

Abstract
The positioning of adhesion molecules relative to the microtopography of the cell surface has a significant influence on the molecule's availability to form adhesive contacts. Measurements of the ratio of fluorescence intensity per unit area in epi-fluorescence images versus total internal reflection fluorescence images provides a means to assess the relative accessibility for bond formation of different fluorescently labeled molecules in cells pressed against a flat substrate. Measurements of the four principal adhesion molecules on human neutrophils reveal that L-selectin has the highest ratio of total internal reflection fluorescence/epi intensity, and that P-selectin glycoprotein ligand-1 (PSGL-1) and the integrins αLβ2 (LFA-1) and αMβ2 (Mac-1) have ratios similar to each other but lower than for L-selectin. All of the ratios increased with increasing impingement, indicating an alteration of surface topography with increasing surface compression. These results are consistent with model predictions for molecules concentrated near the tips of microvilli in the case of L-selectin, and sequestered away from the microvillus tips in the case of LFA-1, Mac-1, and PSGL-1. The results confirm differences among adhesion molecules in their surface distribution and reveal how the availability of specific adhesion molecules is altered by mechanical compression of the surface in live cells.
Introduction
When inflammation occurs, neutrophil recruitment from the blood vessels is initiated via a cascade of events that involves rolling through cell-cell adhesive interaction, cell activation, firm adhesion, and eventual migration through the vascular wall (1). Neutrophil capture from the circulation and attachment to the endothelium requires the formation of specific receptor/ligand bonds. Initial capture and rolling interactions are primarily mediated by selectins and their glycoprotein ligands, whereas cell arrest and firm attachment are mediated by integrins and their immunoglobulin ligands (2). Neutrophil-endothelium adhesion depends not only on the intrinsic kinetic rates of these receptor-ligand interactions but also on how effectively neutrophils present these molecules to their counterparts. In contrast to endothelial cells that are lining blood vessels, neutrophils do not have a flat and smooth surface but instead are covered by ruffles (3). The microvillar protrusions that cover the neutrophil surface limit the proportion of the cell membrane that may come in close contact with an opposing surface (4), thus greatly limiting the number of adhesion molecules available for bond formation. This effect may be enhanced or mitigated by the nonuniform distribution of the receptors over the cell surface. Ultrastructural evidence indicates that the distribution of the major adhesion molecules on the neutrophil surface is, in fact, nonuniform. Immunogold labeling in electron micrographs indicates that neutrophil receptors known to mediate cell rolling, i.e., L-selectin and P-selectin glycoprotein ligand-1 (PSGL-1), are primarily clustered on the tips of the microvillar ruffles (5–8). In contrast, the principal integrin receptors on the neutrophil surface (i.e., LFA-1 and Mac-1) appear to be randomly distributed on the nonvillus cell body (5,9). Thus, the positioning of these molecules relative to the microtopography of the cell surface appears to segregate with their functional role.
The environment in which adhesion between neutrophils and the endothelium occurs is mechanically complex: fluid shear forces act on cells and can have a significant influence on the proximity between receptors and their ligands. After initial contact and adhesion, hydrodynamic forces exert pulling forces at the rear of the neutrophil and compressive forces to the middle and at the front of the cell. These hydrodynamic forces increase the area of contact, thereby increasing the number of receptors and ligands available for bond formation. Mechanical forces may also affect adhesion via alterations of the microtopography of the cell membrane in the contact zone. As shown previously, increasing either the area of contact or the contact stress leads to a linear increase in the formation of bonds at the interface (10,11). Therefore, by compressing the neutrophil at the contact zone, the effect of shear forces may counteract the effect of unfavorable distribution of molecules relative to the cell surface microtopography.
