

Abstract
Voltage-gated K+ channels share a common voltage sensor domain (VSD) consisting of four transmembrane helices, including a highly mobile S4 helix that contains the major gating charges. Activation of ether-a-go-go (EAG) family K+ channels is sensitive to external divalent cations. We show here that divalent cations slow the activation rate of two EAG family channels (Kv12.1 and Kv10.2) by forming a bridge between a residue in the S4 helix and acidic residues in S2. Histidine 328 in the S4 of Kv12.1 favors binding of Zn2+ and Cd2+, whereas the homologous residue Serine 321 in Kv10.2 contributes to effects of Mg2+ and Ni2+. This novel finding provides structural constraints for the position of transmembrane VSD helices in closed, ion-bound EAG family channels. Homology models of Kv12.1 and Kv10.2 VSD structures based on a closed-state model of the Shaker family K+ channel Kv1.2 match these constraints. Our results suggest close conformational conservation between closed EAG and Shaker family channels, despite large differences in voltage sensitivity, activation rates, and activation thresholds.
Introduction
Voltage-gated K+ channels function as tetramers with four independent voltage sensors (VSDs) surrounding a single K+ selective pore (1). Changes in transmembrane voltage drive rearrangement of VSD helices (S1–S4) to gate the pore. A series of basic gating charges on the S4 helix traverse the transmembrane electric field during voltage-dependent gating (2,3) and interact with acidic countercharges on the S2 and S3 helices (4,5). The countercharges themselves may in some cases contribute to gating charge (3,6). At least two major sequential voltage-dependent transitions occur independently in each VSD during activation in Shaker (Kv1) family K+ channels (7,8). A final cooperative transition involving additional VSD movement leads to opening (9,10). Gating models incorporating these three transitions, summarized by the scheme (C1 ↔ C2 ↔ A)4 ↔ O, recapitulate almost all aspects of Kv1 channel gating (7,9).
Crystal structures of Kv1.2 in an open conformation show four S4 arginines, the core gating charges, extracellular to the presumed transmembrane electric field (1,4). Structural representatives of the three major predicted closed conformations (C1, C2, and A) are yet to be determined. Therefore, the VSD motions that lead to channel opening are not fully characterized. Recent attempts to model the resting closed state structure (C1) of Kv1.2 incorporate a new set of constraints derived from mutations that affect ion leakage through the VSD itself. These efforts converge on a model in which S4 translates 6–8 Å orthogonal to the membrane and rotates ∼180° during activation, with gating charges traveling through a “gating canal” at the center of the VSD (6,11,12). The transmembrane electric field is highly focused by aqueous clefts (13–15), allowing this movement to transport gating charges across the electric field. However, it remains to be seen how applicable structural models of Kv1 channels are to other ion channel classes with diverse voltage-gating phenotypes.
We used functional analysis of a divalent binding site in EAG family K+ channels, to develop novel constraints for the positions of VSD helices in a closed state. The EAG family includes the eag (Kv10), erg (Kv11), and elk (Kv12) channel subfamilies, each sharing only ∼20% amino acid identity to Kv1 channels in the VSD. EAG family channels generally activate in a subthreshold voltage range with a relatively low apparent gating valence, but have highly variable gating kinetics (16–21). In contrast, Kv1 channels from diverse species activate rapidly within a narrow voltage range and with a high gating valence (22). Nevertheless, voltage-dependent gating in Kv10 channels shares key features with Kv1 channels. Most notably, two sequential closed-closed transitions have also been detected during Kv10 channel activation. The first movement is rate-limiting and occurs at hyperpolarized voltages; it can be detected with a fluorescent reporter attached to S4 (18,20,23). The second movement is detected in gating currents (18,20). Differences between the QV curve determined from gating currents and the GV curve determined from ionic currents suggests the presence of a third transition required for opening (18,20). Indeed, the (C1 ↔ C2 ↔ A)4 ↔ O gating scheme describes the major features of Kv10 channel gating (20). A unique feature of Kv10 channel gating is that the activation rate is highly sensitive to divalent cations such as Mg2+ and Ni2+ (24,25). These ions preferentially slow the first independent transition and thus bind a closed conformation early in the activation pathway (18,20). The binding site for divalent cations requires acidic residues in S2 and S3 that are unique to the EAG family (26). Silverman et al. (26) proposed that divalent cations electrostatically slow movement of the third S4 gating charge into an extracellular pocket and Schonherr et al. (27) proposed that Mg2+ slows activation sterically by narrowing the gating canal.
We show in this study that divalent cations slow activation in two EAG family channels by serving as a bridge between coordinating residues in S2 and S4 that locks the VSD in a closed conformation. This novel mechanism provides structural constraints for the position of VSD helices in divalent-bound channels. The divalent binding site of EAG family channels is highly compatible with structural models of the Kv1.2 C1 state, suggesting conformational conservation in the VSD of these functionally divergent gene families.
Materials and Methods
Molecular biology and oocyte preparation
Mouse Kv12.1 and Kv10.2 were cloned into the pOX expression vector (28). Mutations were introduced using standard PCR-based mutagenesis and constructs were sequence-confirmed. cRNA transcripts were generated using the T3 mMessage mMachine kit (Ambion, Austin, TX). Mature enzymatically defolliculated Xenopus oocytes were injected with 1–5 ng of cRNA in a 50 nL volume and incubated 1–3 days before recording at 18°C in ND96 (96 mM NaCl, 2 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.2) supplemented with 2.5 mM Na-pyruvate, 100 U/mL penicillin and 100 μg/mL streptomycin. Chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise noted.
Electrophysiology
Recordings were carried out using a Dagan CA-1B amplifier in TEVC mode at room temperature (22–24°C). Microelectrodes were filled with 3 M KCl (<1 MΩ) and 1 M NaCl Agar bridges isolated the bath clamp circuitry. Recording solution consisted of (mM) 98 NaMES, 2 KMES, 2 CaCl2, 5 HEPES, pH 7. Kv10 and Kv12 channels were not affected by 2 mM Ca2+. Deactivation was measured in 20 mM K+. MMTS and MTSEA (Toronto Research Chemicals, Toronto, CA) were diluted to working concentrations from ethanol stocks within 10 min of use. Data collection and analysis were carried out using pCLAMP9/Digidata 1322 acquisition suite (Axon Instruments, Union City, CA). Voltage activation curves (GV) were measured from isochronal tail currents recorded at −40 mV after steps to the indicated voltages. Data were fit with Boltzmann distributions to generate V50 and slope (mV/e-fold change in conductance) values according to the following equation: G/GMAX = 1/(1 + e−(V−V50)/k), where G is the conductance at voltage V, V50 is the voltage for half activation, and k is the slope factor. Activation time constants were measured from single or double exponential fits according to the following equations: I = A[1 − exp(−t/τ)] or I = Afast[1 − exp(−t/τfast)] + Aslow[1 − exp(−t/τslow)], where I is the current at time t, A is the amplitude, and τ is the time constant. Deactivation time constants were measured from single exponential fits of tail currents. Statistical significance was assessed by t-test with a cut off of p < 0.05.
