tryptophan, or disulfide bridges (3):
| (13) |
| (14) |
| (15) |
Upon excitation of Trp and Tyr, generated solvated electrons can subsequently undergo fast geminate recombination with their parent molecule, or they can be captured by electrophilic species like molecular oxygen, H3O+, or cystines:
| (16) |
| (17) |
| (18) |
A hydrogen radical (Scheme 17) can react directly with a nearby disulfide bridge, resulting in bond breakage (15):
| (19) |
Electron capture by cystine can also lead to bond breakage (15):
| (20) |
| (21) |
| (22) |
Solvated electrons can also interact with the peptide chain to create hydroxide ion and ketyl radical, which can propagate along the peptide chain (1):
| (23) |
If trapped by a disulfide bridge, this again results in an RSSR•− radical, possibly leading to bridge disruption. If hydroxyl radicals are created, e.g., through radiolysis of water, they can react with tryptophan to create (16,17) (Table 1). UV excitation of lysozyme leads to photooxidized tryptophan, solvated electrons, and cystine electron adduct (18). UV-induced enzyme inactivation involves fast and very efficient short-range electron transfer between photoexcited tryptophans and nearby disulfide bridges. Zhi Li et al. (19) showed that fast electron transfer is consistent with direct electron transfer between triplet tryptophan and a nearby disulfide bridge (a short-range interaction that decays exponentially as the distance beyond van der Waals overlap between the donor and acceptor increases), leading to RSSR•− radical and likely bridge breakage:
| (24) |
| (25) |
Fusarium solani pisi cutinase contains only one tryptophan within van der Waals contact of a disulfide bridge (∼3.8 Å). The longer the UV excitation of its aromatic pool, the larger the yield of bridge disruption (20,21), resulting in free thiol groups (20–23) that bind to thiol-reactive surfaces (20,21,24–26). We aim to gain insight into the kinetics of UV- induced breakage of disulfide bridges in cutinase (22 KDa, 213 amino acids). Transient absorption data has been acquired for cutinase, with supplemental experimental data on tryptophan and lysozyme (14.7 KDa, 129 amino acids) as reference molecules. The 3D structures of these proteins are shown in Fig. 1. Both proteins have aromatic residues close to disulphide bridges. Cutinase photochemistry has not been previously studied using this technique. Lysozyme photochemistry is partially known. In both proteins, UV illumination leads to disulphide bridge disruption and to free thiol groups. A summary of some of the major photodegradation pathways of proteins upon UV irradiation has been published (27). Many of the referred processes involve molecular oxygen, and can be limited by working with degassed samples. It is a well-known fact in enzymology that pH has a crucial effect on both protein activity and stability. This effect is typically not associated with ultrafast events in the protein. However, pH has a direct influence on the protein electrostatic field (28,29). Thus, charged entities such as solvated electrons will migrate through the protein and its immediate environment immersed in the electrostatic field. We therefore decided to investigate the pH dependence of the transient absorption features of all samples.
Figure 1.
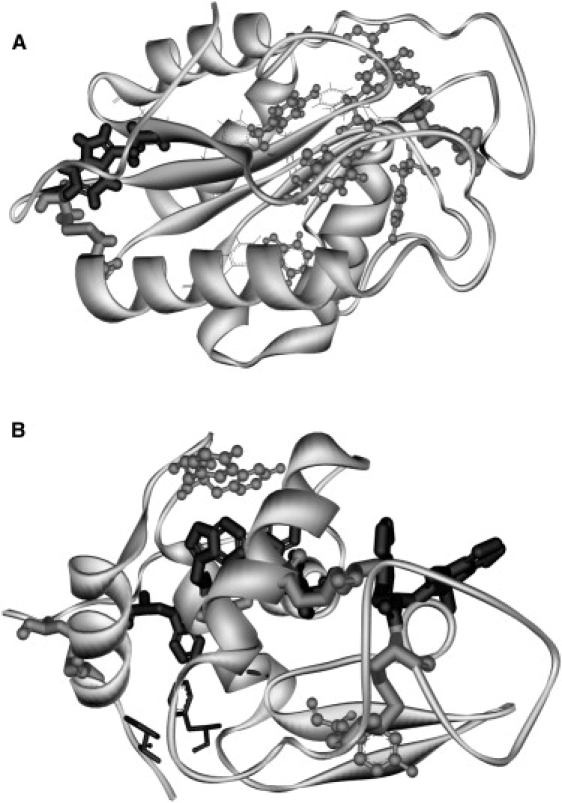
3D structure of cutinase and lysozyme. Secondary structure, aromatic amino acids and disulfide bridges are highlighted: tryptophan (black sticks), tyrosine (gray ball and stick), phenylalanine (black lines) and cystines (gray sticks). Cutinase (A) contains 1 Trp, 6 Tyr, 6 Phe and 2 disulfides, and lysozyme (B) contains 6 Trp, 3 Tyr, 3 Phe and 4 disulfides.
Materials and Methods
Sample preparation
Buffers used were 25 mM acetate, pH 4 (S-2889, Sigma, St. Louis, MO), 25 mM Trizma base, pH 7.5, 25 mM Trizma base, pH 8.5 (T-6066, Sigma), and 25 mM glycine, pH 10 (A1377, Appli Chem, Darmstadt, Germany). Experiments were carried out on l-tryptophan (L-8659, Sigma), lysozyme (L-7651, Sigma), and recombinant cutinase (29) at pH 4, 7.5, 8.5, and 10. Solutions of 147 μM tryptophan (molecular mass 204.2 gmol−1) and 49 μM lysozyme (molecular mass 14,300 gmol−1) were prepared. Cutinase concentrations were estimated using ɛ280 = 13,500 M−1·cm−1: 20 μM at pH 4, 28 μM at pH 8.5, and 30 μM at pH 10. Each sample was placed in a sealable 1-cm cuvette and degassed using cycles of vacuum and saturation with argon gas to minimize oxygen concentration.
Flash photolysis setup
Samples were excited using 266 nm light (third harmonic of a Quanta-Ray 230 Nd:YAG laser, Spectra-Physics, Mountain View, CA). Full width at half-maximum of the excitation pulse was ∼8 ns, and the energy per pulse was 1.5 mJ. The laser beam diameter was 2.5 mm. The cuvette holder was thermostated to 20°C by a computer-controlled Peltier element and the sample was magnetically stirred to reduce photodegradation. The probe light generated by a 75-W xenon arc lamp was focused on the sample collinearly with the excitation beam. Before it reached the sample, the probe light went through high-pass filters mounted on a filter wheel to reduce photodegradation. After the sample, the light was refocused with a spherical concave mirror on the entrance slit of a TRIAX 180 monochromator (SPEX, Reston, VA). Before the monochromator, the light passed two sets of filters mounted on filter wheels. A high-pass filter was used to reduce scattered excitation light, whereas a neutral-density filter was used to reduce probe light intensity before the photomultiplier tube. The signal was detected using a photomultiplier tube (R298, Hamamatsu, Hamamatsu City, Japan).
Data collection and analysis
Transient absorption spectra were collected on different timescales: over 50 μs with a xenon lamp in pulsed mode, and over 1 ms with a xenon lamp in continuous mode. Traces were collected with 10-nm steps over a 280–900 nm range. Data collection started in the least energetic part of the spectrum to minimize photodegradation. Data were collected in a stepwise linear way with more data points in the early parts of the event. Sampling was carried out with constant high frequency during the first part of the sampling time. In the latter parts, sampling frequency decreased stepwise. Data were smoothed with a moving average (window size = 3) and, subsequently, a five-time-slot binning. Absorption intensity decays were fitted to an exponential model (Eq. 26) in OriginPro using the Levenberg-Marquardt routine. The quality of the fit, evaluated by the χ2-value R2 and the residual plots, was used to decide the appropriate exponential model. The governing equations for time-resolved intensity decay are summarized by Eq. 27, in which Abs(t) is the intensity decay, αi is the amplitude (preexponential factor), τi is the lifetime of the ith component, and ∑αi = 1.0.
| (26) |
| (27) |
| (28) |
Data were analyzed using a global analysis approach (30,31). The fractional intensity, fi, of each decay time is given by Eq. 28 and the mean lifetime by Eq. 26.
Data processing
In Figs. 2 A, 3 A, and 4 A, a zero-intensity threshold was applied to suppress the fluorescence afterglow at earlier probe times in the blue edge of the spectrum. Negative intensities observed at early probe times due to sample fluorescence were set to zero intensity. Fluorescence will add to the probe light and the detector will see more light than expected. For this reason, absorption signals cannot be seen in the earliest probe times in some of the data sets (e.g., the “missing” transient absorption below 450 nm in Fig. 2 A close to time zero). The detector was adjusted to high sensitivity to permit detection of weak absorption. This makes the intense fluorescence emission saturate the detector, leading to an oscillating signal while the detector recovers from the overload that is most significant at lower wavelengths. This means that the useful part of the data trace occurs later in the sampling time for the lower wavelengths. Data in Figs. 2 C, 3 C, 4 C, and 5 have been normalized to analyze the formation kinetics of solvated electrons.