In this study, we set out to determine in live, quiescent neutrophils how the relative presence of molecules at a contacting substrate compares for the different types of adhesion molecules on the cell surface, and furthermore, how the presence of molecules changes with the application of force. Total internal reflection fluorescence microscopy (TIRFM) is an ideal technique to determine which receptors reside on the ridges of live neutrophils. TIRFM (also called evanescent wave microscopy) provides a means to selectively excite fluorophores that are very near a surface (within 200 nm) without exciting fluorescence from regions farther from the surface (12). Indeed the evanescent wave created at the glass/cell solution interface decays exponentially, thereby exciting only the fluorescently labeled molecules that are within reach just above the cover glass, leaving all other fluorescent molecules in the cell unexcited. In cellular imaging of neutrophils, which have a surface covered by microvillar ruffles with an average height of 250–350 nm (3,7,13), TIRFM offers the possibility to determine whether fluorescently-labeled receptors reside on the ruffle tips or in the valleys between them (in particular, whether L-selectin and PSGL-1 are localized at the top of the ridges, and if β2 integrins (LFA-1 and Mac-1) are positioned in the valleys (5–9)). Using this approach, coupled with micromanipulation, we also examined the effect of compressive forces between the cell and the substrate on the distribution of these molecules. Observations were compared to a model developed in a companion report (14) in this issue to provide a better understanding of mechanisms leading to the behavior of different molecules in the interface under compression.
Materials and Methods
Antibodies
The following mouse monoclonal anti-human antibodies were used: DREG-56 (eBioscience, San Diego, CA) which reacts with CD62L (L-selectin), clones PL1 (Ancell, Bayport, MN) and PL2 (Santa Cruz Biotechnology, Santa Cruz, CA), which react with CD162 (PSGL-1), clone 38 (Ancell), and HI 111 (eBioscience), which react with CD11a (the α-subunit of LFA-1), and MEM-174 (Abcam, Cambridge, MA) and ICRF44 (eBioscience), which react with CD11b (the α-subunit of Mac-1). All antibodies were conjugated with Alexa Fluor 546 (Molecular Probes/Invitrogen, Eugene, OR/Grand Island, NY). The mean number of fluorophores per antibody (F/P) varied for different preparations: F/P ≈ 2.5 for L-selectin/DREG-56 and PSGL-1/PL1; F/P ≈ 7.4 for PSGL-1/PL2; F/P ≈ 4.0 for Mac-1/MEM-174, LFA-1/clone 38, and LFA-1/HI 111; and F/P ≈ 6 for Mac-1/ICRF44.
Cell preparation (labeling)
We used human neutrophils from four different donors. For each experiment, neutrophils contained in a small drop of whole blood were obtained by finger stick. The whole blood was diluted in Hank's balanced salt solution (BioWhittaker, Walkersville, MD) containing 10 mM N-[2-Hydroxyethyl]piperazine-N′-[2-ethanesulfonic acid] (HEPES, Sigma, St. Louis, MO) and was incubated for 12–15 min with the corresponding anti-human antibody conjugated with Alexa Fluor 546. Labeled cells were washed twice, then resuspended and diluted in the previous buffer supplemented with 4% fetal bovine serum (HyClone, Logan, UT). The cell solution was placed in a glass chamber that had been previously coated with 4% fetal bovine serum buffer solution to prevent cells from sticking to the cover glass. Cells were used within an hour, thus allowing the study of seven-to-nine cells per experiment. Only cells that were passively resting (thus spherical) and not sticking to the glass surface were examined.
Laser epi-fluorescence and TIRF microscopy
The fluorescence microscopy was performed through the objective on an inverted IX 70 Olympus microscope (Olympus, Center Valley, PA) that has been modified to accommodate laser epi-fluorescence and TIRF. A diode laser of wavelength 532 nm and of power 3.40 mW has been incorporated in the microscope. The shift between epi- and TIRF fluorescence was accomplished by rotating a mirror, which was driven by a motor for rapid changeover. A Plan-Apo 60× oil objective of numerical aperture 1.45 was used. Fluorescence emission was filtered between 573 and 645 nm. Images were visualized using a Sensicam electron multiplication (EM) charge-coupled device camera (Cooke, Romulus, MI) and recorded as 12-bit grayscale images. A lens of magnification 2.5× was placed in front of the camera for higher spatial resolution. All bright-field images were captured with an exposure of 40 ms and an EM gain of 2×. All fluorescence images were captured with an EM gain of 20× and a minimum exposure of 200 ms. For some experiments, we used a fluorescence exposure of 300 or 400 ms. In any case, the same camera settings were used for the two types of fluorescence images (epi-fluorescence and TIRF). The operation of the bright-field and laser shutters, the rotation of the epi/TIRF switch mirror, and the camera were automated through the interface of a LabVIEW program (National Instruments, Austin, TX). Automation enabled us to minimize the time interval between two successive captures of image to 2 s. Thus, capturing a series of bright-field (BF), epi, and TIRF images typically required ∼6 s.
