

Abstract
The GM2 activator protein (GM2AP) is an accessory protein required for the enzymatic conversion of GM2 to GM3 by hydrolases in the lysosomal compartments of cells. Here, GM2AP interactions with lipid vesicles are investigated by sucrose-loaded vesicle sedimentation and gel filtration assays, and the effects of pH and lipid composition on membrane binding and lipid extraction are characterized. The sedimentation experiments allow for facile quantification of the percentage of protein in solution and on the bilayer surface, with detailed analysis of the protein:lipid complex that remains in solution. Optimum binding and ligand extraction is found for pH 4.8 where <15% of the protein remains surface associated regardless of the lipid composition. In addition to extracting GM2, we find that GM2AP readily extracts dansyl-headgroup-labeled lipids as well as other phospholipids from vesicles. The ability of GM2AP to extract dansyl-DHPE from vesicles is altered by pH and the specific ligand GM2. Although the unique endosomal lipid, bis(monoacylglycero)phosphate, is not required for ligand extraction, it does enhance the extraction efficiency of GM2 when cholesterol is present in the vesicles.
Introduction
Ganglioside catabolism occurs within the acidic lysosomal compartments of cells (1) and many of the catabolic reactions of gangliosides that contain small oligosaccharide groups require accessory proteins, collectively termed sphingolipid activator proteins (SAPs) (2). In vitro assays demonstrate that SAPs are required for enzymatic activity and the results imply a structural role of orienting the oligosaccharide headgroup for enzymatic hydrolysis (3). Specifically, when hydrolases and ganglioside micelles are mixed, little to no enzymatic cleavage occurs; however, upon addition of the specific accessory protein, the rate of cleavage increases (4). The accessory protein GM2 activator protein (GM2AP) is specific for the hydrolysis of the ganglioside GM2 to GM3, and in vivo, the protein is believed to bind and extract GM2 from the intralysosomal vesicles making the ganglioside headgroup accessible to the hydrolytic enzyme, beta-hexosaminidase A (HexA), for cleavage (5). GM2AP has also been shown to act as a lipid transfer protein (6,7).
The gene encoding GM2AP contains both a pre- and prosequence that direct the expression of GM2AP into the golgi, with a final location in the cell lysosome (8). The mature form of the protein, 18 kD, has been isolated from natural sources (9,10) and expressed as a glycosylated protein from both insect and yeast cells (11,12) and as a nonglycosylated form in Escherichia coli (13,14). The crystal structure of nonglycosylated GM2AP reveals a β-cup topology formed from eight β-strands that form a hydrophobic pocket (Fig. 1). Numerous structures of GM2AP have been deposited into the Protein Data Bank and reveal different modes of lipid binding for various ligands such as the ganglioside GM2 (15) and other phospholipids such as phosphatidylglycerol (PG) (15), phosphatidylcholine, oleic acid/lyso-phosphatidylcholine (16), and platelet-activating factor/lauric acid (17). These various ligand binding modes also correlate with minor conformational changes in the protein structure, as revealed most strikingly in GM2AP crystallized without ligand (Protein Data Bank ID 1GM1), where three different conformations of the protein are found within an 11-monomer unit cell (15). In particular, the most dramatic effects are observed in the putative membrane binding loops, specifically around W131, where in one conformer the tryptophan (TRP) moiety is tucked into the protein, compared with an aqueous exposed conformation in two other conformers. (Our amino acid numbering scheme for the E.coli recombinant construct expressed and utilized within this manuscript designates Ser32 of the proGM2AP sequence as Ser1. Some of the crystallographic assignments and figures within annotated references are shifted 31 amino acids from our numbering scheme.) Although the numerous crystal structures provide molecular level insight into how the protein conformation can adapt to various lipid ligands, a detailed molecular mechanism for how GM2AP interacts with vesicle surfaces, extracts ligands, and transfers lipids between vesicles remains unclear. A fluorescence resonance energy transfer (FRET)-based assay demonstrated the in vitro ability of GM2AP to extract NBD-GM2 from vesicles and to transfer this ligand from donor vesicles to acceptor vesicles containing rhodamine-DOPE (18). In addition, surface plasmon resonance studies of immobilized vesicles indicate that with certain lipid compositions, particularly those containing a unique endosomal lipid, bis(monoacylglycero) phosphate (BMP), GM2AP, and other SAPs can disturb the bilayer structure and mobilize the vesicles from the surface plasmon resonance (SPR) chip (19,20). However, the binding interactions, such as the partioning or kinetics of interaction of GM2AP with liposomes, have not been fully and systematically quantified.
Figure 1.
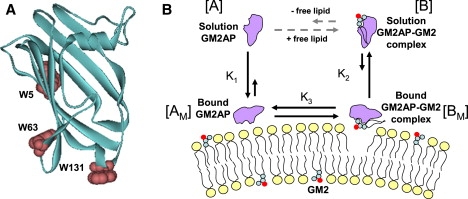
(A) Ribbon diagram of GM2AP (PDB ID 1G13), showing a β-cup topology, with the location of the three TRP residues shown in space filling format. (B) Cartoon showing a putative binding/extraction scheme for the interaction of GM2AP with lipid bilayers.
As reviewed by White (21), there are two general categories of methods for determining the partitioning of proteins or peptides from lipid vesicles: 1), physical separation and direct measurement of protein concentrations or 2), spectroscopic investigations where a given spectral feature correlates with the protein concentration in a given environment (21). Although both techniques find wide-range use in the field, spectroscopic techniques, which often rely on the intrinsic fluorescence emission from a TRP residue or an incorporated fluorophore in the protein or peptide, are highly common. In addition, binding assays have been utilized that exploit the FRET between the donor fluorophore in the protein and an acceptor fluorophore label incorporated into the lipid bilayer. The partitioning coefficient of the glycolipid transfer protein (GLTP) was determined by monitoring the increase in the acceptor fluorescence from FRET with intrinsic TRP fluorescence of the protein and dansyl-labeled DHPE lipids (acceptor) incorporated into the liposomes (22).
GM2AP has three TRP residues in its 162 amino acids. Given that two of these three sites (W63 and W131) are located in the putative membrane binding loops, it was expected that TRP fluorescence would be a useful technique for monitoring the membrane partitioning of GM2AP. However, this assay was of little use in characterizing the membrane partitioning of GM2AP. Ultimately, from physical separation of the protein in solution from that bound to the vesicle surface via ultracentrifugation sedimentation with sucrose-loaded vesicles or via gel filtration (23,24), we show that <15% of GM2AP remains on the surface of liposomes in the presence and absence of specific ligand GM2. From analysis of the protein in solution, it is shown that GM2AP can extract its specific and nonspecific lipid ligands in the absence of BMP. The ability of GM2AP to extract dansyl-DHPE (DDHPE) negates the ability to utilize this probe to monitor membrane partitioning, as was done in previous studies of GLTP (22). A model for GM2AP partitioning with lipid bilayers is presented in Fig. 1 B. This model is similar to that proposed for GLTP; however, a significant difference is that GM2AP can extract nonspecific lipids from the vesicles, thus establishing an equilibrium where the majority of the protein has extracted a ligand forming a protein:lipid complex in solution even in the absence of GM2. Additionally, these findings demonstrate that under acidic conditions, BMP is not required for lipid extraction, although it does enhance extraction efficiency, especially when liposomes contain cholesterol (CHOL). Because GM2AP functions in the acidic lysosomal compartment, the optimum pH for membrane binding and lipid extraction is found to be pH 4.8.