Figure 2.
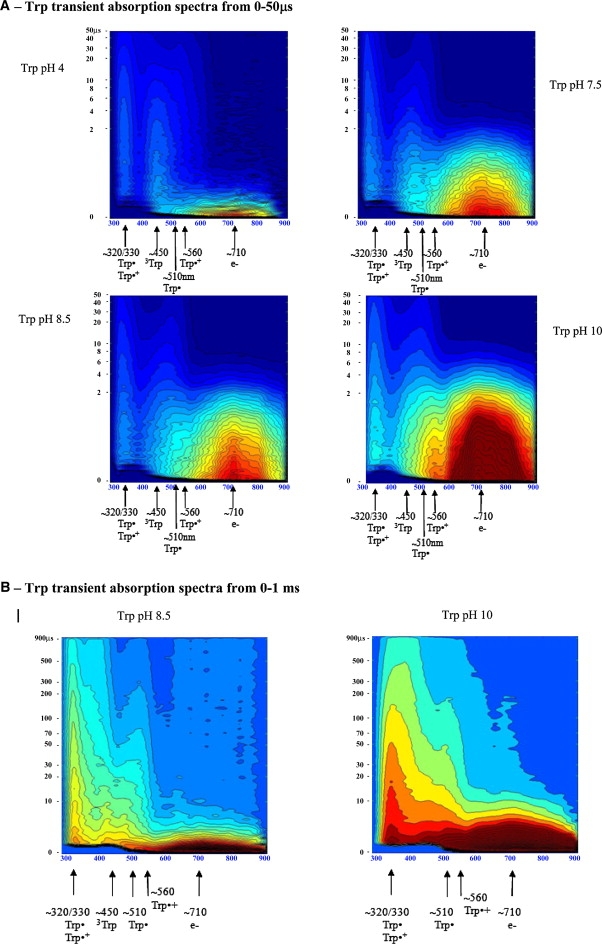
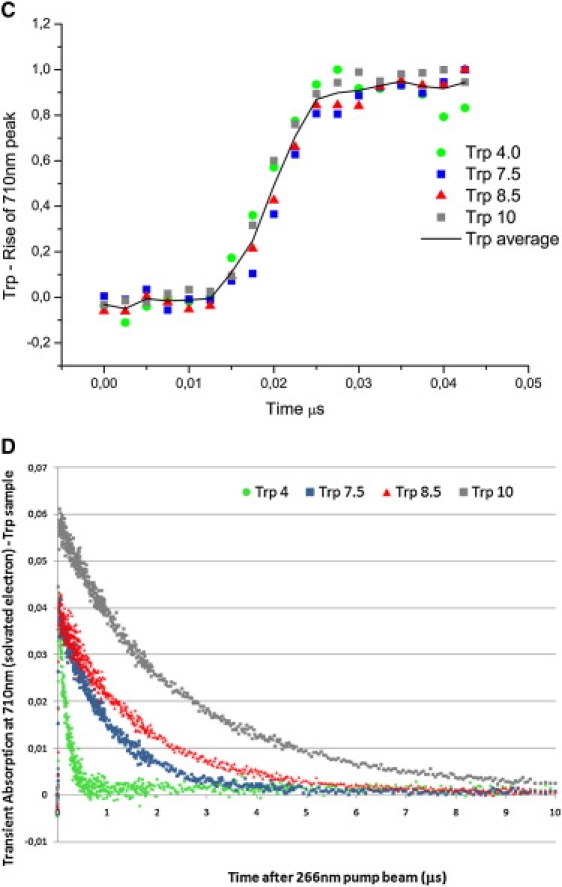
Trp transient absorption data: A shows transient absorption spectra collected at probe times from 0 to 50μs (short timescale); B shows the transient absorption spectra collected at probe times from 0 to 1ms (long timescale). Intensity of each spectrum at a particular probe time is color coded. Red codes for the highest intensity and dark blue for the lowest intensity. Putative transient absorption species assigned to each peak are listed. C displays kinetics of formation of the solvated electron within 45 ns after excitation. D displays decay kinetics of the solvated electron within 10 μs after excitation.
Figure 3.
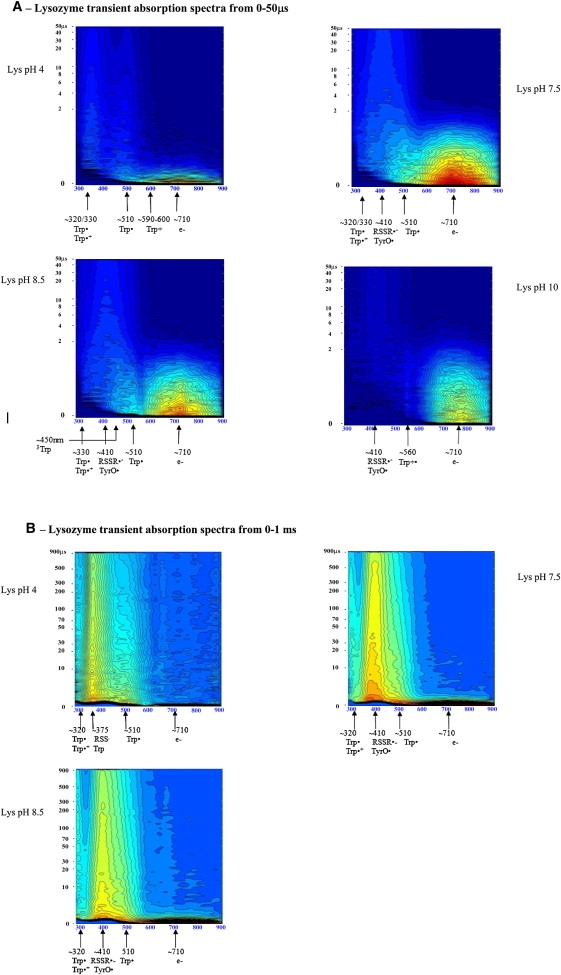
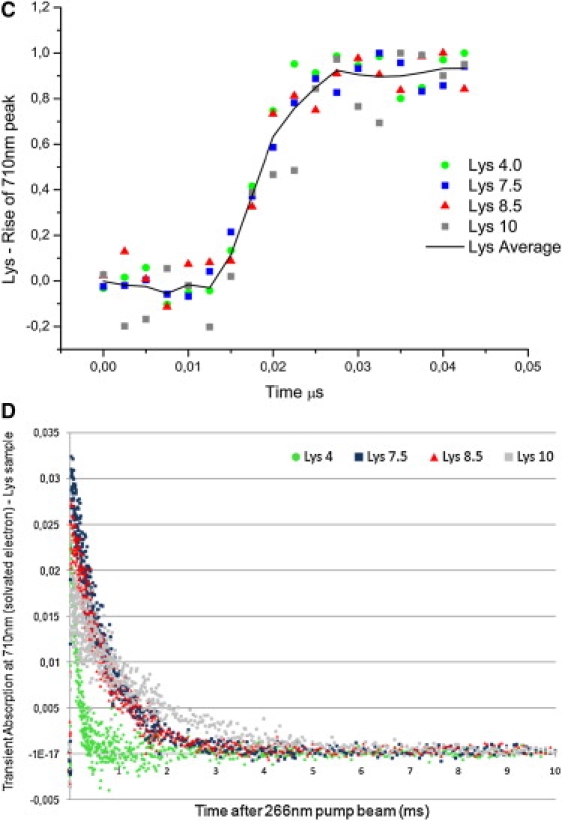
Lysozyme transient absorption data: A shows transient absorption spectra collected at probe times from 0 to 50 μs; B shows transient absorption spectra collected at probe times from 0 to 1 ms. Red codes for the highest intensity and dark blue for the lowest intensity. Putative transient absorption species assigned to each peak are listed. C shows kinetics of formation of the solvated electron within 45 ns after excitation. D displays the decay kinetics of the solvated electron within 10 μs after excitation.
Figure 4.