Micromanipulation
The chamber containing the cell solution had a side opening for the introduction of a pipette used to hold the cells with increasing pressure against the glass surface. The glass pipettes had inside diameters of ∼2.0 μm and were held at a small angle (≈8°) relative to the cover glass surface to avoid contact with it.
Experimental procedure
Fig. 1 illustrates the way we performed our experiments by showing typical experimental images. For each cell, an epi-fluorescence image was captured with the focus positioned at the center of the cell. The purpose was to detect the fluorescence intensity of the molecules throughout the membrane to take into account the different levels of protein expression and label intensity (i.e., mean number of fluorophores per antibody). The focus was then brought to the bottom of the cell just above the cover-glass surface. At least three series of BF (bright-field), epi, and TIRF images were acquired at that position. Finally, a small portion of the cell was sucked in the pipette with a pressure just large enough to maintain a projection length of 2–6 μm. The contact area was increased in successive steps by lowering the pipette and progressively pressing the cell against the glass. At least four, and up to nine, impingement positions were performed, and for each of them, a series of BF, epi, and TIRF images were recorded with the glass-cell interface being in focus. For each cell, the series of BF, epi, and TIRF images were recorded as fast as possible, leading to an average of 11 s between two successive impingement positions.
Figure 1.
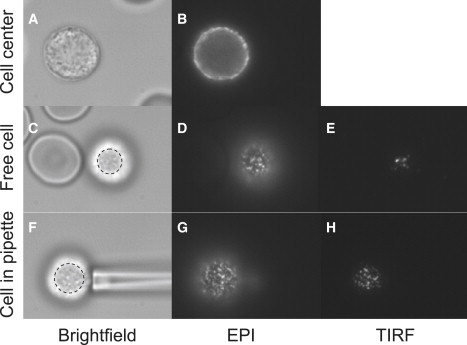
Typical images recorded for one cell. In these images, L-selectin is fluorescently labeled. The images in the first column (A, C, and F) are bright-field images, in the second column (B, D, and G) are epi-fluorescence images, and in the third column (E and H) are TIRF images. The focus was positioned at the center of the cell on the images of the first row and at the bottom of the cell on the images of the last two rows. The cell was held in a pipette for the images of the last row. For presentation, the contrast was adjusted to make the signal clearly visible. Note that the contact area was determined from the bright-field images (C and F). The area was that of a nearly circular ellipse drawn to just include the shaded central region and its boundary (as shown).
Image processing
As a means of assessing the relative proportion of molecules in close contact with the glass substrate, we measured the ratio of intensities per unit area above background for TIRF versus epi-images. This ratio was measured for each individual cell as a function of cell-surface contact area,
| (1) |
For each studied cell, the epi-intensity per unit area, EPIsignal, was extracted from the epi-fluorescence image taken when the focus was at the center of the cell. The EPIsignal was calculated by averaging the intensity of a 1.0-μm-wide band at the bright edge of the cell. The TIRF intensity per unit area, TIRFsignal, was extracted by first selecting the cell-surface contact areas using the bright-field images (see Fig. 1). Then a mask was created so that only fluorescence signal from within the contact region was measured. The corresponding background intensities, EPIbkgd and TIRFbkgd, were obtained by integrating over identical areas in a different (nonfluorescing) region of the same respective images.
Statistical analysis
To account for dependencies between measurements performed on an individual cell, data were described using linear mixed models (15). These models included donor, protein, antibody, contact area, contact area2, and their interactions, as independent variables. Hypothesis testing was conducted using likelihood ratio tests.