Structural modeling
Protein models were built by homology using the Prime 1.6 module from FirstDiscovery suite (29). Kv1.2 models proposed by Pathak et. al. (2007) were used as templates for Kv12.1 models. The Kv10.2 model was built using an optimized Kv12.1 model. Each tetramer was embedded into 160 × 160 Å lipid bilayer composed of POPE lipid molecules and solvated in TIP3P water box in the presence of 0.5 M NaCl. It was then subjected to molecular dynamics simulation under periodic boundary conditions (160 × 160 × 160). The simulation was initiated by minimizing the entire system in which protein backbone atoms were restrained with harmonic force of 10 kcal/mol Å2. That was followed by equilibration simulation that included 1), 50 ps of constant volume simulation in which protein backbone atoms were restrained with harmonic force of 10 kcal/mol Å2 and the temperature was raised gradually to 310 K; 2), 100 ps of constant pressure MD in which backbone carbon atoms were restrained with a harmonic force 10 kcal/mol Å2; and 3), 350 ps of unrestrained constant pressure dynamics. After the equilibrium simulation, a 2-ns simulation was run without restrains. Bonds involving hydrogen atoms were constrained via the SHAKE algorithm. A 12 Å cutoff distance was used for all nonbonded interactions. Time step integration was 1 fs, and the coordinates were saved every 10 ps. Zn2+-bound simulations were setup by placing the ion manually in such a way that it was equidistant (∼3 Å) from D261 and D314 carboxylate oxygen atoms before the final unrestrained simulation. All simulations were carried out using the NAMD software package (30) with a charmm27 force field.
Results
We compared the divalent sensitivity of Kv12.1 and Kv10.2 to gain insights into the structure of the divalent cation binding pocket in EAG family K+ channels. Kv10 subfamily channels are known to have high sensitivity to Mg2+, Mn2+, and Ni2+ (24,25). Kv12.1 was insensitive to Ca2+ and Mg2+ (data not shown) (21). However, we found that Kv12.1 was highly sensitive to Zn2+ (Fig. 1 A). Activation follows a dual exponential time course; both time constants are slowed ∼10-fold by 1 mM Zn2+ (Fig. 1 B), suggesting the gating transitions that produce dual exponential activation occur downstream of Zn2+ binding. Dual exponential activation occurs from a wide range of holding potentials (data not shown); it does not reflect the Cole-Moore gating phenomenon described for fast and slow activation in Kv10 channels (20,31). Zn2+ introduced a pronounced sigmoidicity to the activation time course (Fig. 1 A) implying it slows a closed-closed transition. Zn2+ did not have a significant effect on deactivation time course (Fig. 1 C). Divalent cations similarly modulate a closed-closed transition early in the activation pathway in Kv10 channels (25,32). Hill plots of the fold change in activation time constant at 0 mV produced essentially identical IC50 values for Zn2+ of 980 ± 60 μM and 900 ± 40 μM for measurements of fast and slow time constants, respectively (Fig. 1 D). The effect of other divalent cations on activation of Kv12.1 is shown in Fig. 1 E, and is quantified using the slow time constant of activation. Mn2+ and Ni2+, which potently slow activation of Drosophila eag (25), had little effect on Kv12.1. Cd2+, which is similar to Zn2+ in its coordination preferences (33), slowed activation ∼4-fold at 1 mM. Specific slowing of activation by Zn2+ leads to a large rightward shift in the voltage activation (GV) curve (Fig. 1 F), which provides an alternate measure of divalent effects on channel gating. The Zn2+-induced right shift in GV is significant at 100 μM; both 100 μM and 1 mM Cd2+ also produced significant (but smaller) right shifts in the GV of Kv12.1.
Figure 1.
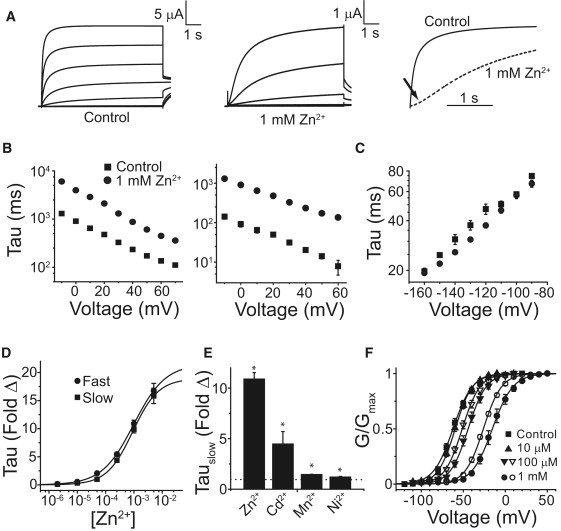
Zn2+ slows activation of Kv12.1. (A) Kv12.1 currents elicited in the absence and presence of 1 mM Zn2+. Eight-second voltage steps were taken in 20 mV increments from −120 mV to +20 mV (Control) or −100 to +20 mV (Zn2+) from a holding potential of −100 mV. Tail currents were recorded at −40 mV. The right panel shows currents elicited at 0 mV for control and 1 mM Zn2+ (dashes) normalized to the maximal current obtained at the end of the 8-s pulse; the arrow indicates sigmoidal delay in the presence of Zn2+. (B) Fast and slow activation time constants from dual exponential fits of the activation time course of Kv12.1 are shown for control (■) and 1 mM Zn2+ (•). (C) Time constants for Kv12.1 deactivation recorded in the presence (•) or absence (■) of 1 mM Zn2+. Tail currents were recorded in 20 mM external K+ after a 0.5-s test pulse to +40 mV. (D) Fold change in the fast (•) and slow (■) activation time constants of Kv12.1 in the presence of increasing concentrations of Zn2+. Lines show Hill equation fits of the data with IC50 values of 980 ± 60 μM and 900 ± 40 μM for the fast and slow time constants, respectively. (E) Fold change in the slow time constant of activation caused by 1 mM Zn2+, Cd2+, and Ni2+ and 5 mM Mn2+. The dotted line at 1 indicates no change. (F) Voltage activation curves (GV) for Kv12.1 shift rightward in the presence of Zn2+ (solid symbols) and Cd2+ (open symbols). Conductance values were measured from isochronal tail currents recorded at −40 mV after 8-s steps to the indicated voltages and normalized to maximal conductance. Lines show Boltzmann fits; parameters are reported in Table S1. Significance (p < 0.05) is indicated with asterisks. Error bars show SE, n = 4–16 for data in B–F.