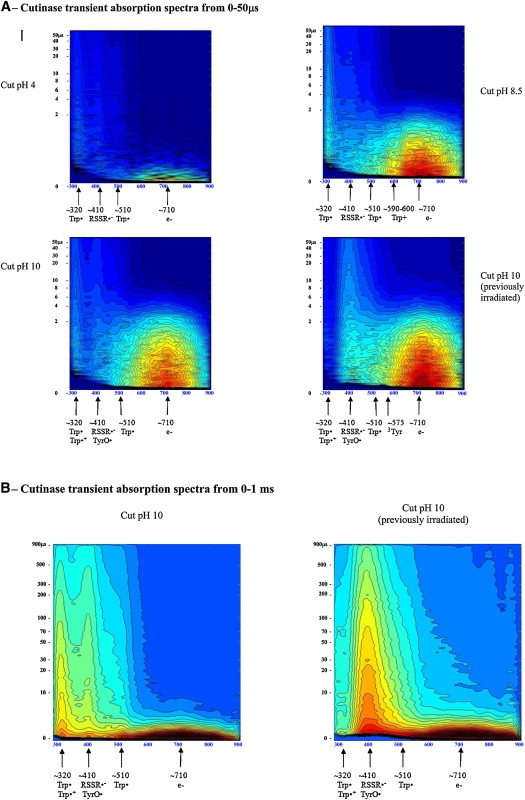
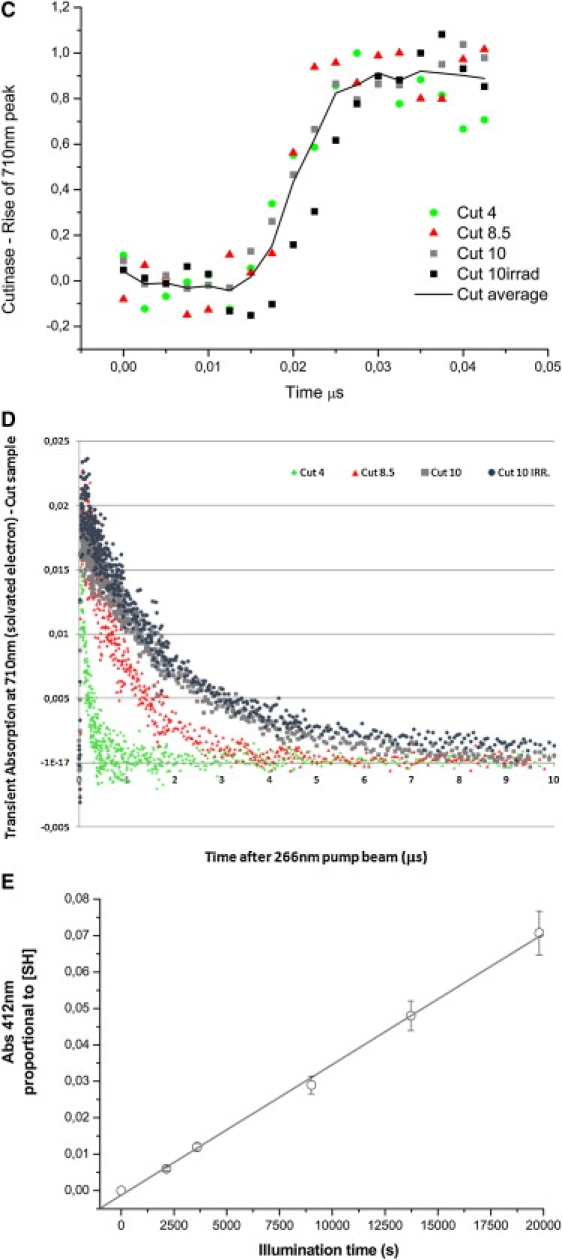
Cutinase transient absorption data: A shows transient absorption spectra collected at probe times from 0 to 50 μs; B displays transient absorption spectra collected at probe times from 0 to 1 ms. Red codes for the highest intensity and dark blue for the lowest intensity. Putative transient absorption species assigned to each peak are listed. C displays the kinetics of formation of the solvated electron within 45 ns after excitation. D shows the decay kinetics of the solvated electron within 10 μs after excitation. (E) Correlation between UV excitation (296 nm) time of cutinase solution and concentration of free thiol groups, detected with the Ellman assay (20). Free thiol concentration, proportional to Abs412nm, is the same as RSSR·− concentration.
Figure 5.
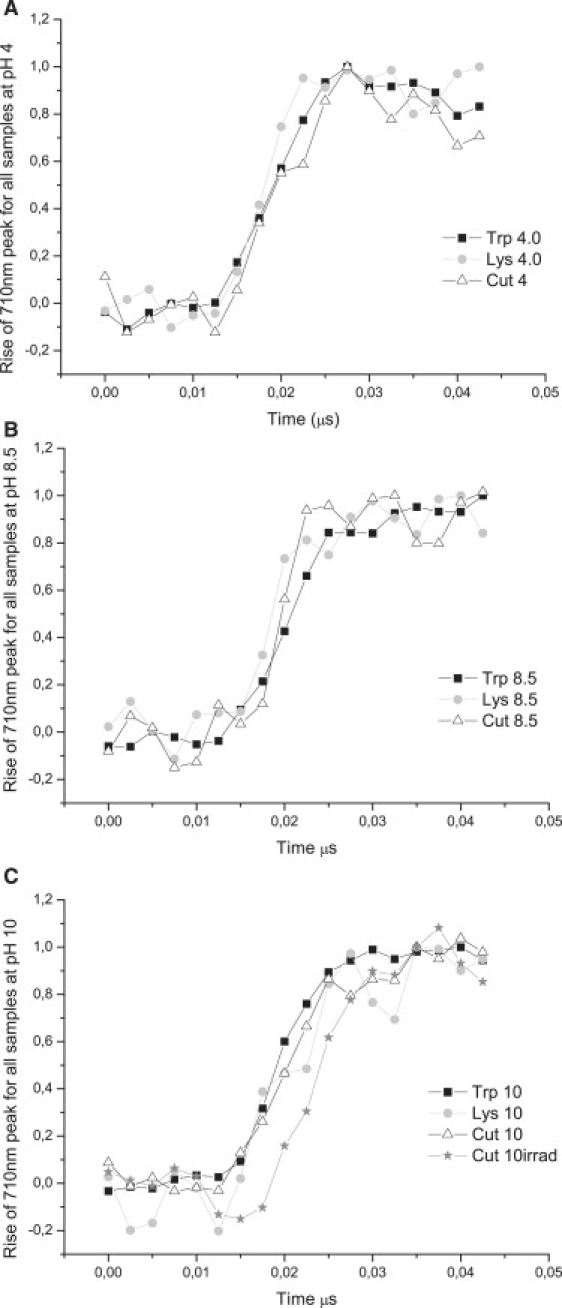
Normalized transient absorption data at 710 nm for 0–50 ns probe times displaying the kinetics of formation of the solvated electron for each sample at each pH value.
Solvated electron concentration
To calculate the yields of solvated electrons relative to the concentration of the parent molecule, solvated electron concentration was determined using absorbance at 710 nm and ɛ710nm = 19,167 M−1 cm−1 (32). Concentrations were normalized by dividing each concentration by the respective parent molecule concentration.
Detection of thiol group concentration
For a description of this process, see Neves-Petersen et al. (20).
Results
The 2D transient absorption spectra (0- to 50-μs time delays) of Trp, lysozyme, and cutinase are shown in Figs. 2 A, 3 A, and 4 A, respectively. Possible transient species, wavelength at maximum absorption, extinction coefficients, and lifetimes are listed in Table 1. Assignments will be discussed based on the experimental data. To observe longer-lived blue-edge absorption peaks, data with time delays from 0 to 1 ms were obtained (Figs. 2 B, 3 B, and 4 B). Lifetimes of each observed species are displayed in Tables 2–5. Formation kinetics of solvated electrons are displayed in Figs. 2 C, 3 C, and 4 C. Decay kinetics of solvated electrons are displayed in Figs. 2 D, 3 D, and 4 D. A comparison of the formation kinetics at each pH value for the different samples is displayed in Fig. 5. As a control, buffer solutions have been irradiated and transient absorption spectra acquired (data not shown). A weak, nondecaying absorption signal is observed at ∼300 nm but nothing above 300 nm, showing that none of the observed species created upon excitation of Trp, lysozyme, and cutinase samples originate from the buffer.
Table 2.