Computation of model predictions and identification of parameter values
The expression for the ratio of TIRF/EPI intensity per unit area (as given by Eq. 2 below) does not have a closed-form expression, and we approximated its values using computer simulations. To determine parameter values yielding model predictions in good agreement with our experimental observations, one possibility would have been to minimize a least-squares criterion computed by summing the squared differences between the model predictions and the individual observations for the TIRF/EPI intensity ratio. Such an approach, however, would have required approximating the mean TIRF intensity at hundreds of values of the contact area, making the approach enormously time-consuming. To reduce the computing time, we therefore proceeded as follows. First, a linear mixed model, including contact area and contact area2 as independent variables, was fitted to the TIRF/EPI intensity ratio. This was done for each protein and for each donor separately. The fitted models allowed random intercepts and random slopes to account for the dependencies between measurements made on the same cell. From these fitted models, we deduced estimates for the average TIRF/EPI intensity ratio at preselected values of the contact area. The area values were spaced at regular intervals of 2.0 μm2, 7–10 points per curve. Parameter values were finally identified by minimizing the sum of the squared differences between these estimated mean ratios and the corresponding mean ratios predicted by the model. This approach avoided approximating (by simulations) the mean TIRF intensity of the proposed model hundreds of times, significantly reducing the computing time, which remained nonetheless substantial. Data from each donor were treated collectively, the assumption being that the microvillus spring constant kv and the factor ξ should be the same for different molecules for a given donor. In addition to these two parameters, one parameter of the beta distribution for each molecule was allowed to vary. Minimization of the optimization function was conducted using the Nelder and Mead simplex algorithm (16), and the sum of the squared residuals was calculated as a measure of fitting accuracy. These are given for each donor in Table 1. This approach made it practical to fit the model to the data, but to the best of our knowledge, the approach does not, unfortunately, allow us to calculate the customary measures of goodness of fit that would be possible in typical nonlinear least-squares regression.
Table 1.
Model parameters obtained for each donor
| Parameter | Experimental constant |
Microvillus spring constant |
Beta distribution |
Sum of squared residuals |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| L-selectin |
PSGL-1 |
Mac-1 |
LFA-1 |
||||||||
| ξ | kv | c | d | c | d | c | d | c | d | (SSR) | |
| Donor 1 | 1.60 | 29.8 | 2.72 | 1 | 1 | 1.90 | 1 | 2.34 | 1 | 3.22 | 0.0036 |
| Donor 2 | 1.24 | 30.5 | 2.23 | 1 | 1 | 2.49 | 1 | 2.29 | 1 | 2.05 | 0.0058 |
| Donor 3 | 1.83 | 27.8 | 2.58 | 1 | 1 | 1.27 | 1 | 1.59 | 1 | 8.40 | 0.0055 |
| Donor 4 | 2.33 | 38.0 | 3.22 | 1 | 1 | 2.57 | 1 | 2.43 | 1 | 15.48 | 0.0053 |
Results
Fluorescence images
Typical examples of fluorescence images obtained for each adhesion molecule are shown in Fig. 2. In each column, one of the molecules is shown for three types of images, when the focus is at the center of the cell (first row, images A–D) and when it is at the cell-glass interface (second and third rows, images E–L). In images E–L, the cells were held in a pipette, and pressed on glass such that the contact areas were similar (15.4 ± 1.3 μm2). All four cells selected for this composite (A–D) exhibited a similar epi-fluorescence intensity averaged over their bright edges (731 ± 25 per pixel). Similarly, the mean fluorescence values for the epi-fluorescence images (E–H) obtained when the focus was brought to the cell-glass interface were similar. For all adhesion molecules, the distribution of fluorescence at the interface was nonuniform (punctate). In both the epi and TIRF images, punctate regions for L-selectin were consistently sharper and more clearly defined than for the other three proteins. This is because the intensity of the L-selectin bright spots is much stronger while the intensity around them is almost at the level of the background. Furthermore, TIRF images of PSGL-1, Mac-1, and LFA-1 contained fewer but seemingly larger bright spots than those of L-selectin, and in contrast to L-selectin, the bright spots on TIRF images of Mac-1 and LFA-1 are surrounded by a diffuse fluorescence, making them look more spread and linked together. Most significantly, there is a quantitative difference in the level of signal obtained in TIRF images of L-selectin versus the other three adhesion molecules, as indicated by the grayscale calibration bars to the right of each panel. To quantify this difference, we calculated the ratios of the TIRF intensities over the epi-fluorescence intensities above background (see next two sections).
Figure 2.