We hypothesized that the divalent binding site of Kv12.1 might include cysteine or histidine, because these amino acid residues often constitute metal binding sites for both Zn2+ and Cd2+ (34). Kv12.1 has a histidine residue (H328) at the outer edge of the S4 helix that is conserved in Kv12 channels (Fig. 2 A). The homologous residue in Kv10 channels is serine. H328 is “in register” with the S4 helix gating charges, and its position upstream of the gating charges (R1–R4) suggests that it could lie in the extracellular aqueous cleft of the VSD in the vicinity of key acidic residues in S2 and S3 in closed channels (Fig. 2, A and B). Substitution of H328 with neutral amino acids virtually eliminated divalent cation sensitivity, regardless of side chain size (Fig. 3 A), arguing that steric issues are not relevant to the loss of sensitivity. These mutations had little effect on activation rate and did not significantly change the V50 or slope of the GV curve (Fig. 3, A and B, Table S1 in Supporting Material). We next examined the effect of charged substitutions. H328R eliminated divalent sensitivity (Fig. 3 C), but mimicked the major effects of divalent cations; it activated slowly with a right-shifted V50 (Fig. 3, C and D, Table S1). H328E, in contrast, caused a dramatic left-shift in V50 and accelerated activation (Fig. 3, C and D, Table S1). H328E retained some sensitivity to divalents, but its predisposition to open may have limited their effect. Divalent cations selectively bind a closed state in Kv10 channels (25). The H328R and H328E phenotypes are best explained by a model in which position 328 is close to S2/S3 acidic residues in the closed channels. The arginine side chain could replace Zn2+ in the divalent binding pocket and might form a salt bridge to S2 or S3 acidic charge(s) to stabilize a closed conformation. Similarly, open state stabilization observed for H328E could arise from electrostatic repulsion. Divalent sensitivity in H328E might then arise from electrostatic screening or direct ion coordination.
Figure 2.
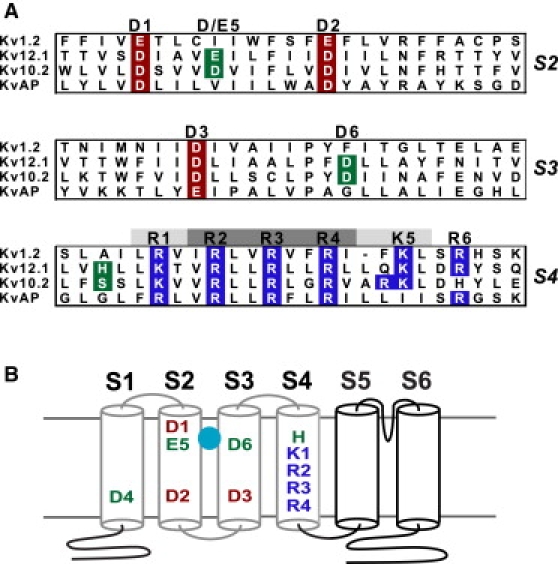
Charged residues of the VSD. (A) Amino acid alignments of VSD transmembrane helices S2–S4 are shown for Kv1.2, Kv12.1, Kv10.2, and the bacterial channel KvAP. Conserved acidic residues in S2 and S3 are highlighted in red and conserved basic residues in S4 are highlighted in blue. H328 of Kv12.1, S321 of Kv10.2, and EAG-specific acidic charges in S2 and S3 are shown in green. The highly conserved acidic residues are designated 1–3 by position; the residues at these three positions in Kv12.1 and Kv1.2 are D261 and E226 (1), D271 and E236 (2), and D306 and D259 (3), respectively. EAG family-specific acidic residues are designated 4–6; in Kv12.1 these residues are D225 in S1 (4), E265 in S2 (5), and D314 in S3 (6). S4 charge positions are labeled R1–R6 based on the nomenclature of Long et al. (4); charge positions that contribute large or small amounts to Kv1 channel gating currents are indicated with dark or light shading, respectively. (B) Schematic drawing of Kv12.1 showing the pattern of charged residues in the VSD. Transmembrane helices are represented by cylinders and the components of the VSD are outlined in gray. Acidic residues at positions 5 and 6 contribute to binding of divalent cations (sphere) in Drosophila eag, and lie within an externally accessible aqueous pocket. The Kv12-specific Histidine residue at the outer edge of S4 is labeled in green, and occupies the site designated as R0 by Long et al. (4).
Figure 3.
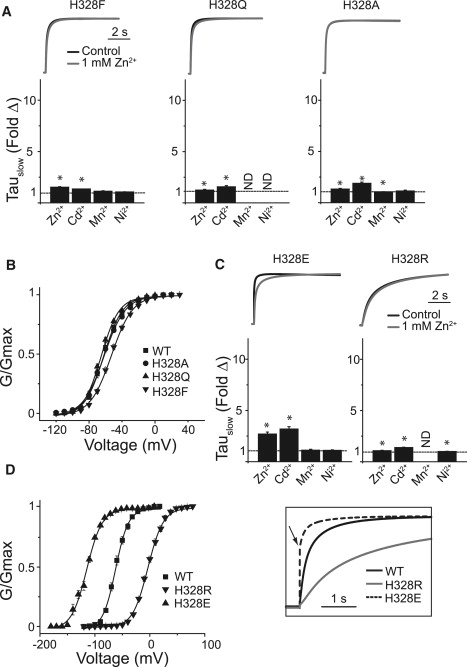
Mutation of H328 in Kv12.1 affects sensitivity to divalent cations. (A) Current traces for H328Q, H328F, and H328A are given for control (black) and 1 mM Zn2+ (gray). Currents were elicited by 8-s steps to 0 mV from a holding potential of −100 mV and were normalized to maximal current amplitude during the step to facilitate comparison of activation rates. The scale bar indicates time. Bar graphs show the fold change in τSlow of activation caused by 1 mM Zn2+, Cd2+, and Ni2+ or 5 mM Mn2+. Asterisks indicate significant difference from WT (p < 0.05). (B) GV curves for Kv12.1 WT, H328A, H328Q, and H328F. Conductance was measured from tail currents recorded at −40 mV after 8-s steps to the indicated voltages. Line show Boltzmann fits; parameters are reported in Table S1. (C and D) Panels show the same series of data as displayed in A and B for the H328R and H328E mutants. The inset shows normalized control currents for WT, H328R, and H328E to allow for comparison of activation rates. Error bars show SE, n = 4–15 for data in A–D.
Does preference for Zn2+ binding in Kv12.1 arise from coordination by H328? If so, we reasoned H328C should retain high Zn2+ sensitivity. H328C activation was indeed highly sensitive to Zn2+ and showed a strong sensitization to Cd2+ (Fig. 4, A–C). Right shifts in the GV were detectable at 10 μM with both ions for H328C, and the magnitude of the GV shift was close to saturation at 100 μM for Cd2+ (Fig. 4 C). Covalent capping of H328C, but not WT, with cysteine-reactive MTS reagents (the methyl donor MMTS and the triethylammonium cation donor MTSEA) greatly reduces sensitivity to Zn2+ (Fig. 4, D–F). MTSEA modification slowed activation and right-shifted the GV of H328C (Fig. 4, G–I) due to the introduction of positive charge. These properties of H328C are exclusively consistent with a model in which the residue at position 328 can directly coordinate an ion occupying the divalent binding site of Kv12.1.