Lifetimes, preexponential factors, and average lifetimes as a function of pH for transient species formed upon Trp excitation
| Wavelength/species | Molecule | pH | Mean lifetime | Lifetimes and preexponential factors for global fitting |
R2 | ||
|---|---|---|---|---|---|---|---|
| τ1 (μs) | τ2 (μs) | ||||||
| 330 nm (0–50 μs data set) | Tryptophan | 1.89 ± 0.06 | 25.1 ± 1.00 | 0.99133 | |||
| Trp· | 〈τ〉 (μs] | α1 | α2 | ||||
| 4.0 | 24.8 | 0.13 ± 0.02 | 0.87 ± 0.01 | ||||
| 7.5 | 23.9 | 0.41 ± 0.01 | 0.59 ± 0.01 | ||||
| 8.5 | 24.0 | 0.40 ± 0.01 | 0.60 ± 0.01 | ||||
| 10.0 | 23.2 | 0.54 ± 0.01 | 0.46 ± 0.01 | ||||
| τ1 (μs) | τ2 (μs) | ||||||
| 550 nm (0–50 μs data set) | Tryptophan | 0.34 ± 0.01 | 2.35 ± 0.03 | 0.98266 | |||
| Trp·+ | 〈τ〉 (μs) | α1 | α2 | ||||
| 4.0 | 34.2 | 0.97 ± 0.03 | 0.03 ± 0.02 | ||||
| 7.5 | 32.5 | 0.94 ± 0.01 | 0.06 ± 0.01 | ||||
| 8.5 | 30.1 | 0.87 ± 0.02 | 0.13 ± 0.01 | ||||
| 10.0 | 20.4 | 0.56 ± 0.01 | 0.44 ± 0.01 | ||||
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| 330 nm (0–1 ms data set) | Tryptophan | 3.02 ± 0.08 | 40.1 ± 0.2 | 316 ± 11 | 0.99709 | ||
| Trp· | 〈τ〉 (μs) | α1 | α2 | α3 | |||
| 8.5 | 292 | 0.45 ± 0.01 | 0.22 ± 0.01 | 0.33 ± 0.01 | |||
| 10.0 | 294 | 0.45 ± 0.01 | 0.20 ± 0.01 | 0.35 ± 0.01 | |||
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| 520 nm (0–1 ms data set) | Tryptophan | 3.13 ± 0.03 | 33.6 ± 2.1 | 291 ± 16 | 0.99702 | ||
| Trp· | 〈τ〉 (μs) | α1 | α2 | α3 | |||
| 8.5 | 260 | 0.71 ± 0.01 | 0.12 ± 0.01 | 0.17 ± 0.01 | |||
| 10.0 | 249 | 0.80 ± 0.01 | 0.09 ± 0.00 | 0.11 ± 0.00 | |||
| τ1 (μs) | τ2 (μs) | ||||||
| 560 nm (0–1 ms data set) | Tryptophan | 2,87 ± 0.02 | 100 ± 4.3 | 0.99701 | |||
| Trp·+ | 〈τ〉 (μs) | α1 | α2 | ||||
| 7.5 | 82.3 | 0.11 ± 0.00 | 0.89 ± 0.01 | ||||
| 8.5 | 78.0 | 0.09 ± 0.00 | 0.91 ± 0.01 | ||||
| 10.0 | 74.8 | 0.08 ± 0.00 | 0.92 ± 0.01 | ||||
| τ1 (μs) | τ2 (μs) | ||||||
| 450 nm (0–50 μs data set) | Tryptophan | 1.51 ± 0.03 | 12.4 ± 0.4 0.99137 | ||||
| 3Trp | 〈τ〉 (μs) | α1 | α2 | ||||
| 4.0 | 11.8 | 0.33 ± 0.01 | 0.67 ± 0.01 | ||||
| 7.5 | 10.6 | 0.62 ± 0.01 | 0.38 ± 0.01 | ||||
| 8.5 | 11.2 | 0.51 ± 0.01 | 0.49 ± 0.01 | ||||
| 10.0 | 10.9 | 0.57 ± 0.01 | 0.43 ± 0.01 | ||||
Table 3.
Lifetimes of the solvated electron in all samples at different pH values
| Molecule | pH | Mean lifetime | Lifetimes and pre-exponential factors for triple exponential global fitting | χ2/DoF | R2 | ||
|---|---|---|---|---|---|---|---|
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| Tryptophan | 0.17 ±0.01 | 1.11 ±0.045 | 2.93 ±0.07 | 1.4126E-6 | 0.99469 | ||
| 〈τ〉(μs) | α1 | α2 | α3 | ||||
| 4.0 | 1.0 | 0.98 ±0.01 | 0.00 ±0.02 | 0.02 ±0.01 | |||
| 7.5 | 1.1 | 0.05 ±0.02 | 0.95 ±0.02 | 0.00 ±0.03 | |||
| 8.5 | 2.3 | 0.00 ±0.01 | 0.61 ±0.02 | 0.39 ±0.02 | |||
| 10.0 | 2.8 | 0.00 ±0.01 | 0.23 ± 0.03 | 0.77 ±0.02 | |||
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| Lysozyme | 0.13 ±0.01 | 0.716 ±0.04 | 1.61 ±0.14 | 1.565E-6 | 0.97039 | ||
| 〈τ〉(μs) | α1 | α2 | α3 | ||||
| 4.0 | 0.3 | 0.99 ±0.02 | 0.00 ±0.04 | 0.01 ±0.02 | |||
| 7.5 | 0.7 | 0.03 ±0.03 | 0.97 ±0.04 | 0.00 ±0.05 | |||
| 8.5 | 0.8 | 0.00 ±0.03 | 0.97 ±0.04 | 0.03 ±0.05 | |||
| 10.0 | 1.6 | 0.03 ±0.05 | 0.00 ± 0.15 | 0.97 ±0.12 | |||
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| Cutinase | 0.15 ±0.01 | 0.97 ±0.07 | 2.11 ±0.13 | 1.0213E-6 | 0.97276 | ||
| 〈τ〉(μs) | α1 | α2 | α3 | ||||
| 4.0 | 0.3 | 1.00 ±0.03 | 0.00 ±0.05 | 0.01 ±0.03 | |||
| 8.5 | 1.0 | 0.00 ±0.03 | 1.00 ±0.06 | 0.00 ±0.07 | |||
| 10.0 | 2.1 | 0.00 ±0.03 | 0.00 ±0.11 | 1.00 ±0.09 | |||
The lifetimes, their normalized weights, and the associated errors were obtained from global fits of the decay data for each molecule. The mean lifetime as a function of pH, as well as goodness-of-fit parameters for each dataset, are presented. The dominant lifetime is in bold.
Table 4.
Lifetimes, pre-exponential factors and average lifetimes as a function of pH for the transient species formed upon lysozyme excitation
| Wavelength/species | Molecule | pH | Mean lifetime | Lifetimes and pre-exponential factors for global fitting | R2 | ||
|---|---|---|---|---|---|---|---|
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| 330 nm (0-50μs data set) | |||||||
| Trp· | Lysozyme | 1.45 ± 0.12 | 17.2 ± 2.24 | 0.89206 | |||
| 〈τ〉(μs) | α1 | α2 | α3 | ||||
| 4.0 | 12.5 | 0.57 ± 0.03 | 0.43 ± 0.02 | ||||
| 7.5 | 14.0 | 0.54 ± 0.03 | 0.46 ± 0.02 | ||||
| 8.5 | 11.5 | 0.59 ± 0.03 | 0.41 ± 0.02 | ||||
| 10.0 | —- | —- | —- | ||||
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| 520 nm (0–50μs data set) | Lysozyme | 0.71 ± 0.01 | 11.0 ± 1.4 | 0.97227 | |||
| Trp· | 〈τ〉(μs) | α1 | α2 | α3 | |||
| 4.0 | 9.4 | 0.74 ± 0.03 | 0.26 ± 0.02 | ||||
| 7.5 | 7.1 | 0.90 ± 0.01 | 0.10 ± 0.01 | ||||
| 8.5 | 7.5 | 0.89 ± 0.01 | 0.11 ± 0.01 | ||||
| 10.0 | —- | —- | —- | —- | |||
| Lysozyme370 nm (0–1 ms data set)(RSS(R)SR)· | τ1 (μs) | τ2 (μs) | |||||
| 3.63 ± 0.18 | 300 ± 40 | ||||||
| 〈τ〉(μs) | α1 | α2 | |||||
| 410 nm (0–1ms data set) | 4.0 | 286 | 0.80 ± 0.04 τ1 (μs) | 0.20 ± 0.01 τ2 (μs) | 0.87372 | ||
| RSSR •− | 7.5 | 476 | 3.81 ± 0.160.79 ± 0.03 | 480 ± 700.21 ± 0.03 | 0.90144 | ||
| 8.5 | 477 | 0.73 ± 0.03 | 0.27 ± 0.02 | ||||
Table 5.