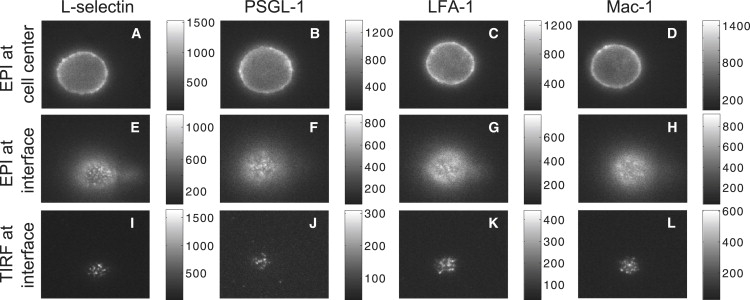
Examples of fluorescence images recorded for one donor. In the first row (A–D) are epi-fluorescence images recorded when the focus was positioned at the center of the cell. In images of the last two rows, cells are held in a pipette and pressed onto the glass to make similar contact areas (15.4 ± 1.3 μm2) and the focus was at the cell-glass interface. In the second row (E–H) are epi-fluorescence images and in the third row (I–L) are the corresponding TIRF images. Images of the first column (A, E, and I), the second column (B, F, and J), the third column (C, G, and K), and the fourth column (D, H, and L) illustrate L-selectin, PSGL-1, LFA-1, and Mac-1 labeling, respectively. For clarity of presentation, the range of grayscales in each image was adjusted, and the shaded values in the original image are given in the grayscale bars to the right of each image. All four cells (A–D) exhibited a similar epi-fluorescence intensity averaged over their bright edges (731 ± 25 per pixel).
Fluorescence ratios: cells resting on glass
The fluorescence images of L-selectin, PSGL-1, LFA-1, and Mac-1 collected when neutrophils were resting on glass were processed, and the ratios obtained are shown in Fig. 3 as a function of the cell-glass contact area. Each of the four graphs contains all the measurements collected on a particular donor. Thus, each panel contains at least three data points per cell and seven-to-nine cells per antibody type. The scatter in the data reflects variability from cell to cell and between successive measurements on the same cell. Clearly, the ratio for L-selectin (stars) falls well above ratios obtained for the other three cell adhesion molecules. This is further documented in Fig. 4. Data points corresponding to the same cell have been averaged into one per cell. Then the mean of all seven-to-nine per cell have been computed for each antibody/molecule and for each donor, and are represented in Fig. 4. Differences between donors were small compared to differences between different molecules. Statistical analysis does not show significant differences between donors or between PSGL-1, Mac-1, and LFA-1, but confirms that for L-selectin is significantly higher (threefold) than data for the other three proteins (P < 0.0001).
Figure 3.
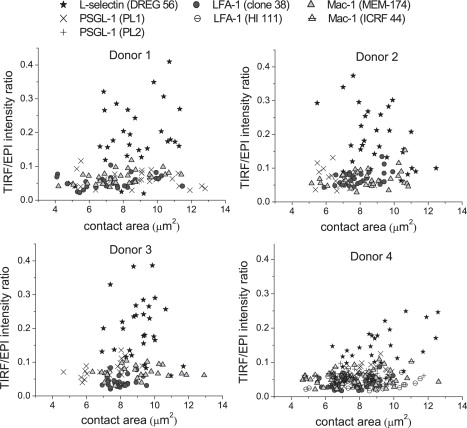
TIRF/EPI intensity ratio as a function of the cell-glass contact area extracted from the fluorescence images of L-selectin, PSGL-1, LFA-1, and Mac-1, when neutrophils were resting on glass. Each panel displays all the data collected for one donor: four proteins, 7–9 cells per antibody, and at least three points per cell. Antibodies DREG-56, PL1, clone 38, and MEM-174 were used against L-selectin, PSGL-1, LFA-1, and Mac-1, respectively. For donor 4 only, antibodies PL2, HI 111, and ICRF 44 were also used against PSGL-1, LFA-1, and Mac-1, respectively.
Figure 4.
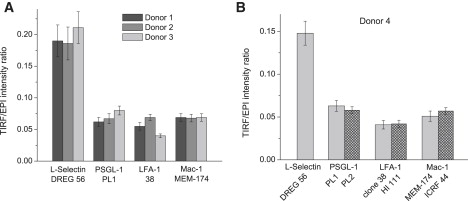
Mean TIRF/EPI intensity ratios obtained for each antibody/protein and for each donor, when neutrophils were freely resting on glass. Error bars represent the standard error of the mean ratios. (A) The mean ratios acquired using the same antibody per cell adhesion molecule, for donor 1, 2, and 3 only. (B) The data gathered from donor 4 only. A second antibody was used against PSGL-1, LFA-1, and Mac-1 for this donor. An exposure of 200 ms was generally used. However donor 4 PSGL-1/PL1 and LFA-1/HI 111 experiments were performed using a 400-ms exposure and donor 4 PSGL-1/PL2 and LFA-1/clone 38 data were obtained using a 300-ms exposure.