Figure 4.
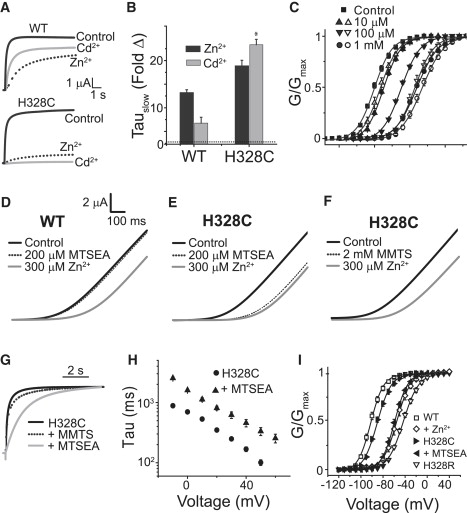
H328C increases the sensitivity of Kv12.1 to Zn2+ and Cd2+. (A) Current traces evoked by 8-s steps to 0 mV from a holding potential of −100 mV are shown for WT and H328C in the presence and absence of Zn2+ or Cd2+. (B) Fold change in the slow time constant of activation in the presence of Zn2+ and Cd2+ for WT Kv12.1 and H328C. Asterisks indicate significant difference from WT (p < 0.05). (C) The GV curve of H328C is robustly right-shifted by Cd2+ (open symbols) and Zn2+ (solid symbols). (D–F) Currents recorded in response to 1-s voltage ramps from −100 to +50 mV for WT Kv12.1 and H328C in the following sequence: control (black), after exposure to the indicated MTS reagent (dotted), and after the subsequent addition of 300 μM Zn2+ (gray). Zn2+ has little to no affect on MTS-modified H328C channels. (G) Normalized currents recorded during 8-s voltage steps to 0 mV for H328C before (solid black) or after modification with 200 μM MTSEA (gray) or 2 mM MMTS (dotted). (H) TauSlow of activation versus voltage for H328C before and after modification with MTSEA. (I) H328C GV curves from isochronal tail currents before and after 200 μM MTSEA application are compared to GV curves for WT, WT + 1 mM Zn2+, and H328R. Lines in C and I are Boltzmann fits of the data; parameters for H328C included in Table S1. Bars indicate SE, n = 4–12 for C, H, and I.
We mutated the acidic charges in S2 and S3 that have previously been shown to influence divalent binding in Drosophila eag and hERG, and confirmed that they also influence divalent binding in Kv12.1. Alanine substitution greatly reduced divalent sensitivity, whereas charge-conserving mutations preserved it, with minor apparent selectivity changes for D261E and E265D (Fig. S1). To test whether these residues affect divalent binding through direct ion coordination, we used cysteine substitution and looked for increases in the affinity for Zn2+ and Cd2+. Like H328C, E265C, and D261C currents showed enhanced sensitivity to both ions (Fig. 5, A and B). In contrast, we observed reduced divalent sensitivity for D314C. We quantified changes in sensitivity in these mutants using GV shifts because it was difficult to measure activation time constants of the sensitized channels at some divalent concentrations. D261C, E265C, and H328C all seem to have an increased affinity for Zn2+ and Cd2+ because they have significantly larger GV shifts at all ion concentrations, except for D261C/1 mM Zn2+ (Figs. 4 C and 5, C–E). These results indicate that D261C and E265C, like H328C, are involved directly in coordination of divalent cations binding in the Kv12.1 VSD. Negative charge at position D6 (D314) seems required for divalent binding, but we found no evidence for direct ion coordination.
Figure 5.
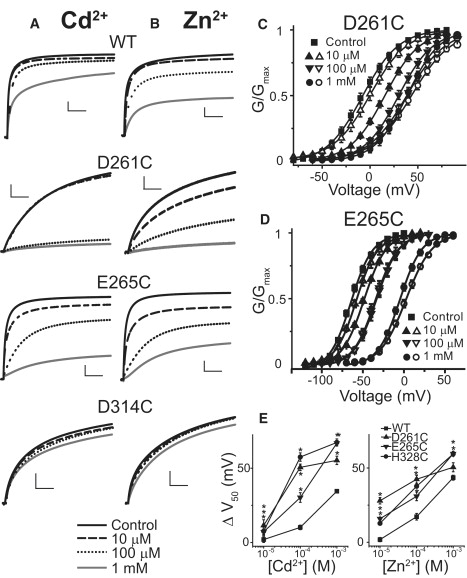
D261 (D1) and E265 (E5) can directly coordinate divalent cations. Current traces recorded in response to steps to 0 mV for D261C, E265C, and D314C in the presence of increasing concentration of (A) Zn2+ and (B) Cd2+. Scale bars show 1 μA/1 s. D261C and E265C show increased sensitivity to Zn2+ and Cd2+, implying direct participation in ion coordination. GV curves of (C) D261C and (D) E265C right shift in the presence of Zn2+ (solid symbols) and Cd2+ (open symbols), even at low concentrations. Lines show Boltzmann fits and parameters for controls are included in Table S1. (E) The magnitude of GV shift caused by increasing concentration of Zn2+ and Cd2+ is plotted for WT Kv12.1, D261C, E265C, and H328C. Error bars show SE, n = 4–6 and asterisks mark values that differ significantly from WT (p < 0.05).
Does S4 contribute to divalent binding in Kv10 channels? In the absence of divalent cations, Kv10.2 activates more rapidly than Kv12.1 (Fig. 6 A, Table S1). We found that Kv10.2 activation was also best described by a dual exponential fit. The relative amplitude of these components, however, was determined by the holding potential, implicating the Cole-Moore shift described for Kv10 channels (20,31). We quantified the slowing of Kv10.2 activation by divalent cations as the fold increase in the slow time constant of activation because: 1), this measurement is equivalent to a single exponential fit of the late phase of activation used by Silverman et al. (25,32); and 2), the amplitude of the fast component was far more sensitive to divalent cations than its time constant, suggesting that fast-activating Kv10.2 channels were not divalent bound. For instance, the amplitude of τFast at +60 mV decreased 5.78 ± 1.39-fold (n = 5) with 100 μM Ni2+, whereas τFast itself increased only 0.34 ± 0.17-fold. Kv10.2 was slowed dramatically by divalent cations (Fig. 6, B and F), but selectivity differed markedly from Kv12.1. Kv10.2 had a relatively similar sensitivity to 5 mM Mg2+, 1 mM Mn2+, 1 mM Cd2+, and 1 mM Zn2+. It was much more sensitive to 1 mM Ni2+ (>100-fold slowing). The sensitivity of Kv10.2 to Mn2+, Mg2+, and Ni2+ is very similar to that reported for Drosophila eag (25). Larger fold changes in activation time constants for Kv10.2 versus Kv12.1 are attributable to the differences in activation time constants in the absence rather than presence of divalent cations. For instance, τSlow for Kv10.2 + 1 mM Ni2+ and Kv12.1 + 1 mM Zn2+ was similar at 4157 ± 205 ms and 3971 ± 139 ms, respectively (n = 6–10).