Lifetimes, preexponential factors, and average lifetimes as a function of pH for the transient species formed upon cutinase excitation
| Wavelength/species | Molecule | pH | Mean lifetime | Lifetimes and preexponential factors for global fitting |
R2 | ||
|---|---|---|---|---|---|---|---|
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| 330 nm (0–50 μs data set) | Cutinase | 0.73 ± 0.06 | 14.8 ± 1.1 | 0.91229 | |||
| Trp· | 〈τ〉 | α1 | α2 | α3 | |||
| (μs) | |||||||
| 4.0 | 13.3 | 0.70 ± 0.10 | 0.30 ± 0.01 | ||||
| 8.5 | 13.1 | 0.74 ± 0.10 | 0.26 ± 0.01 | ||||
| 10.0 | 13.3 | 0.70 ± 0.09 | 0.30 ± 0.01 | ||||
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| 520 nm (0–50 μs data set) | Cutinase | 0.69 ± 0.03 | 2,5 ± 0.08 | 0.97832 | |||
| Trp· | 〈τ〉 | α1 | α2 | α3 | |||
| (μs) | |||||||
| 4.0 | 1.0 | 0.95 ± 0.08 | 0.06 ± 0.06 | ||||
| 8.5 | 1.5 | 0.81 ± 0.02 | 0.19 ± 0.02 | ||||
| 10.0 | 2.4 | 0.10 ± 0.03 | 0.90 ± 0.03 | ||||
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| 310 nm (0–1 ms data set) | Cutinase | 3.76 ± 0.20 | 48.8 ± 4.9 | 398 ± 40 | 0.99131 | ||
| Trp· | 〈τ〉 | α1 | α2 | α3 | |||
| (μs) | |||||||
| 10.0 | 368 | 0.43 ± 0.01 | 0.23 ± 0.01 | 0.35 ± 0.01 | |||
| τ1 (μs) | τ2 (μs) | τ3 (μs) | |||||
| 500 nm (0–1 ms data set) | Cutinase | 1.95 ± 0.03 | 25.0 ± 2.7 | 330 ± 26 | 0.98486 | ||
| Trp· | 〈τ〉 | α1 | α2 | α3 | |||
| (μs) | |||||||
| 4.0 | 316 | 0.21 ± 0.03 | 0.50 ± 0.01 | 0.30 ± 0.01 | |||
| 10.0 | 304 | 0.85 ± 0.01 | 0.10 ± 0.00 | 0.05 ± 0.00 | |||
| 310 nm (0–1 ms data set) | Cutinase | τ1 (μs) | τ2 (μs) | τ3 (μs) | |||
| Trp· | 3.76 ± 0.31 | 50 ± 7.8 | 400 ± 60 | 0.97841 | |||
| Nonirradiated | 〈τ〉 | α1 | α2 | α3 | |||
| (μs) | 0.43 ± 0.02 | 0.23 ± 0.02 | 0.35 ± 0.02 | ||||
| Irradiated | 10.0 | 370 | τ1 (μs) | τ2 (μs) | τ3 (μs) | 0.87159 | |
| 10.0 | 206 | 3.86 ± 0.42 | 210 ± 20 | ||||
| α1 | α2 | ||||||
| 0.54 ± 0.01 | 0.46 ± 0.01 | ||||||
| 400 nm (0–1 ms data set) | Cutinase | τ1 (μs) | τ2 (μs) | τ3 (μs) | |||
| RSSR·− | 1.83 ± 0.32 | 10.0 ± 2.0 | 390 ± 40 | ||||
| Nonirradiated | 〈τ〉 | α1 | α2 | α3 | 0.94340 | ||
| (μs) | 0.59 ± 0.08 | 0.18 ± 0.03 | 0.23 ± 0.01 | ||||
| 385 | τ1 (μs) | τ2 (μs) | τ3 (μs) | ||||
| Irradiated | 10.0 | 341 | 2.07 ± 0.17 | 30.0 ± 3.7 | 360 ± 30 | 0.97312 | |
| α1 | α2 | α3 | |||||
| 0.58 ± 0.05 | 0.15 ± 0.01 | 0.27 ± 0.01 | |||||
Trp data
A short-lived peak centered at ∼710 nm assigned to solvated electrons (1) and longer-lived peaks centered at ∼330, 450, 510, and 560 nm are observed (Fig. 2, A and B). According to the literature, the 330-nm peak can be assigned to Trp· and Trp•+ (1,2), the 450-nm peak to 3Trp (1), the 510-nm peak to Trp• (2), and the 560-nm peak to Trp•+ (1). To confirm these assignments, the decay kinetics of each peak were analyzed, and lifetimes were recovered and compared with those from the literature (Tables 2 and 3). To conclude whether the 330-nm peak could be assigned to Trp•+, its lifetime was compared with that at 560 nm (the peak assigned to Trp·+). If the peaks at 560 nm and 330 nm were to be assigned to the same transient species, the recovered lifetimes should be similar. It can be seen clearly from Fig. 2 B and Table 2 that a faster decay is observed at 560 nm (20–34 μs in the 0–50 μs data set, 550 nm data, and 82 μs in the 0–1 ms data set) than at 330 nm (∼293 μs in the 0–1 ms data set). A 560-nm peak is clearly observed at pH 10 (Fig. 2 A). To conclude whether the 330-nm peak is to be assigned to Trp•, we compared these peak lifetimes with the lifetime of the 510-nm peak, known to be assigned to Trp· (lifetime in the hundreds of microseconds range (2,33,34)). The recovered lifetime is ∼250 μs (see Table 2, 520 nm data). The magnitude of the lifetime at 510 nm is very similar to the recovered lifetime at 330 nm (Table 2, 0–1 ms data set at 330 and 510/520 nm), indicating that the species absorbing at 510 nm and 330 nm are the same. From Fig. 2 B, we can see that the decay kinetics of the 560-nm peak (74–82 μs) is clearly faster than the 510-nm and 330-nm kinetics (249–294 μs). Thus, the 330-nm and 510-nm peaks are assigned to Trp· and the 560-nm peak to Trp•+. The 330-nm peak is dominated by the contribution from Trp•. The average lifetimes of the 330-nm and 560-nm traces are displayed in Fig. 6, B and C, respectively, and Table 2 (520 nm). The average lifetime of Trp•+ (560 nm) decreases with increasing pH (Table 2 and Fig. 6 C, circles).
Figure 6.
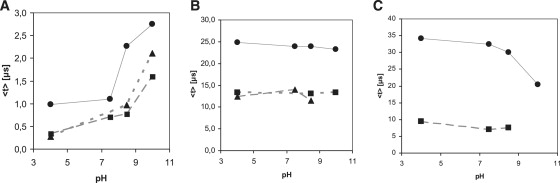
pH dependence of the recovered mean lifetimes of specific transient species. (A) Mean lifetime of solvated electrons in tryptophan (solid circles), cutinase (solid triangles), and lysozyme (solid squares). (B) Mean lifetime of Trp· species at 330 nm in tryptophan (solid circles), cutinase (solid triangles), and lysozyme (solid squares). (C) Mean lifetime of Trp·+ at 560 nm (Trp sample, solid circles), and of Trp· species at 510 nm (Lys sample, solid squares).
Absorption at 450 nm is assigned to 3Trp (1). Lifetime analysis at 450 nm shows that 3Trp has lifetimes from 10 to 12 μs, depending on pH (Table 2, 0–50 μs data set), in agreement with previously reported values (11–16 μs in neutral solution (1)). The 710-nm data show that the solvated electron has microsecond and submicrosecond decay lifetimes (Table 3). Solvated electron peak intensity is clearly pH-sensitive: the higher the pH value, the larger the yield of solvated electrons formed after excitation of the molecules with UV light (Fig. 2 A). The decay of the 710-nm peak is faster at pH 4 than at higher pH values for all samples (Figs. 2, A and D, and 6 A, and Table 3). Solvated electron lifetime increases nonlinearly with pH from 0.2 μs at pH 4 to 2.5 μs at pH 10.0 (Fig. 6 A, circles). Solvated electrons are observed ∼12 ns after the pump pulse, and the 710-nm peak reaches its maximum intensity ∼40 ns after excitation (Fig. 2 C). The rate of formation of the solvated electron is similar at the different pH values (Fig. 2 C) and is similar for all samples at a particular pH (Fig. 5).
Lysozyme data
Characteristic solvated electron absorption at ∼710 nm is observed as well as longer-lived peaks centered ∼320/330 nm, 510/520 nm, 560 nm (Fig. 3, A and B). For the reasons discussed above for Trp, 330-nm and 510-nm peaks are assigned to Trp• (Tables 3 and 4). A 560-nm peak is only observed at pH 10 (Fig. 3 A, pH 10) and is assigned to Trp•+. The Trp· average lifetime at 510 nm is not pH-sensitive (Fig. 6 C (lysozyme) and Table 4). Lifetimes recovered at 330 nm and 520 nm are very similar, spanning 7–9 μs at 520 nm and 11–14 μs at 330 nm (Table 4), although shorter than the ones observed with Trp alone. The yield of formation of solvated electrons drops from neutral to alkaline pH values (data not shown). Solvated electron has microsecond and submicrosecond decay lifetimes (Table 3, Fig. 6 A, squares, and Fig. 3 D), with mean lifetime increasing exponentially with pH (Fig. 6 A, squares), from 0.3 μs at pH 4 to 1.6 μs at pH 10.0. Solvated electrons are observed ∼12 ns after pump pulse, and reach maximum intensity ∼40 ns after excitation (Fig. 3 C). Rate of formation of solvated electron is similar at different pH values (Fig. 3 C) and is similar for all samples at a particular pH (Fig. 5).