To test whether the antibody chosen has an effect on the ratio obtained, two different antibodies were used against PSGL-1, LFA-1, and Mac-1 for one donor (donor 4). Results are presented in Fig. 4 B. No significant difference (P = 0.06) was found between ratio values obtained with both PSGL-1 antibodies (clones PL1 and PL2). Likewise, ratios obtained with both LFA-1 antibodies (clones 38 and HI 111) are very similar (P = 0.24), as are those obtained with both Mac-1 antibodies (MEM-174 and ICRF44, P = 0.10).
Fluorescence ratios: cells pressed on glass
Data collected when the cells were held in a pipette and pressed against the glass surface with increasing force are shown in Fig. 5. All ratios, , increased with increasing contact area. Greater variability between donors was observed for the dependence of on increasing impingement than was observed for cells simply resting on the glass. Nevertheless, L-selectin clearly exhibits the greatest increase in with increasing compression on the surface (P < 0.0001). The relationships between the intensity ratio and the contact area were not affected significantly by using different antibodies against the proteins. Tests were performed on cells from one donor (donor 4) using the same pairs of antibodies as previously used for cells resting on glass. No significant differences were detected between the two antibodies against PSGL-1 (P = 0.10), or LFA-1 (P = 0.78). The very close agreement between the latter two experiments may have been due in part to the fact that they were performed on the same day, and therefore strongly confirmed that the results were specific to the protein and independent of the label used. A slight difference was observed for antibodies against Mac-1 (P = 0.03) (measured on different days). The ratio obtained with ICRF44 increased at a slightly lower rate with the contact area than the ratio obtained with MEM-174, although both displayed similar behavior compared to L-selectin.
Figure 5.
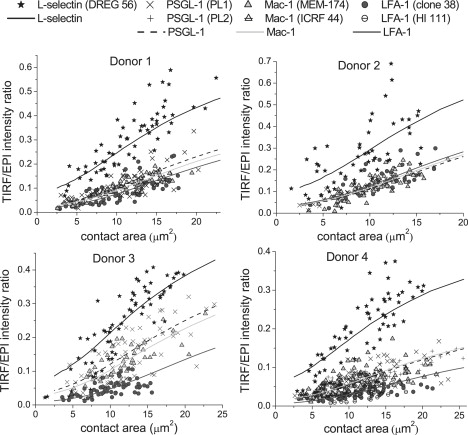
TIRF/EPI intensity ratio as a function of the cell-glass contact area for L-selectin, PSGL-1, LFA-1, and Mac-1, when neutrophils were pressed on glass. Each panel displays all the data collected for one donor: four proteins, 7–9 cells per antibody, and 4–9 points per cell. Antibodies DREG-56, PL1, clone 38, and MEM-174 were used against L-selectin, PSGL-1, LFA-1, and Mac-1, respectively. For donor 4 only, second antibodies, namely PL2, HI 111, and ICRF 44 were also used against PSGL-1, LFA-1, and Mac-1, respectively. Curves correspond to model predictions corresponding to the different molecules as indicated in the legend and for the parameters shown in Table 1.
Comparison with model predictions
In a companion report (14), we developed a model of a cell being pressed onto a smooth substrate. The model accounts for the presence of microvilli on the cell surface, the deformation of the microvilli, changes in the overall cell surface contour as the cell is compressed against a substrate, and a nonuniform distribution of molecular concentrations relative to the position on the microvillar surface. As described in the companion report (14), the ratio of the TIRF intensity per unit area to the epi-intensity per unit area, , for a given compression of the cell can be expressed as
| (2) |
where r is the radial coordinate measured perpendicular to the axis of symmetry passing through the center of the contact zone; R is the radius of the contact area; ℓ0 is the separation distance between the cell body and the substrate at the center of the contact zone; ℓ(r) is the axial separation distance at position r between the spherical contour of the cell and the substrate (thus ℓ(r) depends implicitly on ℓ0); L represents the length of the microvilli at rest; D and Dc are the distances of the protein relative to the cell body when the microvillus is not compressed and when the microvillus is compressed, respectively; β(D/L;c,d) denotes the probability density function of the beta distribution with parameters c and d describing the distribution of the positions of the proteins along the microvillus length, g(L;a,b) is the probability density function of the gamma distribution with shape and scale parameters a and b describing the distribution of microvillus lengths; dp denotes the penetration depth of the evanescent wave of the TIRF excitation light; and ξ denotes an unknown experimental constant that depends on the depth of focus and reflects the difference in amplitude of the incident epi-illumination compared to the TIRF illumination at the interface. Note that the TIRF intensity per unit area corresponds to the subtraction of the measured values and likewise the epi-intensity per unit area corresponds to . The compression of the cell is expressed through the value of ℓ0: the smaller the ℓ0, the more compressed the cell. The TIRF intensity per unit area is calculated by integrating over the whole area of contact of radius R. This radius is deduced when ℓ(R), the vertical distance between the substrate and the cell body, reaches a specified maximum value at radial position r = R.