Matierials and Methods
Lipids
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG) and BMPdi18:1 or BMP in chloroform were purchased from Avanti Polar Lipids (Alabaster, AL) and used without further purification. N-(5-dimethylaminonaphthalene-1-sulfonyl)-1 and 2-dihexadecanoyl-sn- glycero-3-phosphoethanolamine (DDHPE) were obtained from Molecular Probes (Eugene, OR) in the form of powder. Monosialoganglioside GM2 and CHOL were purchased from Sigma-Aldrich (St. Louis, MO) as powders. All other reagents were obtained from Fisher Scientific (Pittsburg, PA) and used as received.
GM2AP
Recombinant GM2AP was prepared using an E. coli expression system as described in an earlier report (25) and is a modified procedure originally published by Wright (26). The structural integrity and purity of final protein samples were verified by circular dichroism spectra and SDS-PAGE. The circular dichroism spectra are consistent with the GM2AP generated from insect expression system as reported (20). The protein concentration was determined by Bradford assay and by absorption at 280 nm using an extinction coefficient of 23,000 M−1cm−1.
Preparation of lipid vesicles
Lipid vesicles were prepared by mixing the desired molar ratios of lipids in chloroform or other solvent. The lipid mixtures were dried under a stream of nitrogen to produce a dry film. The film was then subjected to at least six hours of vacuum desiccation. Unless otherwise stated, the lipids were hydrated in an appropriate volume of sodium acetate (50 mM NaOAc) buffer at desired pH and were subjected to a couple of freeze-thaw cycles using liquid nitrogen. Large unilamellar vesicles of the above lipid samples were prepared by extrusion, consisting of 55 passes through 100 nm polycarbonate filters using an Avanti hand-held miniextruder (Avanti Polar Lipids). Stock solutions of sucrose-loaded vesicle were prepared as described before (23,24) but hydrated with 176 mM sucrose, 50 mM NaOAC, pH 4.8. The vesicle size distributions were determined to be 100–160 nm by dynamic light scattering (Brookhaven Instruments, Holtsville, NY). Final phospholipid concentrations were determined on the basis of total phosphate determination by Malachite Green Phosphate Assay Kit (BioAssay Systems, Hayward, CA). Lipid percentages given throughout are in units of mol %.
Spectroscopic measurements
Fluorescence spectra were acquired on a FluoroMax-3 fluorimeter (Horiba Jobin Yvon, Edison, NJ) with a temperature-controlled cell holder. All experiments were performed at 20 °C by using a HAAKE K20 water bath circulator (Thermo Electron Corporation, Waltham, MA). Measurements were made using a 4 mm × 4 mm light path quartz cuvette (Starna, Atascadero, CA) with excitation and emission polarizers set to 90° and 0° orientations, respectively (27). Absorption spectra were collected on a Cary 50 Bio UV–Visible Spectrophotometer (Varian, Palo Alto, CA) using a 1 cm light path microvolume cuvette. All UV absorption measurements were performed at room temperature.
Fluorescence measurements
The extraction of DDHPE from lipid vesicles by GM2AP as a function of pH was monitored by fluorescence spectroscopy. For these experiments, the excitation wavelength was set to 280 nm and the emission spectra were recorded from 300 nm to 550 nm to include the TRP fluorescence emission of GM2AP and the emission of DDHPE. The DDHPE-labeled vesicles (POPC:DDHPE = 9:1, POPC:POPG:DDHPE = 6:3:1 or POPC:GM2:DDHPE = 8:1:1) were prepared in 50 mM NaOAc buffer (pH 4.8) at a final concentration of 100 μM. Fluorescence spectra were acquired as a function of titrating GM2AP into the vesicle solution. Samples were allowed to equilibrate for 8 min before spectra were acquired. The DDHPE emission intensities at 518 nm were used to monitor protein-lipid interactions. Signal intensities were corrected for dilution before subtracting the intensity in the absence of GM2AP.
Sedimentation assays for membrane partitioning and DDHPE extraction
The sedimentation procedure is similar to that reported by Buser (24). GM2AP protein concentration was determined by fluorescamine labeling (28). The ability of GM2AP to extract DDHPE was determined by quantification of the fluorescence intensity of DDHPE in the supernatant and pellet fractions of samples containing the same total amount of sucrose-loaded vesicles with varying concentrations of GM2AP (25). Further details are given in the Supporting Material. The measured fluorescence intensity of each sample was corrected for the appropriate dilution factor to give Isup (signal in the supernatant) and Ipel (signal of the pellet). Given a 200 μL volume of 100 μM lipid (POPC:DDHPE 2:1), the total concentration of DDHPE in the sample was known to be 33.3 μM. Therefore, the concentration of DDHPE in the supernatant Csup (in μM) was calculated by:
| (1) |
The residual lipid that remained in the supernatant from unpelleted vesicles was determined from control experiments (no protein), and these values were set as Cctr. Therefore, we define the change in DDHPE concentration in the supernatant, ΔDDHPE, as:
| (2) |
Model for membrane partitioning
Results from sedimentation assays were analyzed according to the equilibria shown in Fig. 1 B, where the fraction bound to the vesicle surface was determined quantitatively from knowing the total protein concentration and the fraction that remained in the supernatant. From the equilibria shown in the model, an expression that describes the relationship of the fraction of protein bound, fb, and the accessible lipid concentration [L] can be determined:
| (3) |
where K1, K2 and K3 are defined as follows:
| (4) |
| (5) |
| (6) |
Rearranging and substituting gives an expression in terms of the equilibrium constants and accessible lipid concentration as follows:
| (7) |
Quantification of GM2 extraction
GM2 extraction was quantified with an absorption resorcinol assay (29). For these experiments, 7.5 nmol GM2AP and 200 nmol vesicles containing 10 mol % GM2 with varying concentrations of POPC, Chol, and/or BMP were mixed in NaOAc buffer (50 mM, pH 4.8) with a total volume of 100 μL and incubated for 20 min at room temperature. Then the mixture was loaded onto a self-packed column (1.6 mm × 500 mm) with sephacryl S-200. The elution fractions were collected every two drops. The fraction volume was determined by weight or micro syringe to be on average 45 μL in our experiments. For fractions not containing vesicles, GM2AP concentration was determined from the optical density at 280 nm (OD280) with a 1 cm light path microcell. The GM2 concentration in each fraction (with or without vesicles) was measured by the resorcinol assay (29,30). Further details are given in the Supporting Material.