A 3Trp characteristic absorption peak at 450 nm is likely observed (Fig. 3 A, Lys pH 8.5). The assignment of this peak in protein spectra is not as trivial as in the Trp sample due to the fact that other peaks populate nearby spectral regions. The characteristic 575-nm peak of 3Tyr is likely to be observed, though this peak is convolved with a neighboring broad and intense solvated electron peak (Fig. 3 A). A peak centered around 410 nm is observed at neutral and basic pH (Fig. 3, A and B). Two known species absorb at this wavelength (Table 1): RSSR·− and TyrO·. The latter has two characteristic absorption bands, at 410 nm and 390 nm (35,36). The 410-nm peak has lifetimes in the microsecond timescale (∼476 μs, Table 4, 0–1 ms data set). The extinction coefficient at 420 nm of RSSR·− is 3.3 times larger than the extinction coefficient of TyrO· (Table 1). RSSR·− is formed upon reaction of the triplet state of Trp or Tyr and a nearby disulphide bridge. Both lysozyme and cutinase have disulphide bridges near aromatic residues. These triplets are rapidly quenched by disulfides with kq = 2 − 4 × l09 M−1 s−1 (3). RSSR·− is known to dissociate into thiol compounds (15,27). UV excitation of lysozyme leads to the formation of free thiol groups (data not shown). The only sources of SH groups in this protein are its disulphide bridges. Native protein has no free SH groups. This indirectly confirms the presence of the precursor adduct RSSR·− formed upon UV excitation of our sample. On the other hand, TyrO· is known to be involved in other reactions that lead to its depletion (27). Also, the 290-nm characteristic peak of TyrO· is not clearly observed. Based on the above considerations, we conclude that the 410-nm peak (Fig. 3 A) could be assigned to both species, however most likely to the disulphide electron adduct RSSR·−. The observed average lifetimes at pH 7.5 and pH 8.5 are identical (see Table 4).
A peak at 370 nm is observed at pH 4, with an average lifetime of 286 μs (Table 4, Fig. 3 B, pH 4), previously assigned to the formation of a transient adduct radical (37):
| (29) |
This species is suggested to be sulfuranyl radical with the unpaired electron located in an antibonding σ∗ orbital within a trisulphide bridge. On the other hand, the same band has been assigned by Morine and Kuntz to RSS·, produced in a primary photolytic step by breaking C-S bond. This radical is formed in degassed solutions, e.g., achieved through argon/vaccum degassing like in our case. Protonation of the cyclic disulphide bridge anion RSSR·− leads to the disulphide-H atom adduct, which can rapidly decay leading to the formation of both RS· and RSS· (Scheme 30), the latter absorbing at 380 nm (38–40). Ito and Matsuda (41) photolysed di-t-butyl disulfide in hexane and attributed the 370 nm to thiyl radicals (RS·).
| (30) |
Therefore, we conclude that the 370-nm peak should be assigned to perthyil radicals, RSS·. A peak with absorption at ∼390 nm has also been attributed to an indole OH· adduct formed when, e.g., electron quenchers like N2O are present (42), which is not the case. The 295-nm and 575-nm peaks attributed to 3Tyr (3) are not observed.
Cutinase data
As in the case of lysozyme and Trp, a short-lived positive peak centered around 710 nm and longer-lived peaks centered around 320/330 nm and 510 nm are observed in cutinase. Trp·+ 560 nm is not clearly observed. The average lifetime of the Trp· radical at 330 nm and 520 nm, displayed in Table 5, is significantly shorter than those observed with Trp alone. At all pH values, the largest yield of formation of solvated electrons has been found upon excitation of cutinase, followed by lysozyme and Trp (data not shown). For cutinase at pH 4, compared to Trp, a 325% increase in the formation of solvated electrons was observed. For both lysozyme and cutinase, the yield of formation of solvated electrons drops from neutral to alkaline pH, whereas the opposite is observed for Trp. For cutinase, the mean lifetime of the solvated electron increases from 0.17 μs at pH 4 to 2.1 μs at pH 10.0 (Table 2). Solvated electrons are observed ∼12 ns after the pump pulse and the 710-nm peak, reaching its maximum intensity at ∼40 ns (Fig. 4 C). Fig. 4 D shows the decay kinetics of the solvated electron. The formation kinetics of solvated electron are similar at all pH values.
The 450 nm 3Trp peak is likely to be observed, though this peak is convolved with other peaks. The characteristic 3Tyr 575-nm peak is likely to be convolved with the solvated electron peak (Fig. 4 A). A peak at ∼410 nm is observed. For the reasons stated above, this peak is likely to be assigned to RSSR·−, formed according to
| (31) |
| (32) |
RSSR·− is the precursor of thiol groups according to
| (33) |
In Fig. 4 E, it can be seen that the longer the UV excitation time, the higher the concentration of free SH groups in cutinase, which is correlated with an increase of cutinase fluorescence (20). Transient absorption of cutinase at pH 10 was obtained both before and after excitation until its fluorescence emission intensity reached a plateau (Fig. 4, A and B). The UV-excited sample before flash photolysis shows a more intense solvated electron peak. Furthermore, a larger yield of solvated electrons is seen (data not shown). In addition, illumination of the sample before flash photolysis leads to a more intense RSSR·− peak and to the near disappearance of the 320-nm Trp· peak. Recovered lifetimes are summarized in Table 5: the average lifetime at 310 nm has dropped from 370 μs to 206 μs due to preillumination of the sample before flash photolysis. The distribution of preexponential factors associated with solvated electron lifetimes (0–50 μs data) is shown in Table 3, and mean lifetimes are displayed in Fig. 6. For all samples it can be observed that the preexponential factor associated with the shortest lifetime dominates at low pH, whereas the preexponential factor associated with the longest lifetime takes over at the highest pH value (Table 3). Solvated-electron mean lifetime increases for all samples as a function of pH (Fig. 6 A).
Discussion
In the experiments presented here, we collected near-UV, visible, and near-infrared transient absorption data for aqueous Trp, lysozyme, and cutinase. The peaks observed in the near-UV and visible regions for Trp, lysozyme, and cutinase in solution exhibited clear dynamics within the time window of the experiment. As a control, buffer solutions were irradiated at 266 nm and transient absorption spectra were acquired (data not shown). A weak, nondecaying absorption signal around 300 nm is observed, but nothing is observed above 300 nm. This shows that no species observed upon 266-nm illumination of Trp, lysozyme, and cutinase samples are created upon 266-nm illumination of buffer alone. The 3Trp recovered lifetimes (Trp sample) agree with the literature (1). Due to the close distance between disulphide bridges and aromatic residues in the proteins, it is very likely that RSSR is the quencher of 3Trp and 3Tyr triplet states, since it will lead to efficient electron transfer from donor to acceptor molecule, according to
| (34) |
| (35) |
The pH dependence of the Trp·+ lifetime is in agreement with earlier findings (1). As found for indole, its lifetime in neutral solutions is relatively short (τ ∼ 1 × 10−6 s), and in alkaline solutions, it is much shorter-lived, due, presumably, to loss of a proton to form the neutral radical (1). For all samples, transient absorption data shows an absorption peak centered around 710 nm, assigned to transient absorption of solvated electrons. Ejected electron becomes solvated by water molecules showing the characteristic broad absorption at ∼710 nm (1). The observed microsecond and submicrosecond decay lifetimes (Table 3) can be attributed to different recombination pathways of this species: recombination with the parent molecule (geminate recombination), with a hydronium ion, or with another electron acceptor, such as disulphide bridges and positively charged groups. The intensity of the solvated electron peak is clearly pH-dependent and correlates with H3O+ being an electron scavenger. Recombination happens according to (43)
| (36) |
Therefore, the lower the pH, the faster the rate of decay of solvated electrons formed upon UV excitation of Trp molecules. In a protein, different groups can act as electron scavengers, e.g., positively charged residues, the carbonyl group of the peptide chain (44), and disulphide bridges:
| (37) |
Data show that the higher the pH, the longer time it takes for the solvated electron to recombine with the parent molecule (geminate recombination) or another electron scavenger molecule, such as H3O+. The observed lifetime increase with pH is easily explained, since the lower the pH, the higher the concentration of H3O+, and therefore the larger the probability of recombination of the solvated electron with hydronium ion. Furthermore, for proteins, the higher the pH, the larger the number of basic residues that have lost their positive charge and become neutral (His, Lys, Arg) and the larger the number of acidic residues that have acquired a negative charge (Asp, Glu, Tyr). Therefore, pH increase leads to a gain of negative charge in the protein and to an increase of the protein area that carries negative electrostatic potential. Therefore, pH increase will decrease the efficiency of electron recombination with the molecule due to electrostatic repulsion. This will lead to an increase of the solvated electron lifetime (Figs. 3 D and 4 D). As observed in Figs. 2 C, 3 C, and 4 C, the rate of formation of the solvated electron is similar at the different pH values. Furthermore, the rate of formation of the solvated electron at a particular pH value is similar for Trp, lysozyme, and cutinase (Figs. 5). We conclude, therefore, that the kinetics of electron ejection and solvation are similar, and independent of the solvent accessibility of the electron donor.