Some parameters of the model depend on the experimental setup, whereas others reflect characteristics of the protein, the cell, and/or the donor under consideration. Table 1 of the companion report (14) summarizes the values of the optical and cellular parameters that were kept constant. These include the radius of the cell (4.1 μm), the density of microvilli on the cell surface (1.5/μm2), and the cortical tension of the cell (20 μN/m). Several of the model parameters (listed in Table 2 of the companion report (14)) were varied to obtain agreement between the model predictions and our measurements, subject to the following assumptions:
-
1.
The same gamma distribution for the microvillus length was used for all donors.
-
2.
The same value for ℓ(R), which defines the contact area πR2, was used for all donors.
-
3.
The value of the coefficient ξ was allowed to vary across donors, but was kept constant for different molecules within the same donor.
-
4.
The value of the microvillus spring constant kv was allowed to vary across donors.
-
5.
The parameters of the beta distribution for the localization of the proteins were allowed to vary across proteins and donors.
While trying to match the model predictions to our data, we found that a gamma distribution corresponding to the microvilli lengths measured by Bruehl et al. (7), provided an excellent agreement with the data (14). Therefore, we used a gamma distribution with shape and scale parameters and , corresponding to a mean microvillus length of 265 ± 165 nm. In our simulations, the radius of the whole area of contact, R, was defined such that the vertical distance from the substrate to the cell body at the edge of the contact zone, ℓ(R), was equal to 615 nm. The companion report (14) explains the rationale leading to the selection of this value.
Table 1 of this report summarizes the values of the coefficients ξ and of the microvillus spring constants kv that were optimized for each donor. We found that coefficients ξ ranging from 1.24 to 2.33 and microvillus spring constant values varying between ∼28 and 38 pN/μm gave the best agreement between model predictions and measurements. Also listed in Table 1 are the optimized values for the parameters c and d of the beta distributions. To limit the range of possibilities and facilitate the comparison afterwards, we have arbitrarily chosen the smaller of the two parameters to be 1.0, and varied the other to obtain agreement between model predictions and observations. Such a restriction, however, still allows the model to capture the most salient features of our experimental data because beta distributions with parameter values satisfying would describe proteins being more concentrated toward the tips of the microvilli, whereas beta distributions with parameters satisfying would describe proteins tending to be more localized toward the planar cell body. Thus, as seen in Table 1, we obtained and for L-selectin for every donor. In comparison, we obtained and for both LFA-1 and Mac-1, and this for every donor also. Measurements for PSGL-1 closely resembled those observed for Mac-1, and we obtained and for PSGL-1 for all donors as well.
The expected ratio values calculated under these circumstances are displayed in Fig. 5. For each donor and for each of the four proteins (L-selectin, PSGL-1, Mac-1, and LFA-1), the model prediction is superimposed over the corresponding experimental data. The expected is in good agreement with our experimental results, which indicate first that obtained for L-selectin is always higher than for PSGL-1, Mac-1, or LFA-1 for a given contact area, and second that values increase faster with increasing compression of the cell for L-selectin than for PSGL-1, Mac-1, or LFA-1. Ultimately, the model confirms that L-selectin should be mainly distributed toward the tips of the microvilli, because the parameters of the beta distributions describing its localization along the length of the microvilli satisfy c > d. The model also corroborates the fact that Mac-1 and LFA-1 should be mainly distributed around the planar cell body (because we obtained c < d in these two cases). The optimized parameter values for the beta distributions of LFA-1 and Mac-1 further suggest that LFA-1 might be more segregated toward the cell body than Mac-1. Fitted coefficients for PSGL-1 also suggest that it is more concentrated toward the planar cell body, and that the localization of this protein most resembles that of Mac-1.