Figure 6.
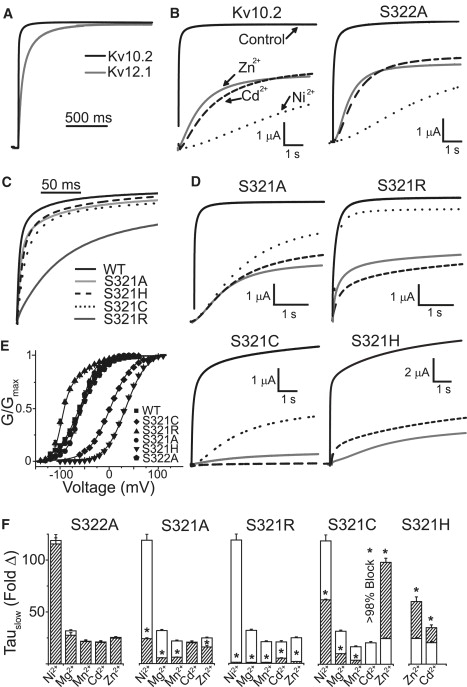
High efficacy slowing of activation by divalents involves coordination at position 321 in the S4 of Kv10.2. (A) Normalized Kv12.1 and Kv10.2 currents recorded in response to a voltage step to 0 mV show comparatively fast activation in Kv10.2. (B) Kv10.2 WT and S322A respond to divalent cations in a similar manner; traces show control currents and currents after addition of 1 mM Cd2+ (dashes), Zn2+ (gray), and Ni2+ (dots); currents were recorded at 0 mV. (C) Comparison of the activation rates of Kv10.2 WT, S322A, and S321 mutants at 0 mV; traces were normalized to current amplitude at 4 s. (D) Effects of 1 mM of Ni2+, Zn2+, or Cd2+ on S321 mutant currents recorded in response to voltage steps to 0 mV (+60 mV for S321H). See B for key to traces. (E) GV curves for Kv10.2 WT and S4 mutants derived from isochronal tail current measurements. Parameters of Boltzmann fits (lines) are given in Table S1. S321R was fit by a dual Boltzmann. The major left-shifted component is given in Table S1; the second component was indistinguishable from WT. Fold changes in τSlow caused by the divalent cations (1 mM Ni2+, Mn2+, Cd2+, and Zn2+; 5 mM Mg2+) for each mutant are shown in F. Open bars show effects on WT and dashed bars show effects on the mutants. Fold change was not measured for Cd2+ on S321C because the current was blocked almost completely; this indicates a large increase in Cd2+ sensitivity. Measurements were made at 0 mV for all mutants except S321H, which was measured at +60 mV. Asterisks indicate significant difference (p < 0.05) from WT. Error bars show SE, (n = 3–6) for data in E and F.
We mutated S321 in Kv10.2 (equivalent to H328 in Kv12.1) to alanine, arginine, cysteine, and histidine. We neutralized S322 to alanine in all the S321 mutants to match the SA sequence found at this site in Drosophila eag to facilitate comparison. We found no significant differences between the activation or divalent sensitivity of WT Kv10.2 and S322A (Fig. 6, B,E,F). Effects of divalents on currents and activation time courses of the Kv10.2 S321 mutants are shown in Fig. 6, D and F. Control activation properties are compared in Fig. 6, C and E (parameters in Table S1). S321A had little effect on activation, but produced major changes in divalent sensitivity. Effects of ions to which Kv10 channels are uniquely sensitive (Mg2+, Mn2+, and Ni2+) were reduced ∼5-fold. However, unlike Kv12.1 H328A, Kv10.2 S321A retained sensitivity to Cd2+ and Zn2+, indicating that these ions can still bind in the VSD. S321R virtually eliminated effects of Mg2+, Mn2+, and Ni2+, but Cd2+ and Zn2+ still reduced peak current amplitudes significantly. Like Kv12.1 H328R, Kv10.2 S321R had significantly slower activation than WT, but it was surprisingly accompanied by a left shift in the GV. The shift seemed to be caused by a slowing of deactivation that was not observed for Kv12.1 H328R (data not shown). The ions most affected by the S321A and S321R mutations (Mg2+, Mn2+, and Ni2+) can all be coordinated by serine (34). We reasoned that if coordination at position 321 is important to divalent effects in Kv10.2, then S321C and S321H mutants should have a gain-of-sensitivity to Zn2+ and Cd2+. These mutants did indeed significantly sensitize Kv10.2 to both ions (Fig. 6, D and F). S321C current was blocked almost completely by 1 mM Cd2+ at 0 mV and τSlow could not be measured; the effect of Zn2+ on τSlow was significantly increased ∼5-fold. In contrast S321C had a reduced sensitivity to Ni2+ and Mg2+, which prefer coordination by serine (33). S321H also produced significant increases in sensitivity to Zn2+ and Cd2+. These phenotypes can only be explained by direct ion coordination. Therefore, we conclude that the side chain of residue 321 must face the divalent binding pocket in a closed state. Some divalent ions seem to fit into the Kv10.2 VSD divalent binding site without coordination by 321, but their ability to slow Kv10.2 activation is greatly increased by this interaction. Thus a mechanism in which divalent ions slow activation by directly binding to S4 seems to be common to both Kv10.2 and Kv12.1.