The lifetime of the solvated electron created upon illumination of the proteins is shorter than the solvated electron lifetime in Trp solution, possibly because in proteins several groups can act as electron scavengers (disulphide bridges, positively charged groups, regions with positive electrostatic potential ≥1 kT/e), including solvent. This will result in the observed shorter lifetime of this transient species. Data also show that prolonged UV excitation of cutinase before flash photolysis increases the concentration of solvated electrons. This could indicate that the efficiency of recombination of the ejected electron with electron acceptors such as disulphide bridges has been reduced. This would be the case if UV preillumination induced structural changes in the protein leading to the increase of the distance between the aromatic residues and the disulphide bridges in cutinase, and if it created a pool of molecules with broken disulphide bridges. Indeed, it is known that UV illumination induces conformational changes in cutinase that lead to changes in the secondary structural content of the protein (21), leading to Trp fluorescence dequenching due to induced disruption of the nearby disulphide bridge upon excitation of the Trp residue ((20) and Fig. 4 E). Since cutinase has no free thiol groups, and since free thiol groups can only emerge due to the breakage of disulphide bridges, the precursor of these groups is likely to be the disulphide electron adduct RSSR·− (Scheme 33).
The UV-light-induced reactions that we are trying to unravel in proteins involve disruption of disulphide bridges upon illuminating the spatially nearby aromatic residues, believed to be induced by UV-induced electron ejection from the side chains of the aromatic residues and subsequent capture by the nearby good electron acceptor, the disulphide bridge. Indeed, our data here show that for the cutinase molecules preilluminated with UV light, the concentration of ejected electrons from the aromatic residues present in the 3D structure of cutinase increases. As explained above, this could be due to the disruption of the nearby disulphide bridge that no longer can act as an electron acceptor. When observing Fig. 4 B, we can clearly see that upon preillumination of the sample, the RSSR•− peak at ∼410 nm becomes more intense, whereas the 330-nm peak seems to almost disappear. Furthermore, data show that the 310–330 nm peak decays faster due to sample preillumination. The increase in signal intensity at 410 nm is likely due to the larger concentration of the disulphide electron adduct RSSR•− and/or the formation of another species, the TyrO· radical, according to (27)
| (38) |
The decay of Tyr·+ to Tyr· is much faster than the time resolution of our experiments. This possible mechanism explains the increase in intensity of the 410-nm peak concomitant with the decay of the intensity of the 330-nm peak assigned to Trp· and the shorter lifetime of the Trp· species after preillumination of the sample. Tyr· has longer decay kinetics (lifetime on the millisecond timescale) not observed in our data. However, we cannot completely rule out the presence of this species, since the time window of our experiments prevents reliable lifetime assignment on the millisecond timescale.
There could be several other reasons for the observed decrease of the Trp· radical peak. 1), This radical can propagate along the protein backbone (27) and recombine with other groups. If caught by RSSR, this would also explain the observed increase in the intensity at 410 nm. 2), Photo-induced degradation of the parent Trp molecule may occur. 3), UV-illumination-induced conformational changes in the protein (21), which likely lead to larger distances between aromatic residues and RSSR, mean that electron transfer (ET) will not be efficient or will disappear (Scheme 39). The decay of Trp·+ to Trp· is much faster than the time resolution of our experiments.
| (39) |
Upon ET, the excited-state electron on the donor molecule is transferred to a vacant excited-state orbital on the acceptor molecule. However, this is a very short-range interaction that decays rapidly as the distance beyond van der Waals overlap between donor and acceptor increases (19).
Trp· lifetimes are shorter in lysozyme and cutinase compared with those of Trp, likely due to additional reaction pathways: propagation of radicals through the protein backbone and recombination with other groups, and reaction with Tyr residues according to
| (40) |
The lysozyme 370/380-nm peak has been previously assigned to the formation of a transient H-atom adduct (18). The same band was assigned to RSS· (38), formed in degassed solutions as in our case. This radical is said to be formed by protonation of the disulphide anion with pK of 5–6 (15,41). Ito and Matsuda photolysed di-t-butyl disulfide in hexane and attributed the 370 nm to perthyil radicals (RSS·) and showed that it could not be due to RS· radicals (41). Disulphide-hydrogen atom adducts decay rapidly (38–40) with a possible mechanism displayed in Scheme 30. Therefore, we conclude that the 370-nm peak should be assigned to perthyil radicals, RSS·.
Acknowledgments
The authors declare that they have no competing financial interests.
References
- 1.Bent D.V., Hayon E. Excited-state chemistry of aromatic amino-acids and related peptides .3. tryptophan. J. Am. Chem. Soc. 1975;97:2612–2619. doi: 10.1021/ja00843a004. [DOI] [PubMed] [Google Scholar]
- 2.Creed D. The photophysics and photochemistry of the near-UV absorbing amino-acids .1. Tryptophan and its simple derivatives. Photochem. Photobiol. 1984;39:537–562. [Google Scholar]
- 3.Bent D.V., Hayon E. Excited-state chemistry of aromatic amino-acids and related peptides .1.tyrosine. J. Am. Chem. Soc. 1975;97:2599–2606. doi: 10.1021/ja00843a002. [DOI] [PubMed] [Google Scholar]
- 4.Creed D. The photophysics and photochemistry of the near-uv absorbing amino-acids .2. Tyrosine and its simple derivatives. Photochem. Photobiol. 1984;39:563–575. [Google Scholar]
- 5.Bent D.V., Hayon E. Excited-state chemistry of aromatic amino-acids and related peptides .2.phenylalanine. J. Am. Chem. Soc. 1975;97:2606–2612. doi: 10.1021/ja00843a003. [DOI] [PubMed] [Google Scholar]
- 6.Creed D. The photophysics and photochemistry of the near-UV absorbing amino-acids.3. Cystine and its simple derivatives. Photochem. Photobiol. 1984;39:577–583. [Google Scholar]
- 7.Hart E.J., Boag J.W. Absorption spectrum of the hydrated electron in water and in aqueous solutions. J. Am. Chem. Soc. 1962;84:4090–4095. [Google Scholar]
- 8.Wiesenfeld J.M., Ippen E.P. Dynamics of electron solvation in liquid water. Chem. Phys. Lett. 1980;73:47–50. [Google Scholar]
- 9.Crowell R.A., Bartels D.M. Multiphoton ionization of liquid water with 3.0–5.0 eV photons. J. Phys. Chem. 1996;100:17940–17949. [Google Scholar]
- 10.Sander M.U., Luther K., Troe J. On the photoionization mechanism of liquid water. Ber. Bunsenges. Phys. Chem. 1993;97:953–961. [Google Scholar]
- 11.Migus A., Gauduel Y., Martin J.L., Antonetti A. Excess electrons in liquid water: first evidence of a prehydrated state with femtosecond lifetime. Phys. Rev. Lett. 1987;58:1559–1562. doi: 10.1103/PhysRevLett.58.1559. [DOI] [PubMed] [Google Scholar]
- 12.Long F.H., Lu H., Eisenthal K.B. Femtosecond studies of the presolvated electrons: an excited state of the solvated electron? Phys. Rev. Lett. 1990;64:1469–1472. doi: 10.1103/PhysRevLett.64.1469. [DOI] [PubMed] [Google Scholar]
- 13.Tsentalovich Y.P., Snytnikova O.A., Sagdeev R.Z. Properties of excited states of aqueous tryptophan. Photochem. Photobiol. Chem. 2004;162:371–379. [Google Scholar]
- 14.Armstrong R.C., Swallow A.J. Pulse- and gamma-radiolysis of aqueous solutions of tryptophan. Radiat. Res. 1969;40:563–579. [PubMed] [Google Scholar]