Results
Results from sucrose-loaded sedimentation assays
GM2AP is known to extract GM2 from intralysosomal vesicles, forming a GM2AP:GM2 complex for further reaction with HexA (31). Here, the partitioning of GM2AP with liposomes was monitored via sucrose-loaded vesicle sedimentation assays (24). This methodology is based on physical separation of protein free in solution from that bound to the vesicle surface, and it easily allows for direct quantification of the protein in both environments. From these experiments, the percentage of protein that remained in solution for a given lipid composition was measured. Because the total protein concentration was known, the fraction of protein bound, fb, can easily be calculated from measurements of protein remaining in solution after centrifugation. Fig. 2 plots the results from a series of sedimentation experiments performed at pH 4.8, where GM2AP was added to POPC liposomes of varied composition, including anionic lipids (PG or BMP) and functional ligand (GM2). These results show that the fraction of GM2AP bound to the liposomes reaches a maximum of 15 mol % for lipid concentrations >150 μM irrespective of the lipid compositions investigated. The top panel of Fig. 2 shows results from POPC vesicles. When protein that was isolated in solution was reequilibrated with a new set of lipid vesicles for >30 min, the same partitioning was obtained, with only 15% of the total protein pelletted with the sucrose-loaded vesicles. The bottom panel of Fig. 2 shows results for sedimentation experiments using POPC:POPG (7:3), POPC:GM2 (9:1) or POPC:BMP:GM2 (7:2:1) vesicles.
Figure 2.
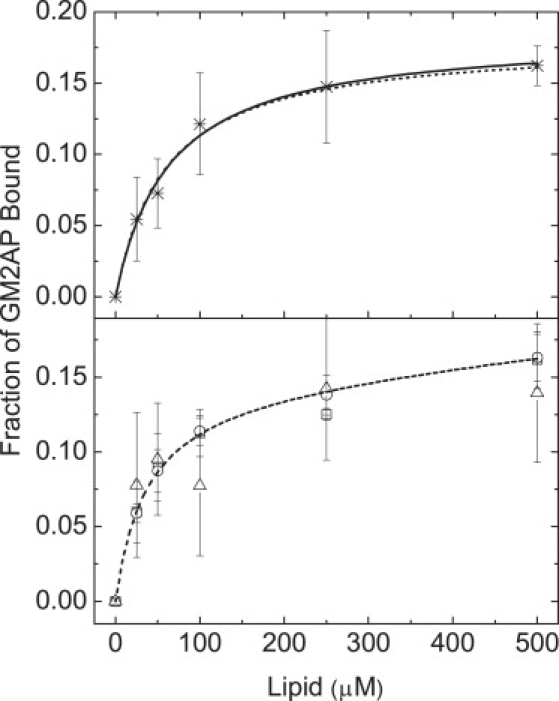
Membrane partitioning isotherms of GM2AP from sedimentation experiments. All experiments were performed in 50 mM NaOAc buffer, pH 4.8, with final GM2AP concentration of 10 μM. Figures plot the fraction of GM2AP bound to the lipid vesicles. Protein was incubated with 0 to 500 μM large unilamellar vesicles for 20 min and followed by ultracentrifugation at 100,000 × g for 1 h. The protein concentration in the supernatant was measured by fluorescamine labeling. The top panel shows results for POPC vesicles. The bottom panel shows results for POPC:POPG (7:3), circles; POPC:GM2 (9:1), squares; POPC:BMP:GM2 (7:2:1), down triangles. Lines are fits to the data as described within the text. Error bars represent standard deviations of three separate measurements.
The finding that only 15% of GM2AP remains associated with the vesicle and that lipid composition does not alter the amount of GM2AP that sediments with the vesicles is surprising. SPR studies have shown that when the endosomal lipid, BMP, is incorporated into vesicles, an addition of GM2AP caused a lowering of the signal below that in the absence of protein, which was interpreted as mobilization of the bilayer away from the chip surface (19). From SPR experiments, it is generally assumed that BMP is required for lipid extraction. Based on this assumption and other results from the GM2AP literature (4,18,19,32), it was anticipated that the amount of protein that sediments with the vesicles would follow a trend where the greatest “binding” would be observed for vesicles containing the functional ligand GM2, followed by those containing the negatively charged lipid PG, followed by POPC vesicles, and finally, the least binding was expected for vesicles containing BMP because of extraction of lipid expected in the presence of BMP. This anticipated trend relied on the assumption of a simple two-state model of protein in solution and protein bound to the surface, where changing the electrostatic charge of the bilayer or the addition of GM2, would alter the binding affinity. However, a two-state model of membrane binding cannot account for these data. An alternative explanation is that GM2AP can extract both nonspecific and specific ligands from vesicle surfaces in the absence of BMP. A model for how GM2AP interacts with lipid vesicles that includes lipid extraction is depicted in Fig. 1 B. This model is similar to that proposed for GLTP (22); however, a significant difference is the ability of GM2AP to extract lipids other than the ganglioside GM2, so the four-state equilibrium exists even when only POPC lipids are used. The solid lines in the data in Fig. 2 are fits to a model (Eq. 7) that takes into account three equilibrium constants between a), apo-protein in solution and the vesicle surface, defined as K1, b), protein and protein:lipid complex on the bilayer surface, defined as K3 and c), protein:lipid complex in solution in equilibrium with the vesicle surface, defined as K2. Fits to the data can be obtained for K2 < K3 < K1. This model implies that apo-GM2AP (protein without lipid ligand) and the GM2AP:lipid complex have different membrane binding affinities, with the complex having a lower affinity (K2 < K1), which may result from conformational changes in the protein upon binding of ligands; thus making the GM2AP:lipid complex less able to bind to the vesicle surface.
GM2AP extracts Dansyl-DHPE from vesicles forming a complex in solution
GM2AP has been crystallized with numerous lipid ligands (15–17) and we have previously shown that GM2AP can form a 1:1 complex with DDHPE when injected from an ethanol solution (25). Because the fluorescence intensity of DDHPE blue shifts to a wavelength comparable to that in benzene solution, we infer that DDHPE binds into the GM2AP pocket in the mode consistent with that seen for POPG (15). Various crystal structures for GM2AP show two distinct lipid binding pockets, one for phosphoglycerol lipids and one for the gangioside, GM2, where the sugar headgroup protrudes into solution. The alternative orientation for phosphoglycerol lipids is consistent with the in vitro finding that GM2AP protects phosphoglycerol lipids from the action of phospholipase D (33). It is likely that the DDHPE molecule is oriented within the hydrophobic pocket of GM2AP similarly to other phospholipids studied.
From sucrose-loaded sedimentation assays of POPC:DDHPE (3:1) vesicles, we find that the concentration of DDHPE in the supernatant increases in a linearly dependent manner as a function of GM2AP concentration, thus indicating that GM2AP can extract the DDHPE ligand from the vesicles. In addition, the presence of POPG, GM2 or CHOL does not alter the amount of DDHPE extracted by GM2AP. Fig. 3 A plots the change in DDHPE concentration in the supernatant as a function of GM2AP concentration for POPC vesicles and POPC vesicles containing POPG, GM2, or CHOL. Analysis of total protein and DDHPE concentration in the supernatant fractions shows that 85% of the GM2AP has formed a 1:1 complex with DDHPE; indicating that GM2AP has a preference of extraction of DDHPE over POPC, POPG, or CHOL. Thin layer chromatography reveals that the concentrations of both POPC and DDPHE increase as a function of GM2AP concentration (Supporting Material), indicating that GM2AP extracts not only DDHPE but also POPC from the liposomes. Note that BMP was not included in these vesicles and the data present direct evidence that BMP is not required for the extraction of DDHPE or POPC.