We built structural homology models of the Kv12.1 VSD to understand how VSD helices might arrange to form the divalent binding site. Because there are no representative crystal structures of a closed VSD, we chose to model the VSD by homology to the Kv1.2 closed-state model proposed by Pathak et al. (6). This model is consistent with a wide variety of Kv1 channel gating data and maintains compatibility with the open state crystal structure (4). Conservation of key residues between Kv12.1 and Kv1.2 allow for helical alignment. Only the S1–S6 region was included in the homology models because there is no conservation outside this region. After homology alignment, sequential molecular dynamics simulations were carried out in a lipid environment (that seems to be critical to voltage-sensor conformation (4,35)) to minimize energy and optimize the model in the presence and absence of Zn2+ (see Materials and Methods for details). The Zn2+-bound Kv12.1 channel model is shown in Fig. 7 A. We obtained four largely independent simulations of VSD structure from the tetramer. Zn2+ binds within the extracellular aqueous cleft and is coordinated by D261 (D1), E265 (E5), and H328 in all four VSDs. This model is consistent with functional data: we obtained evidence for direct ion coordination for each of these three residues. The short simulation run (2 ns) precluded major changes from the VSD conformation found in the Kv1.2 closed model, showing that the divalent bound VSD conformation of Kv12.1 is probably quite similar to the C1 conformation of Kv1.2. A control simulation run in the absence of Zn2+ shows similar conformation with only modest changes in the positioning of VSD helices and coordinating side chains (Fig. S2). We next built a homology model of Kv10.2 + Zn2+ based on the Kv12.1 model (Fig. 7, B–D). Zn2+ was coordinated by S321 in only two of four VSDs, supporting functional analysis suggesting that Zn2+ can enter the VSD pocket in the S321A mutant. The Kv10.2 simulation shows much greater variation in coordination of Zn2+ by acidic residues, including D6 in S3. This flexibility supports observations that various divalent cations may sit differently within the VSD pocket of Drosophila eag, in part because of the presence of D instead of E at the D/E5 position (26). We were technically unable to model Ni2+ docking on Kv10.2 in the simulation software.
Figure 7.
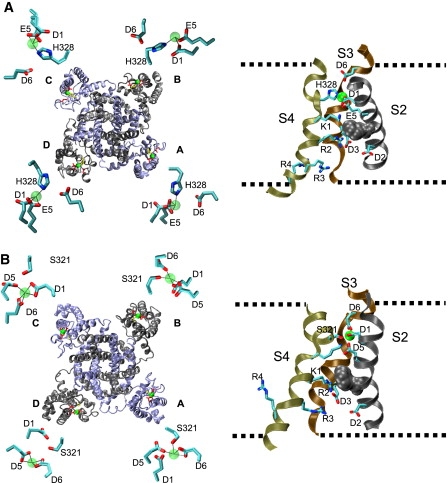
Structural models of Zn2+-bound VSDs from Kv12.1 and Kv10.2. (A) Structural homology model of Kv12.1 with Zn2+ bound based on the closed-state model of Kv1.2 proposed by Pathak et al. (6). The left view shows a ribbon diagram of the entire tetramer viewed from the extracellular side of the membrane. Subunits alternate gray and blue color. Side chains are shown for H328 (yellow) and acidic residues D261 (D1), E265 (E5), and D314 (D6) (red). Zn2+ ions are represented as green spheres. The S1–S2 and S5–P-domain linkers have been removed to facilitate viewing. Insets show close-up views of Zn2+ coordinating side chains in each VSD of the model. Bonds are depicted as dotted lines and bond lengths are provided in Table S2. The right view shows a side view of the VSD from chain D with the S1 (front) and linkers removed to allow viewing of key residues. Side chains are shown for acidic residues at positions D1–D3, D5 and D6, H328, and R1–R4. Nitrogen atoms in the side chains are blue and oxygen atoms are red. The approximate outline of the lipid membrane is depicted with dashed lines and a conserved phenylalanine residue that marks the approximate location of a narrow hydrophobic plug separating extracellular and intracellular aqueous crevices is shown in gray space fill. The electric field sensed by VSD gating charges is likely to be focused across this region (13,15). Note that the Zn2+ binding site is located extracellular to the plug and that K1 sits near the plug similar to the position or the homologous residue R1 in Kv1.2. The bound Zn2+ ion is represented as a green sphere. (B) Identical views of a structural model of Kv10.2 + Zn2+. S321 is the yellow side chain in the full tetramer view. Note the variable coordination systems for Zn2+ that can include S321 and involve all three acidic residues at positions D1, D5, and D6. Bond lengths are given in Table S2.
It is important to note that our divalent binding data only provides constraints for the relative positions of VSD helices S2–S4. Other aspects of these structural models are speculative and should not be overinterpreted. The S1–S2, S2–S3, and S4–S5 linker sequences are poorly conserved and their positioning is entirely speculative. S1 is also unconstrained by divalent binding data. The largest variations between VSDs in our Zn2+-docking simulation occurred in these regions. We did not analyze pore structure or details of VSD/pore coupling because our functional data does not provide constraint for these regions. However, we did build a hypothetical model of the Kv12.1 open state based on the Pathak et al. (6), which is highly similar to crystal structures (1,4) (Fig. S2). This open state model is speculative, but it does provide a simple explanation for why Kv12.1 is much less sensitive to divalent cations in the open state: H328 has moved away from the acidic residues in S2 and S3, and the binding site is occupied by the positive S4 gating charges R3 and R4 that could electrostatically repel divalent cations.
Discussion
Our data suggest divalent cations slow the activation of Kv12.1 by serving as a bridge between S4 and S2. These results provide structural constraints for the position of S4 in Kv12.1 in the divalent-bound conformation: the S4 residue H328 must be located within the extracellular aqueous pocket of the VSD at the level of (and facing) in the S2 acidic residues D261 and E265. Modeling shows this position is highly compatible with structural models of closed Kv1.2 channels proposed by Pathak et al. (6) and Campos et al. (11). It places the first potential S4 gating charge of EAG and Kv1 family channels in a similar position near the putative hydrophobic plug across which the membrane electric field is thought to be focused (4,11,13–15). It implies that S4 gating charges of EAG family channels travel through a protein-lined gating canal rather than the membrane lipid core, as suggested by omega current data and histidine-scanning mutagenesis from Kv1 channels (6,11,12,36,37). This aspect of our model is consistent with previous models of Kv10 channel gating derived from divalent block data, which postulated the presence of the gating canal (27,32). However, those studies proposed that divalent cations slowed activation through steric or electrostatic block of the gating canal rather than direct interaction between S4 and the gating canal “wall”.
Mutations in the outer edge of S4 have been found previously to affect Mg2+ sensitivity of Kv10 channels, but divalent binding to this region was not explored. A mutation equivalent to L319H in Kv10.2 reduces Mg2+ efficacy in Drosophila eag and Kv10.1, whereas equivalent mutations to S318C and S321C in Kv10.1 seem to reduce Mg2+ sensitivity modestly (20,23,27). We identified a direct role of S321 in divalent coordination by examining sensitivity of Kv10.2 S321C and S321H to Zn2+ and Cd2+. Other mutations in this region may affect divalent sensitivity by altering the position of S321. Our data implies that Kv10 channels differ from Kv12.1 in that divalent cations can enter the VSD pocket and have modest effects on channel activation even in the absence of formation of a “bridge” between S4 and S2 and/or S3. However, the efficacy of Zn2+ and Cd2+ seems to be increased greatly by formation of this bridge in the S321C and S321H mutants. Furthermore, Mg2+, Mn2+, and Ni2+ all had their greatest effect in WT that has serine at position 321. Serine is the most favorable residue for coordination of these ions among those that we tested (33,34), implying that bridge formation is important to the efficacy of these ions as well.