- 15.Hoffman M.Z., Hayon E. One-electron reduction of the disulfide linkage in aqueous solution. Formation, protonation, and decay kinetics of the RSSR- radical. J. Am. Chem. Soc. 1972;94:7950–7957. [Google Scholar]
- 16.Adams G.E., Willson R.L., Aldrich J.E., Cundall R.B. On the mechanism of the radiation-induced inactivation of lysozyme in dilute aqueous solution. Int. J. Radiat. Biol. 1969;16:333–342. doi: 10.1080/09553006914551361. [DOI] [PubMed] [Google Scholar]
- 17.Redpath J.L., Santus R., Ovadia J., Grossweiner L.I. Oxidation of tryptophan by radical-anions. Int. J. Radiat. Biol. 1975;27:201–204. doi: 10.1080/09553007514550211. [DOI] [PubMed] [Google Scholar]
- 18.Grossweiner L.I., Usui Y. Flash photolysis and inactivation of aqueous lysozyme. Photochem. Photobiol. 1971;13:195–214. doi: 10.1111/j.1751-1097.1971.tb06106.x. [DOI] [PubMed] [Google Scholar]
- 19.Li Z., Lee W.E., Galley W.C. Distance dependence of the tryptophan-disulfide interaction at the triplet level from pulsed phosphorescence studies on a model system. Biophys. J. 1989;56:361–367. doi: 10.1016/S0006-3495(89)82682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neves-Petersen M.T., Gryczynski Z., Lakowicz J., Fojan P., Pedersen S. High probability of disrupting a disulphide bridge mediated by an endogenous excited tryptophan residue. Protein Sci. 2002;11:588–600. doi: 10.1110/ps.06002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neves-Petersen M.T., Snabe T., Klitgaard S., Duroux M., Petersen S.B. Photonic activation of disulfide bridges achieves oriented protein immobilization on biosensor surfaces. Protein Sci. 2006;15:343–351. doi: 10.1110/ps.051885306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prompers J.J., Hilbers C.W., Perpermans H.A.M. Tryptophan mediated photoreduction of disulfide bond causes unusual fluorescence behaviour of Fusarium solani pisi cutinase. FEBS Lett. 1999;45:409–416. doi: 10.1016/s0014-5793(99)00990-4. [DOI] [PubMed] [Google Scholar]
- 23.Vanhooren A., Devreese B., Vanhee K., Van Beeumen J., Hanssens I.A. Photoexcitation of tryptophan groups induces reduction of two disulfide bonds in goat alpha-lactalbumin. Biochemistry. 2002;41:11035–11043. doi: 10.1021/bi0258851. [DOI] [PubMed] [Google Scholar]
- 24.Duroux M., Skovsen E., Neves-Petersen M.T., Duroux L., Gurevich L. Light-induced immobilisation of biomolecules as a replacement for present nano/micro droplet dispensing based arraying technologies. Proteomics. 2007;7:3491–3499. doi: 10.1002/pmic.200700472. [DOI] [PubMed] [Google Scholar]
- 25.Neves-Petersen M.T., Duroux M., Skovsen E., Duroux L., Petersen S.B. Printing novel architectures of nanosized molecules with micrometer resolution using light. J. Nanosci. Nanotech. 2009;9:3372–3381. doi: 10.1166/jnn.2009.ns04. [DOI] [PubMed] [Google Scholar]
- 26.Skovsen E., Neves-Petersen M.T., Kold A., Duroux L., Petersen S.B. Immobilizing biomolecules near the diffraction limit. J. Nanosci. Nanotech. 2009;9:4333–4337. doi: 10.1166/jnn.2009.m55. [DOI] [PubMed] [Google Scholar]
- 27.Kerwin B.A., Remmele R.L. Protect from light: photodegradation and protein biologics. J. Pharm. Sci. 2007;96:1468–1479. doi: 10.1002/jps.20815. [DOI] [PubMed] [Google Scholar]
- 28.Neves-Petersen M.T., Petersen S.B. Protein electrostatics: a review of the equations and methods used to model electrostatic equations in biomolecules—applications in biotechnology. Biotechnol. Annu. Rev. 2003;9:315–395. doi: 10.1016/s1387-2656(03)09010-0. [DOI] [PubMed] [Google Scholar]
- 29.Neves-Petersen M.T., Petersen E.I., Fojan P., Noronha M., Madsen R.G. Engineering the pH-optimum of a triglyceride lipase: from predictions based on electrostatic computations to experimental results. J. Biotechnol. 2001;87:225–254. doi: 10.1016/s0168-1656(01)00240-1. [DOI] [PubMed] [Google Scholar]
- 30.Janssens L.D., Boens N., Ameloot M., De Schryver F.C. A systematic study of the global analysis of multiexponential fluorescence decay surfaces using reference convolution. J. Phys. Chem. 1990;94:3564–3576. [Google Scholar]
- 31.Verveer P.J., Squire A., Bastiaens P.I. Global analysis of fluorescence lifetime imaging microscopy data. Biophys. J. 2000;78:2127–2137. doi: 10.1016/S0006-3495(00)76759-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kloepfer J.A., Vilchiz V.H., Lenchenkov V.A., Germaine A.C., Bradforth S.E. The ejection distribution of solvated electrons generated by the one-photon photodetachment of aqueous I- and two-photon ionization of the solvent. J. Chem. Phys. 2000;113:6288–6307. [Google Scholar]
- 33.Mazul V.M., Scherbin D.G., Shashilov V.A., Sukhodola A.A., Zaitseva E.M. Recombination prolonged luminescence of indole and tryptophan in a solution at room temperature. J. Appl. Spectrosc. 2003;70:270–274. [Google Scholar]
- 34.Burdi D., Aveline B.M., Wood P.D., Stubbe J., Redmond R.W. Generation of a tryptophan radical in high quantum yield from a novel amino acid analog using near-UV/visible light. J. Am. Chem. Soc. 1997;119:6457–6460. [Google Scholar]
- 35.Grossweiner L.I., Swenson G.W., Zwicker E.F. Photochemical generation of the hydrated electron. Science. 1963;141:805–806. doi: 10.1126/science.141.3583.805. [DOI] [PubMed] [Google Scholar]
- 36.Grossweiner L.I., Kaluskar A.G., Baugher J.F. Flash photolysis of enzymes. Int. J. Radiat. Biol. 1976;29:1–16. doi: 10.1080/09553007614551511. [DOI] [PubMed] [Google Scholar]
- 37.Bonifacic M., Asmus K.-D. Adduct formation and absolute rate constants in the displacement reaction of thiyl radicals with disulfides. J. Phys. Chem. 1984;88:6286–6290. [Google Scholar]
- 38.Morine G.H., Kuntz R.R. Observations of c-s and s-s bond cleavage in the photolysis of disulfides in solution. Photochem. Photobiol. 1981;33:1–5. [Google Scholar]
- 39.Purdie J.W., Gillis H.A., Klassen N.V. The pulse radiolysis of penicillamine and penicillamine disulfide in aqueous solution. Can. J. Chem. 1973;51:3132–3142. [Google Scholar]
- 40.Neta P., Schuler R.H. Rate constants for reaction of hydrogen atoms with compounds of biochemical interest. Radiat. Res. 1971;47:612–627. [Google Scholar]
- 41.Ito O., Matsuda M. Flash photolysis study on the addition reaction of alkylthiyl radical to olefins. Bull. Chem. Soc. Jpn. 1978;51:427–430. [Google Scholar]
- 42.Grossweiner L.I., Usui Y. The role of the hydrated electron in photoreduction of cystine in the presence of indole. Photochem. Photobiol. 1970;11:53–56. doi: 10.1111/j.1751-1097.1970.tb05717.x. [DOI] [PubMed] [Google Scholar]
- 43.Spanel P., Smith D. Recent studies of electron attachment and electron-ion recombination at thermal energies. Plasma Sources Sci. Technol. 1995;4:302–306. [Google Scholar]
- 44.Faraggi M., Bettelheim A. The reaction of the hydrated electron with amino acids, peptides, and proteins in aqueous solutions: tryptophyl peptides. Radiat. Res. 1977;72:81–88. [PubMed] [Google Scholar]
- 45.Jovanovic S.V., Steenken S.J. Substituent effects on the spectral, acid-base, and redox properties of indolyl radicals: a pulse radiolysis study. J. Phys. Chem. 1992;96:6674–6679. [Google Scholar]
- 46.Dudley Bryant F., Santus R., Grossweiner L.I. Laser flash photolysis of aqueous tryptophan. J. Phys. Chem. 1975;79:2711–2716. [Google Scholar]
- 47.Feitelson J., Hayon E. Electron ejection and electron capture by phenolic compounds. J. Phys. Chem. 1973;77:10–15. [Google Scholar]
- 48.Feitelson J., Hayon E. Electron transfer from the excited state of tyrosine to compounds containing disulfide linkages. Photochem. Photobiol. 1973;17:265–274. [Google Scholar]
- 49.Bansal K.M., Fessenden R.W. Pulse radiolysis studies of the oxidation of phenols by s04- and br2- in aqueous solutions' 2. Rad. Res. 1976;67:1–8. [PubMed] [Google Scholar]
- 50.Fielden E.M., Hart E.J. Primary radical yields in pulse-irradiated alkaline aqueous solution. Radiat. Res. 1967;32:564–580. [PubMed] [Google Scholar]