Figure 3.
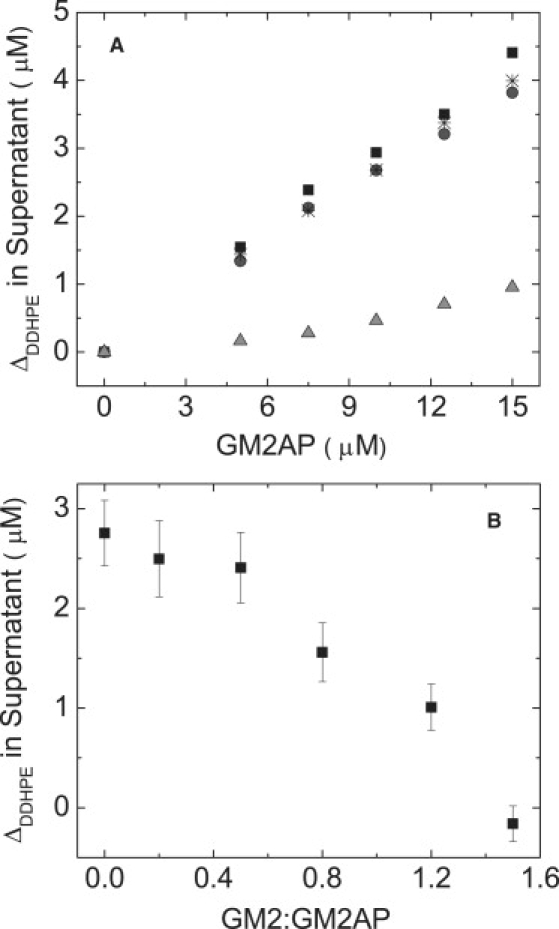
(A) GM2AP extracts DDHPE from liposomes. POPC:DDHPE (2:1), black squares; POPC:POPG:DDHPE (1:1:1), dark gray circles; POPC:CHOL:DDHPE (47:20:33), black asterisks; POPC:GM2:DDHPE (47:20:33), light gray up triangles. (B) Preincubation of GM2AP with GM2 micelles inhibits the extraction of DDHPE. 10 μM GM2AP was preincubated for 20 min with varying concentrations of GM2 micelles, which ranged from 0 to 15 μM. The GM2AP-GM2 mixture was then allowed to incubate with POPC:DDHPE (2:1) vesicles for 20 min, followed by ultracentrifugation at room temperature. For both A and B, the change in the DDHPE concentration in the supernatant was determined from fluorescence measurements as described in the Materials and Methods section. Data points represent the average value from three separate experiments with data points representing the standard deviation in A and error bars showing the standard deviation in B. All experiments were performed in 50 mM NaOAc, 100 mM KCl, pH 4.8 with 100 μM final lipid concentration.
Given that DDHPE is not the functional lysosomal ligand for GM2AP, it can be predicted that GM2AP should extract GM2 more readily than DDHPE from vesicles. Detailed studies have been performed for quantifying the affinity of GM2AP for various glycosphingolipids (4,18,19,32), and GM2 micelles have been shown to prevent GM2AP from binding a rhodamine labeled fatty acid, R18, and an assay based on the displacement of R18 by GM2 has been utilized to test function of GM2AP constructs (6,7). Fig. 3 A shows that when both DDHPE and GM2 are incorporated into POPC vesicles, the amount of DDHPE extracted is lowered when GM2 in present, indicating that GM2AP extracts GM2 from the liposome in preference to DDHPE. In addition, when GM2AP is preincubated with GM2 micelles, the extraction of DDHPE is lowered in a dose-dependent manner (Fig. 3 B). From these experiments, we can infer that GM2AP has a higher affinity for GM2 than DDHPE. When GM2AP is preincubated with a concentration of GM2 micelles equal to that of protein (10 μM, corresponding to a 1:1 GM2:GM2AP ratio), only 1 μM DDHPE was detected in the supernatant. For this experiment, 100 μM POPC:DDHPE (2:1) vesicles were utilized. Assuming an equal distribution of DDHPE in both lipid leaflets, this gives an accessible DDHPE concentration of 16.5 μM. When GM2AP was incubated with a concentration of GM2 micelles that equals that of accessible DDHPE (value of 1.6 on x-axis in Fig. 3 B), no DDHPE could be detected in the supernatant. Clearly, the presence of GM2, whether added to GM2AP before mixing with vesicles or when incorporated into the vesicles, mitigates the extraction of DDHPE, as expected.
BMP is not required for GM2 extraction
It has been reported that the anionic lipid BMP stimulates sphingolipid degradation and the ability of GM2AP to mobilize lipids from the bilayer surface (19,34). The results of the previous section directly show that GM2AP can extract DDHPE from vesicles in the absence of BMP. GM2AP can also extract GM2 from vesicles that do not contain BMP, but BMP enhances the extraction efficiency. Extraction efficiency is defined as the ratio of amount of GM2 relative to GM2AP recovered after gel-filtration in the nonvesicle fractions. It is a measure of how many GM2AP protein molecules extracted GM2 in the experiment. The gel filtration assay was chosen over the sucrose-loaded vesicle sedimentation assay because of interference of the remaining sucrose in solution with the method of detection of GM2. Each of the fractions from the gel-filtration column were analyzed for protein concentration and for GM2 concentration. Fig. 4 A shows an example of results from analysis of gel filtration fractions. For these experiments, GM2AP was allowed to incubate for 20 min with vesicles of varying composition before separation. The data are shown for POPC:GM2:CHOL:BMP (50:10:20:20) vesicles in 50 mM NaOAc, pH 4.8 buffer. Fig. 4 B plots the extraction efficiency (GM2/GM2AP) determined from this assay for varying lipid compositions. Both wild-type GM2AP and the AB variant (C107R) construct (35,36) were characterized. From these data, we can clearly see that GM2AP can extract GM2 from POPC:GM2 (9:1) vesicles without the presence of BMP. Specifically, a ratio of 43 ± 7% is found, indicating that this percentage of GM2AP in solution has formed a complex with GM2. A ratio of 100% would indicate that every GM2AP protein contains one GM2 lipid. The ability of GM2AP to extract GM2 is lessened to 25 ± 5% when CHOL is present (POPC:GM2:CHOL = 70:10:20). When BMP was introduced (POPC:GM2:CHOL:BMP = 50:10:20:20), 74 ± 12% of GM2AP formed a complex with GM2. The AB variant construct (C107R) also possesses GM2 extraction ability as a function of lipid concentration roughly equal to that of the wild-type GM2AP. This finding that the AB variant extracts GM2 in a manner similar to the wild-type protein is consistent with the published results of Xie et al. (37), using R18 dequenching assays, which claims that the mutant C107R does not alter the membrane binding and extraction function but rather, prevents interactions with HexA.