The selectivity of divalent binding sites in Kv12 and Kv10 channels seems to be determined by both the coordinating residue on S4 (histidine and cysteine favor Zn2+ and Cd2+) and acidic residues in S2. The presence of glutamate at the D/E5 position has been proposed as a reason for of the insensitivity of Kv12 channels to hard ions because the E5 substitution in Drosophila eag eliminates Mg2+ block (25). We find that placing D5 (E265D) in Kv12.1 is unable to establish sensitivity to Mg2+; perhaps both D5 and serine at the S4 coordinating position are required for sensitivity to hard ions, as suggested by the reduced Mg2+ sensitivity in S321 mutants of Kv10.2. Kv11 channels have D5 but a glycine residue in the position equivalent to H328 and S321. Thus they are relatively similar to Kv10.2 S321A, which, like hERG, has moderate sensitivity to Cd2+ and low sensitivity to hard ions (38). Another major difference between the divalent binding sites of Kv10 and Kv12 channels is the effect of neutralization of the D1 position. Substitution of D1 with alanine in Drosophila eag dramatically increases sensitivity to Ni2+ (32), whereas the same mutation in Kv12.1 reduces sensitivity to all divalents. In Kv10.2, the D1 side chain might actually compete with S321 for an ion such as Ni2+, which can be coordinated by both acidic residues and serine but requires fewer coordination points than hard ions such as Mg2+ (33,34). If so, then binding of Ni2+ to S321 could be stabilized with D1 neutralized. Such an effect would be absent in Kv12.1 where D1 may be required for divalent binding because D6 in S3 may not be able to make up for the lost coordinating residue.
Our divalent binding constraints suggest a strong similarity in S4 position between the divalent-bound conformation of EAG family channel VSDs and the C1 state represented in recent Kv1.2 structural models (6,11). But what part of the Kv12.1/Kv10.2 activation pathway does the divalent-bound VSD conformation represent? It is likely that only a single conformation binds divalents with high affinity in Kv12.1 because binding requires precise arrangement of residues in both S2 and S4. The same is likely to be true for ions that coordinate to the S4 residue S321 in Kv10.2. We cannot rule out the possibility that Zn2+, Cd2+, or Ni2+ can bind to multiple closed conformations in Kv10.2 because they still can affect activation (with reduced efficacy) in the absence of coordination by the residue at position 321. It is clear that divalent cations slow a closed-closed transition because of the sigmoidicity they introduce in the activation time course. Comparison of gating currents, ionic currents and fluorescent movements all suggest that divalent binding primarily affects the first of two identified closed-closed transitions that occur sequentially during voltage-dependent gating (18,20). Slowing of the Drosophila eag ionic current by Mg2+ correlates directly with slowing of the first S4 movement as measured with a fluorescent reporter (18), but some caution may be warranted because the gating current produced by this first transition has yet to be measured. Assuming the (C1 ↔ C2 ↔ A)4 ↔ O gating scheme, which can effectively describe most aspects of Drosophila eag gating (20), the implication is that divalent binding would occur in the basal resting state (C1) and slow the transition to the intermediate resting state (C2). Binding to C2 itself would also create sigmoidicity, but is not supported by the small effect of divalent cations on the second identified gating transition in Drosophila eag (18). The mechanism we propose, in which divalent ions interact directly with S4, is not able to distinguish binding to C1 from capture of the VSD in transition between C1 and C2. Given these constraints, we can assume the position of the S4 helix in the Zn2+-bound structural models represents an upper bound for the S4 position in the C1 state and a lower bound for the S4 position in the C2 state. Therefore, it seems likely that S4 gating charges sit in similar positions in Eag and Kv1 family channels in the resting state. A diagram of the full gating scheme with the proposed point of divalent action can be found in Fig. S4. Analysis of a distinct Cu2+-binding site in the VSD of BK channels also points to conformational conservation with Kv1 channels (39), although differences in Kv1 and BK channel gating models make the identity of shared states less clear.
The relevance of divalent binding to the biological functions of EAG family channels is yet to be resolved. Kv10 channels are sufficiently Mg2+ sensitive to assume that physiological Mg2+ concentrations will affect their gating behavior. Our data suggest that formation of a bridge between S321 in S4 and acidic residues in S2 and/or S3 by Mg2+ may be a factor in providing this high sensitivity. However, there is no evidence that Mg2+ serves as a dynamic modulator of Kv10 channel function in vivo. Zn2+ is a putative cotransmitter at some central synapses (40), but it is not yet known if Kv12 channels are present at these synapses. Nevertheless, the structural constraints and models presented in this study could prove useful for efforts to determine the basis for the distinctive voltage-gating phenotypes of Kv10 and Kv12 channels.
Supporting Material
Four figures and two tables are available at http://www.biophysj.org/biophysj/supplemental/S0006-3495(09)00897-2.
Supporting Material
Acknowledgments
We thank Sinead Clancy and Federica Bertaso for helpful discussions.
Funding was provided by the Genomics Institute of the Novartis Research Foundation and the National Institutes of Health (1R21NS057000-01).