Figure 4.
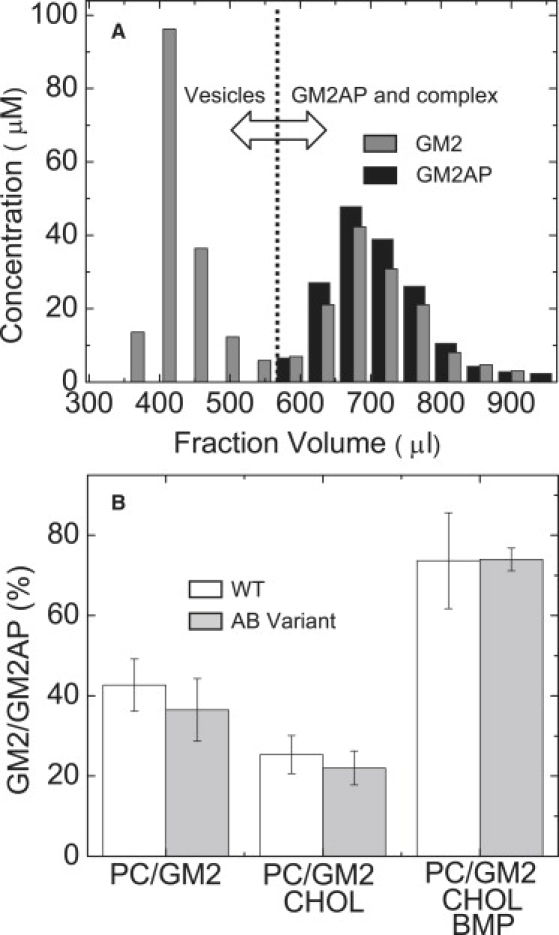
Gel filtration assay for GM2 extraction. (A) Elution profile of the mixture of 7.5 nmol GM2AP with 200 nmol POPC:GM2:CHOL:BMP (50:10:20:20) vesicles in 50 mM NaOAc pH 4.8 buffer with total volume of 100 μl. The average fraction size was 45 μl. The gray columns show the concentration of GM2 (determined from resorcinol assay) in each fraction. The black columns show the GM2AP concentration (determined from UV–VIS) in each fraction. Fractions that contained vesicles were determined by light scattering at 550 nm. Typically, fractions for elution volumes >500 μL contained no vesicles. (B) Efficiency of GM2 extraction as a function of lipid composition for both GM2AP (white) and the AB variant of GM2AP (light gray). The extraction efficiency is defined as the percent ratio of GM2 to GM2AP (100∗GM2/GM2AP). Error bars represent the standard deviations of three separate measurements. Lipid compositions: POPC:GM2 (9:1), POPC:GM2:CHOL (70:10:20), POPC:GM2:CHOL:BMP (50:10:20:20).
Monitoring the formation of GM2AP:DDHPE complex as a function of solution pH
Fig. 5 shows the effects of pH on the ability of GM2AP to extract DDHPE from POPC vesicles. Two different analytical assays were used. The top panel of Fig. 5 shows results from direct measurement of the concentration of DDHPE that remained in the supernatant after sucrose-loaded vesicle sedimentation via centrifugation, where POPC:DDHPE (2:1) vesicles were utilized. The largest change in DDHPE concentration was observed for pH 4.8. The bottom panel of Fig. 5 shows the change in fluorescence signal detected at 518 nm (the emission maximum of DDHPE in the GM2AP:DDHPE complex) with excitation at 280 nm, as a function of solution pH with POPC:DDHPE (3:1) vesicles. Here, no sedimentation was utilized, only direct spectroscopic measurement of changes in fluorescence intensity. The change in intensity at 518 nm arises from numerous factors including a change in the fluorescence quantum yield of DDHPE upon moving into the more hydrophobic environment of the protein, from resonance energy transfer from the GM2AP on the surface of the vesicles and from the GM2AP:DDHPE complex in solution. The quantum yield of dansyl fluorescence is known to be dependent on the polarity of the environment and the effects of FRET can be seen on the decrease of the TRP fluorescence upon addition of lipid vesicles, which is not seen when POPC vesicles are added (data not shown). The largest increase in fluorescence signal is also detected at pH 4.8. Interestingly, if the His-tag is left on the protein, the pH profile for maximum fluorescence signal is altered. This finding can indicate that the His-tag alters the pH- dependent membrane binding profile of GM2AP or simply quenches the fluorescence signal differently at various pH values. The effect was not investigated further by sucrose-loaded sedimentation experiments.
Figure 5.
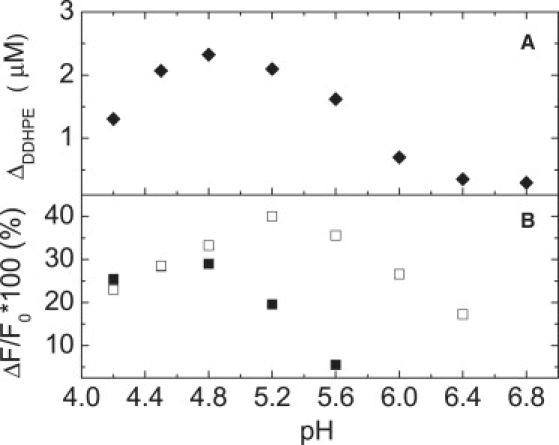
Effects of pH and His-tag on membrane binding and lipid extraction of GM2AP. (A) Plot of the change in DDHPE concentration in the supernatant as a function of solution pH for GM2AP extraction determined from sucrose-loaded vesicle sedimentation assays. For these experiments, 10 μM GM2AP was allowed to incubate for 20 min with 100 μM POPC:DDHPE (2:1) vesicles before separation by ultracentrifugation. For each pH value, the change in DDHPE concentration was referenced to samples that did not contain protein. (B) Percentage change in the fluorescence intensity detected at 518 nm with excitation wavelength of 280 nm for 10 μM GM2AP (solid) or 10 μM GM2AP10His-tag (open) with 50 μM POPC:DDHPE (9:1) vesicles as solution pH was varied. Initial fluorescence, F0, of vesicles was taken before addition of protein. The ΔF signal was determined by subtracting F0 from the value obtained (corrected for dilution) after the protein was added and allowed to incubate for 8 min. For both A and B, the solution buffers contained 25 mM NaOAc and 25 mM phosphate and the pH was adjusted by acetic acid. Each experiment was performed in triplicate and data point sizes are indicative of the error.
The 10-His tag alters lipid extraction properties of GM2AP
Because the fluorescence assay described above has contributions from both the membrane bound state of GM2AP and the complex formed upon extraction of DDHPE, the change in the fluorescence intensity at 518 nm can be used to monitor the kinetics of DDHPE extraction as a function of solution variables such as salt and pH. Fig. 6 shows results from kinetic assays as a function of pH and moderate ionic strength (100 mM NaCl) for GM2AP and without with His-tag. Fig. 6, A and C show the results without and with NaCl; respectively, at pH 4.8 for protein with and without the His-tag. It is clear that although the His-tag does not change the equilibrium signal (i.e., the change in fluorescence intensity reaches the same value within error for times longer than 10 minutes), the early kinetics of the function are clearly different. Additionally, the time course profile of GM2AP containing the His-tag, open symbols, changes only slightly when 100 mM NaCl is added, whereas for GM2AP that has had the tag cleaved, a stronger dependence on ionic strength for extraction of DDHPE is observed and the kinetics of interaction are slowed, although after 10 min, the change in total fluorescence signal for experiments with and without salt are nearly the same. Fig. 6 B shows how the pH profile is altered by the His-tag. For experiments performed at pH 6.4, no increase in fluorescence signal is detected for GM2AP that has the His-tag cleaved. However, a moderate increase in fluorescence intensity is seen for GM2AP that retains the His-tag, indicating some “function” over the 10-minute period. These findings indicate that care must be taken to ensure that the presence of the His-tag used during purification does not alter the function of the protein. Interestingly, the His-tag does not alter the circular dichroism spectrum of GM2AP (Supporting Material).
Figure 6.
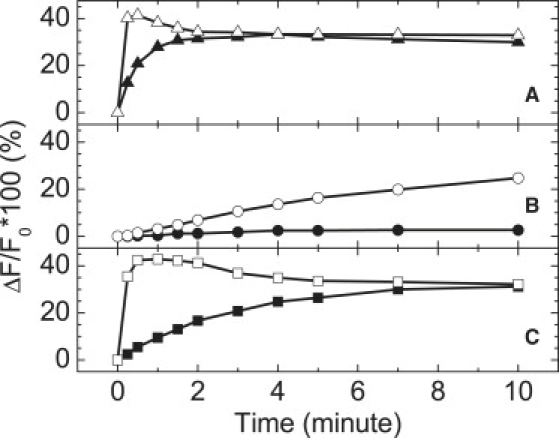
Effects of His-tag on membrane binding and lipid extraction monitored by time course fluorescence intensity at 518 nm from 10 μM GM2AP (solid) or 10 μM GM2AP10His-tag (open) with 50 μM POPC:DDHPE (9:1) in different conditions. (A) Results obtained for 50 mM NaOAc, pH 4.8, (B) 50 mM NaOAc, pH 6.4; (C) 50 mM NaOAc, 100 mM NaCl, pH 4.8. Results are discussed in the text. Excitation wavelength was 280 nm.
Discussion
GM2AP extracts nonspecific lipids with low selectivity compared to GM2 and BMP is not required for lipid extraction
The two reported functions of GM2AP, to present GM2 to HexA for hydrolytic cleavage and to transfer GM2 to acceptors, both start with the same initial step: extraction of GM2 from intralysosomal vesicle surfaces. As such, it is of interest to characterize the membrane binding interactions and lipid extraction properties in the absence of either HexA or other acceptor biomolecules. Our findings show that at equilibrium, ∼85% of GM2AP remains in solution as a complex with lipid ligands after it has extracted these from donor vesicles. Other reports have shown that GM2AP can bind varied ligands in vitro, and in those cases, the ligands were introduced in a concentrated ethanolic solution into a purified solution of GM2AP in acidic buffer (15). It has also been shown that GM2AP can extract rhodamine-labeled fatty acids from vesicles (18). Here, it is shown that GM2AP extracts various lipid ligands from vesicle surfaces; and for the case of DDHPE with selectivity lower than GM2; as shown by the inhibitory effect of GM2 on the extraction of DDHPE when either introduced into the membrane or preincubated with GM2AP in the form of micelles before exposure to POPC:DDHPE vesicles. Interestingly, GM2AP can also extract POPC from vesicles, an unexpected finding; although consistent with x-ray structures showing phospholipids bound when introduced from ethanolic solution (15,16,17). In addition, the data reported here clearly demonstrates the ability of GM2AP to extract lipids from vesicles without BMP. Both GM2 and DDHPE were extracted by GM2 when incorporated into POPC vesicles. The presence of CHOL lowered the ability of GM2AP to extract GM2. Addition of BMP did significantly enhance the extraction efficiency when CHOL was present.
Intrinsic fluorescence of GM2AP and Dansyl-DHPE fluorescence cannot be used to monitor membrane partitioning
As described in the introduction, two of the three TRP residues of GM2AP (W63 and W131) are located in two loops predicted to interact with the membrane surface. A change in the environment (polarity, hydrophobicity) upon membrane binding is expected to influence the fluorescence emission properties of the TRP residues. For GM2AP, only a 1 nm blue shift in the TRP fluorescence is seen when incubated with excess POPC vesicles at acidic pH (Supporting Material). This blue shift is not seen when the experiment is performed with excess POPC vesicles at neutral pH (data not shown), indicating that the shift does arise from interactions with the membrane. But, this variation is too small to use for partitioning studies or binding kinetics. Additionally, from the sedimentation and gel-filtration studies, the origin of this minor shift is now clear; only a small population of GM2AP is bound to the membrane surface. Clearly, GM2AP is more properly classified as a lipid extraction or lipid transfer protein as opposed to a membrane binding protein. Because GM2AP can extract DDHPE and other phospholipids from vesicle surfaces, a common FRET-based assay with DDHPE cannot be used to provide membrane-partitioning coefficients. Nevertheless, DDHPE vesicles offer a unique route to study “function” when defined as both binding and extraction of lipids, as well as providing an assay to study the kinetics of ligand extraction and release as solution pH and lipid composition are varied (25).
The existence of GM2AP-ligand complexes in solution also implies that care should be taken when analyzing other fluorescence-based assays with this protein, specifically those based upon dequenching assays (6,18). Analysis of the numerous x-ray structures of GM2AP shows that this protein contains a rather large binding pocket, capable of binding various and numerous ligands (16). In hindsight, it is not surprising that GM2AP can form complexes with DDHPE (25) and rhodamine-labeled fatty acids (6,18). However, not all lipid extraction/binding proteins are able to form complexes with fluorescent lipids. For example, the membrane partitioning of the GLTP has been determined using DDHPE FRET in the presence of 20% specific ligand galactosylceramide in POPC membranes (22). Additionally, the Sacchromyces cerviciae phosphotidylinositol transfer protein, Sec14p, has been shown to bind spin-labeled fatty acids and phospholipids in addition to its specific substrates, but appears unable to bind fluorescently labeled lipids (38).