References
- 1.Long S.B., Campbell E.B., Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal S.K., MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 3.Seoh S.A., Sigg D., Papazian D.M., Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 4.Long S.B., Tao X., Campbell E.B., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 5.Papazian D.M., Shao X.M., Seoh S.A., Mock A.F., Huang Y. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 6.Pathak M.M., Yarov-Yarovoy V., Agarwal G., Roux B., Barth P. Closing in on the resting state of the Shaker K(+) channel. Neuron. 2007;56:124–140. doi: 10.1016/j.neuron.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 7.Zagotta W.N., Hoshi T., Aldrich R.W. Shaker potassium channel gating. III: evaluation of kinetic models for activation. J. Gen. Physiol. 1994;103:321–362. doi: 10.1085/jgp.103.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker O.S., Larsson H.P., Mannuzzu L.M., Isacoff E.Y. Three transmembrane conformations and sequence-dependent displacement of the S4 domain in Shaker K+ channel gating. Neuron. 1998;20:1283–1294. doi: 10.1016/s0896-6273(00)80507-3. [DOI] [PubMed] [Google Scholar]
- 9.Ledwell J.L., Aldrich R.W. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 1999;113:389–414. doi: 10.1085/jgp.113.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pathak M., Kurtz L., Tombola F., Isacoff E. The cooperative voltage sensor motion that gates a potassium channel. J. Gen. Physiol. 2005;125:57–69. doi: 10.1085/jgp.200409197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campos F.V., Chanda B., Roux B., Bezanilla F. Two atomic constraints unambiguously position the S4 segment relative to S1 and S2 segments in the closed state of Shaker K channel. Proc. Natl. Acad. Sci. USA. 2007;104:7904–7909. doi: 10.1073/pnas.0702638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tombola F., Pathak M.M., Gorostiza P., Isacoff E.Y. The twisted ion-permeation pathway of a resting voltage-sensing domain. Nature. 2007;445:546–549. doi: 10.1038/nature05396. [DOI] [PubMed] [Google Scholar]
- 13.Ahern C.A., Horn R. Focused electric field across the voltage sensor of potassium channels. Neuron. 2005;48:25–29. doi: 10.1016/j.neuron.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 14.Treptow W., Tarek M. Environment of the gating charges in the Kv1.2 Shaker potassium channel. Biophys. J. 2006;90:L64–L66. doi: 10.1529/biophysj.106.080754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sands Z.A., Sansom M.S. How does a voltage sensor interact with a lipid bilayer? Simulations of a potassium channel domain. Structure. 2007;15:235–244. doi: 10.1016/j.str.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jegla T., Zmasek C., Batalov S., Nayak S.K. Evolution of the human ion channel set. Comb. Chem. High Throughput Screen. 2009;12:2–23. doi: 10.2174/138620709787047957. [DOI] [PubMed] [Google Scholar]
- 17.Zou A., Lin Z., Humble M., Creech C.D., Wagoner P.K. Distribution and functional properties of human KCNH8 (Elk1) potassium channels. Am. J. Physiol. 2003;285:C1356–C1366. doi: 10.1152/ajpcell.00179.2003. [DOI] [PubMed] [Google Scholar]
- 18.Bannister J.P., Chanda B., Bezanilla F., Papazian D.M. Optical detection of rate-determining ion-modulated conformational changes of the ether-a-go-go K+ channel voltage sensor. Proc. Natl. Acad. Sci. USA. 2005;102:18718–18723. doi: 10.1073/pnas.0505766102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engeland B., Neu A., Ludwig J., Roeper J., Pongs O. Cloning and functional expression of rat ether-a-go-go-like K+ channel genes. J. Physiol. 1998;513:647–654. doi: 10.1111/j.1469-7793.1998.647ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang C.Y., Bezanilla F., Papazian D.M. Extracellular Mg(2+) modulates slow gating transitions and the opening of Drosophila ether-a-Go-Go potassium channels. J. Gen. Physiol. 2000;115:319–338. doi: 10.1085/jgp.115.3.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trudeau M.C., Titus S.A., Branchaw J.L., Ganetzky B., Robertson G.A. Functional analysis of a mouse brain Elk-type K+ channel. J. Neurosci. 1999;19:2906–2918. doi: 10.1523/JNEUROSCI.19-08-02906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Islas L.D., Sigworth F.J. Voltage sensitivity and gating charge in Shaker and Shab family potassium channels. J. Gen. Physiol. 1999;114:723–742. doi: 10.1085/jgp.114.5.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schonherr R., Hehl S., Terlau H., Baumann A., Heinemann S.H. Individual subunits contribute independently to slow gating of bovine EAG potassium channels. J. Biol. Chem. 1999;274:5362–5369. doi: 10.1074/jbc.274.9.5362. [DOI] [PubMed] [Google Scholar]
- 24.Terlau H., Ludwig J., Steffan R., Pongs O., Stuhmer W. Extracellular Mg2+ regulates activation of rat eag potassium channel. Pflugers Arch. 1996;432:301–312. doi: 10.1007/s004240050137. [DOI] [PubMed] [Google Scholar]
- 25.Silverman W.R., Bannister J.P., Papazian D.M. Binding site in eag voltage sensor accommodates a variety of ions and is accessible in closed channel. Biophys. J. 2004;87:3110–3121. doi: 10.1529/biophysj.104.044602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silverman W.R., Tang C.Y., Mock A.F., Huh K.B., Papazian D.M. Mg(2+) modulates voltage-dependent activation in ether-a-go-go potassium channels by binding between transmembrane segments S2 and S3. J. Gen. Physiol. 2000;116:663–678. doi: 10.1085/jgp.116.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schonherr R., Mannuzzu L.M., Isacoff E.Y., Heinemann S.H. Conformational switch between slow and fast gating modes: allosteric regulation of voltage sensor mobility in the EAG K+ channel. Neuron. 2002;35:935–949. doi: 10.1016/s0896-6273(02)00869-3. [DOI] [PubMed] [Google Scholar]
- 28.Jegla T., Salkoff L. A novel subunit for shal K+ channels radically alters activation and inactivation. J. Neurosci. 1997;17:32–44. doi: 10.1523/JNEUROSCI.17-01-00032.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prime 1.6. Schrodinger, LLC, Portland, OR.
- 30.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ludwig J., Terlau H., Wunder F., Bruggemann A., Pardo L.A. Functional expression of a rat homologue of the voltage gated either a go-go potassium channel reveals differences in selectivity and activation kinetics between the Drosophila channel and its mammalian counterpart. EMBO J. 1994;13:4451–4458. doi: 10.1002/j.1460-2075.1994.tb06767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silverman W.R., Roux B., Papazian D.M. Structural basis of two-stage voltage-dependent activation in K+ channels. Proc. Natl. Acad. Sci. USA. 2003;100:2935–2940. doi: 10.1073/pnas.0636603100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seebeck B., Reulecke I., Kamper A., Rarey M. Modeling of metal interaction geometries for protein-ligand docking. Proteins. 2008;71:1237–1254. doi: 10.1002/prot.21818. [DOI] [PubMed] [Google Scholar]
- 34.Rulisek L., Vondrasek J. Coordination geometries of selected transition metal ions (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) in metalloproteins. J. Inorg. Biochem. 1998;71:115–127. doi: 10.1016/s0162-0134(98)10042-9. [DOI] [PubMed] [Google Scholar]
- 35.Lee S.Y., Lee A., Chen J., MacKinnon R. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. USA. 2005;102:15441–15446. doi: 10.1073/pnas.0507651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Starace D.M., Bezanilla F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 2004;427:548–553. doi: 10.1038/nature02270. [DOI] [PubMed] [Google Scholar]
- 37.Tombola F., Pathak M.M., Isacoff E.Y. Voltage-sensing arginines in a potassium channel permeate and occlude cation-selective pores. Neuron. 2005;45:379–388. doi: 10.1016/j.neuron.2004.12.047. [DOI] [PubMed] [Google Scholar]
- 38.Fernandez D., Ghanta A., Kinard K.I., Sanguinetti M.C. Molecular mapping of a site for Cd2+-induced modification of human ether-a-go-go-related gene (hERG) channel activation. J. Physiol. 2005;567:737–755. doi: 10.1113/jphysiol.2005.089094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma Z., Wong K.Y., Horrigan F.T. An extracellular Cu2+ binding site in the voltage sensor of BK and Shaker potassium channels. J. Gen. Physiol. 2008;131:483–502. doi: 10.1085/jgp.200809980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qian J., Noebels J.L. Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J. Physiol. 2005;566:747–758. doi: 10.1113/jphysiol.2005.089276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.