The His-tag must be removed for proper biophysical characterization measurements
It has been documented that the interactions between histidine residues and aromatic residues can change fluorescence emission profiles as well as protein structure (39–42). For purification convenience, the N-terminus of GM2AP is fused with a 10 histidine tag and a Factor Xa cleavage site: MGHHH HHHHH HHSSG HIEGR-. For GM2AP, the coexpressed His-tag does not affect protein conformation, as determined from circular dichroism spectroscopy, or protein stability, determined from a urea induced unfolding experiment (Supporting Material). However, its presence does alter the TRP fluorescence emission spectra under acidic conditions (data not shown). Most significant are the effects the His-tag has on the pH profile of DDHPE binding and extraction kinetics of GM2AP. Thus, the His-tag on GM2AP must be removed for proper biophysical characterization measurements. These results indicate that, in general, care must be taken when considering the effects that His-tags have upon protein structure and function. Given that GM2AP has numerous aspartic acid (ASP) and glutamic acid (GLU) residues lining the rim of the hydrophobic lipid binding pocket, it is possible that the His-tag, when protonated for pH < 6.4, interacts through H-bonding with these residues that surround the binding pocket, thus changing the electrostatic interactions with the bilayers and interactions with lipid ligands. Without the His-tag, optimum function is determined for pH 4.8, which is near the expected pKa values of ASP and GLU residues in proteins. Binding to bilayers upon a pH trigger can be understood by considering that under acidic conditions, the ASP and GLU residues can become protonated, lessening the charge and the Born repulsion energy that can promote a more energetically favorable interaction with the bilayer surface. It is interesting that the GM2AP “function” is titratable near the pKa of histidine residues when the His-tag is present, thus supporting our speculation that the His-tag is interacting with the numerous acidic residues in GM2AP, whose partial neutralization is likely important for membrane binding and ligand extraction.
Supporting Material
Additional experimental details including five figures are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00895-9.
Supporting Material
Acknowledgments
We are thankful to S. A. McLaughlin for providing details regarding analytical centrifugation sedimentation assays of sucrose-loaded vesicles and to J. D. Mathias for careful reading of the manuscript.
The research herein was funded by National Institutes of Health R01GM077232 (G.E.F).
References
- 1.Meier E.M., Schwarzmann G., Furst W., Sandhoff K. The human GM2 activator protein. A substrate specific cofactor of beta-hexosaminidase. A.J. Biol. Chem. 1991;266:1879–1887. [PubMed] [Google Scholar]
- 2.Furst W., Sandhoff K. Activator proteins and topology of lysosomal sphingolipid catabolism. Biochim. Biophys. Acta. 1992;1126:1–16. doi: 10.1016/0005-2760(92)90210-m. [DOI] [PubMed] [Google Scholar]
- 3.Kolter T., Winau F., Schaible U.E., Leippe M., Sandhoff K. Lipid-binding proteins in membrane digestion, antigen presentation, and antimicrobial defense. J. Biol. Chem. 2005;280:41125–41128. doi: 10.1074/jbc.R500015200. [DOI] [PubMed] [Google Scholar]
- 4.Mahuran D.J. The GM2 activator protein, its roles as a co-factor in GM2 hydrolysis and as a general glycolipid transport protein. Biochim. Biophys. Acta. 1998;1393:1–18. doi: 10.1016/s0005-2760(98)00057-5. [DOI] [PubMed] [Google Scholar]
- 5.Li S.C., Wu Y.Y., Sugiyama E., Taki T., Kasama T. Specific recognition of N-acetylneuraminic acid in the GM2 epitope by human GM2 activator protein. J. Biol. Chem. 1995;270:24246–24251. doi: 10.1074/jbc.270.41.24246. [DOI] [PubMed] [Google Scholar]
- 6.Smiljanic-Georgijev N., Rigat B., Xie B., Wang W., Mahuran D.J. Characterization of the affinity of the G(M2) activator protein for glycolipids by a fluorescence dequenching assay. Biochim. Biophys. Acta. 1997;1339:192–202. doi: 10.1016/s0167-4838(97)00002-2. [DOI] [PubMed] [Google Scholar]
- 7.Kuwana T., Mullock B.M., Luzio J.P. Identification of a lysosomal protein causing lipid transfer, using a fluorescence assay designed to monitor membrane fusion between rat liver endosomes and lysosomes. Biochem. J. 1995;308:937–946. doi: 10.1042/bj3080937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glombitza G.J., Becker E., Kaiser H.W., Sandhoff K. Biosynthesis, processing, and intracellular transport of GM2 activator protein in human epidermal keratinocytes. The lysosomal targeting of the GM2 activator is independent of a mannose-6-phosphate signal. J. Biol. Chem. 1997;272:5199–5207. doi: 10.1074/jbc.272.8.5199. [DOI] [PubMed] [Google Scholar]
- 9.Conzelmann E., Sandhoff K. Purification and characterization of an activator protein for the degradation of glycolipids GM2 and GA2 by hexosaminidase A. Hoppe. Seylers Z. Physiol. Chem. 1979;360:1837–1849. doi: 10.1515/bchm2.1979.360.2.1837. [DOI] [PubMed] [Google Scholar]
- 10.Hechtman P., LeBlanc D. Purification and properties of the hexosaminidase A-activating protein from human liver. Biochem. J. 1977;167:693–701. doi: 10.1042/bj1670693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendeler M., Lemm T., Weisgerber J., Hoernschemeyer J., Bartelsen O. Expression of recombinant human GM2-activator protein in insect cells: purification and characterization by mass spectrometry. Protein Expr. Purif. 2003;27:259–266. doi: 10.1016/s1046-5928(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 12.Wendeler M., Hoernschemeyer J., John M., Werth N., Schoeniger M. Expression of the GM2-activator protein in the methylotrophic yeast Pichia pastoris, purification, isotopic labeling, and biophysical characterization. Protein Expr. Purif. 2004;34:147–157. doi: 10.1016/j.pep.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Klima H., Klein A., van Echten G., Schwarzmann G., Suzuki K. Over-expression of a functionally active human GM2-activator protein in Escherichia coli. Biochem. J. 1993;292:571–576. doi: 10.1042/bj2920571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y.Y., Lockyer J.M., Sugiyama E., Pavlova N.V., Li Y.T. Expression and specificity of human GM2 activator protein. J. Biol. Chem. 1994;269:16276–16283. [PubMed] [Google Scholar]
- 15.Wright C.S., Zhao Q., Rastinejad F. Structural analysis of lipid complexes of GM2-activator protein. J. Mol. Biol. 2003;331:951–964. doi: 10.1016/s0022-2836(03)00794-0. [DOI] [PubMed] [Google Scholar]
- 16.Wright C.S., Mi L.Z., Lee S., Rastinejad F. Crystal structure analysis of phosphatidylcholine-GM2-activator product complexes: evidence for hydrolase activity. Biochemistry. 2005;44:13510–13521. doi: 10.1021/bi050668w. [DOI] [PubMed] [Google Scholar]
- 17.Wright C.S., Mi L.Z., Rastinejad F. Evidence for lipid packaging in the crystal structure of the GM2-activator complex with platelet activating factor. J. Mol. Biol. 2004;342:585–592. doi: 10.1016/j.jmb.2004.07.063. [DOI] [PubMed] [Google Scholar]
- 18.Schwarzmann G., Wendeler M., Sandhoff K. Synthesis of novel NBD-GM1 and NBD-GM2 for the transfer activity of GM2-activator protein by a FRET-based assay system. Glycobiology. 2005;15:1302–1311. doi: 10.1093/glycob/cwj018. [DOI] [PubMed] [Google Scholar]
- 19.Werth N., Schuette C.G., Wilkening G., Lemm T., Sandhoff K. Degradation of membrane-bound ganglioside GM2 by beta -hexosaminidase A. Stimulation by GM2 activator protein and lysosomal lipids. J. Biol. Chem. 2001;276:12685–12690. doi: 10.1074/jbc.M007970200. [DOI] [PubMed] [Google Scholar]
- 20.Wendeler M., Werth N., Maier T., Schwarzmann G., Kolter T. The enzyme-binding region of human GM2-activator protein. FEBS J. 2006;273:982–991. doi: 10.1111/j.1742-4658.2006.05126.x. [DOI] [PubMed] [Google Scholar]
- 21.White S.H., Wimley W.C., Ladokhin A.S., Hristova K. Protein folding in membranes: determining energetics of peptide-bilayer interactions. Methods Enzymol. 1998;295:62–87. doi: 10.1016/s0076-6879(98)95035-2. [DOI] [PubMed] [Google Scholar]
- 22.Rao C.S., Chung T., Pike H.M., Brown R.E. Glycolipid transfer protein interaction with bilayer vesicles: modulation by changing lipid composition. Biophys. J. 2005;89:4017–4028. doi: 10.1529/biophysj.105.070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buser C.A., Sigal C.T., Resh M.D., McLaughlin S. Membrane binding of myristylated peptides corresponding to the NH2 terminus of Src. Biochemistry. 1994;33:13093–13101. doi: 10.1021/bi00248a019. [DOI] [PubMed] [Google Scholar]
- 24.Buser C.A., McLaughlin S. Ultracentrifugation technique for measuring the binding of peptides and proteins to sucrose-loaded phospholipid vesicles. Methods Mol. Biol. 1998;84:267–281. doi: 10.1385/0-89603-488-7:267. [DOI] [PubMed] [Google Scholar]
- 25.Ran Y., Fanucci G.E. A dansyl fluorescence-based assay for monitoring kinetics of lipid extraction and transfer. Anal. Biochem. 2008;382:132–134. doi: 10.1016/j.ab.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wright C.S., Li S.C., Rastinejad F. Crystal structure of human GM2-activator protein with a novel beta-cup topology. J. Mol. Biol. 2000;304:411–422. doi: 10.1006/jmbi.2000.4225. [DOI] [PubMed] [Google Scholar]
- 27.Ladokhin A.S., Jayasinghe S., White S.H. How to measure and analyze tryptophan fluorescence in membranes properly, and why bother? Anal. Biochem. 2000;285:235–245. doi: 10.1006/abio.2000.4773. [DOI] [PubMed] [Google Scholar]
- 28.De Bernardo S., Weigele M., Toome V., Manhart K., Leimgruber W. Studies on the reaction of fluorescamine with primary amines. Arch. Biochem. Biophys. 1974;163:390–399. doi: 10.1016/0003-9861(74)90490-1. [DOI] [PubMed] [Google Scholar]
- 29.Svennerholm L. Quantitative estimation of sialic acids. II. A colorimetric resorcinol-hydrochloric acid method. Biochim. Biophys. Acta. 1957;24:604–611. doi: 10.1016/0006-3002(57)90254-8. [DOI] [PubMed] [Google Scholar]
- 30.Christie W. Pergamon Press; UK: 1982. Lipid Analysis. [Google Scholar]
- 31.Kolter T., Sandhoff K. Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 2005;21:81–103. doi: 10.1146/annurev.cellbio.21.122303.120013. [DOI] [PubMed] [Google Scholar]
- 32.Li S.C., Hama Y., Li Y.T. Interaction of GM2 activator protein with glycosphingolipids. Methods Enzymol. 2003;363:230–241. doi: 10.1016/S0076-6879(03)01055-3. [DOI] [PubMed] [Google Scholar]
- 33.Shimada Y., Li Y.T., Li S.C. Effect of GM2 activator protein on the enzymatic hydrolysis of phospholipids and sphingomyelin. J. Lipid Res. 2003;44:342–348. doi: 10.1194/jlr.M200234-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Remmel N., Locatelli-Hoops S., Breiden B., Schwarzmann G., Sandhoff K. Saposin B mobilizes lipids from cholesterol-poor and bis(monoacylglycero)phosphate-rich membranes at acidic pH. Unglycosylated patient variant saposin B lacks lipid-extraction capacity. FEBS J. 2007;274:3405–3420. doi: 10.1111/j.1742-4658.2007.05873.x. [DOI] [PubMed] [Google Scholar]
- 35.Schroder M., Schnabel D., Suzuki K., Sandhoff K. A mutation in the gene of a glycolipid-binding protein (GM2 activator) that causes GM2-gangliosidosis variant AB. FEBS Lett. 1991;290:1–3. doi: 10.1016/0014-5793(91)81211-p. [DOI] [PubMed] [Google Scholar]
- 36.Xie B., Wang W., Mahuran D.J. A Cys138-to-Arg substitution in the GM2 activator protein is associated with the AB variant form of GM2 gangliosidosis. Am. J. Hum. Genet. 1992;50:1046–1052. [PMC free article] [PubMed] [Google Scholar]
- 37.Xie B., Rigat B., Smiljanic-Georgijev N., Deng H., Mahuran D. Biochemical characterization of the Cys138Arg substitution associated with the AB variant form of GM2 gangliosidosis: evidence that Cys138 is required for the recognition of the GM2 activator/GM2 ganglioside complex by beta-hexosaminidase A. Biochemistry. 1998;37:814–821. doi: 10.1021/bi971211s. [DOI] [PubMed] [Google Scholar]
- 38.Smirnova T.I., Chadwick T.G., MacArthur R., Poluektov O., Song L. The chemistry of phospholipid binding by the Saccharomyces cerevisiae phosphatidylinositol transfer protein Sec14p as determined by EPR spectroscopy. J. Biol. Chem. 2006;281:34897–34908. doi: 10.1074/jbc.M603054200. [DOI] [PubMed] [Google Scholar]
- 39.Shinitzky M., Goldman R. Fluorometric detection of histiine-tryptophan complexes in peptides and proteins. Eur. J. Biochem. 1967;3:139–144. doi: 10.1111/j.1432-1033.1967.tb19508.x. [DOI] [PubMed] [Google Scholar]
- 40.Loewenthal R., Sancho J., Fersht A.R. Histidine-aromatic interactions in barnase. Elevation of histidine pKa and contribution to protein stability. J. Mol. Biol. 1992;224:759–770. doi: 10.1016/0022-2836(92)90560-7. [DOI] [PubMed] [Google Scholar]
- 41.Fernandez-Recio J., Vazquez A., Civera C., Sevilla P., Sancho J. The tryptophan/histidine interaction in alpha-helices. J. Mol. Biol. 1997;267:184–197. doi: 10.1006/jmbi.1996.0831. [DOI] [PubMed] [Google Scholar]
- 42.Matthews J.M., Ward L.D., Hammacher A., Norton R.S., Simpson R.J. Roles of histidine 31 and tryptophan 34 in the structure, self-association, and folding of murine interleukin-6. Biochemistry. 1997;36:6187–6196. doi: 10.1021/bi962939w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.