Discussion
The relative accessibility of adhesion molecules at the interface between a cell and a substrate makes a critical difference in the effectiveness with which adhesive bonds can form (4,17). Indeed a number of studies have examined the distribution of different adhesion molecules on the neutrophil surface relative to the cell surface topography (e.g., (5,7)), but all of these prior studies have examined fixed specimens in electron microscopy. To our knowledge, this report is the first to provide a measure of molecular distribution relative to surface topography in a living neutrophil.
For all donors, results were unequivocal as far as L-selectin is concerned. Compared to the other three proteins, L-selectin was approximately three times higher when neutrophils were freely resting on glass, and they increased faster with compression of the neutrophils on the cover glass. As confirmed by model calculations (14), this indicates that L-selectin must be distributed preferentially toward the tips of microvilli on the cell surface, such that they are more readily available at the points of contact. This is consistent with prior studies (5,7), and makes teleological sense in view of the role of L-selectin as an initiator of cell adhesion and rolling (18). Even when L-selectin is transfected in L-selectin-deficient leukocytes (like murine L1-2 and 300.19 pre-B cells), it is usually observed to localize on the microvilli tips (18–20). The same is observed for the integrin α4β7, which is confined to the tips of microvillar protrusions in mouse TK1 lymphoma cells (21) and localized at microvilli tips when transfected into K562 cells (22).
In contrast to L-selectin, integrins LFA-1 (αLβ2) and Mac-1 (αMβ2) have been observed to be distributed mainly on the cell body of human neutrophils (5), as well as other leukocytes, such as mouse TK1 lymphoma cells (21). More particularly when α4β7, αLβ2, and αMβ2 integrins are transfected in K562 cells, they are found where expected, which is on the microvilli tips for the α4β7 integrin and on the cell body for αLβ2 and αMβ2 integrins (22). The present results are completely consistent with these findings. The ratio of LFA-1 in particular is consistent with a large proportion of the molecules distributed well away from microvillus tips. LFA-1 and Mac-1 ratio values appear slightly different overall (P = 0.0005), with Mac-1 tending toward a more uniform distribution compared to LFA-1. In the case of Mac-1, the more uniform distribution and the greater variability observed between individuals might be attributable to slight but different activation levels of the cells. Neutrophil activation leads to the upregulation of Mac-1 and to its redistribution randomly over the whole cell surface (5,23). Previously, we have measured different expression levels of Mac-1 on cells from different donors and showed that higher levels of Mac-1 correlated with more rapid and robust responses of the cells to IL-8 stimulus (24), even though the cells appeared morphologically normal. Thus, variability in Mac-1 expression and localization is to be expected for different donors and at different times. As indicated by model calculations (14), a uniform distribution of the adhesion molecules would lead to intermediate and an intermediate rate of increase as a function of the contact area, consistent with what we have observed for Mac-1.
That PSGL-1 exhibits behavior more similar to the β2 integrins than L-selectin, is the most surprising result of this study. Electron micrographs showing immunogold labeling of PSGL-1 indicate that PSGL-1 is, like L-selectin, primarily confined to the tips of microvillar protrusions (6,8). Nonetheless obtained for PSGL-1 was not at all like that obtained for L-selectin, but was very similar to those obtained for the β2 integrins. Moreover, this result was confirmed with two different antibody labels, the same ones used in the ultrastructural studies (6,8). It is possible that some contribution to the lower-than-expected fluorescence ratios for PSGL-1 could be due to internalization of the molecules and the generation of a submembrane pool that contributes to the denominator (epi) but not the numerator (TIRF) of the fluorescence ratio, but the degree of internalization observed for PSGL-1 was not substantially different than what was observed for the other proteins, making this explanation appear unlikely. Studies of PSGL-1 reconstituted in other cell types support the finding of a more-uniform distribution of PSGL-1 than is indicated in earlier studies on neutrophils. Unlike the other proteins examined in reconstituted systems, which exhibit the distribution observed in native systems, PSGL-1 when transfected in K562 cells is not concentrated at the tips of projections, but exhibits a uniform distribution over the cell surface (25,26). Experimental procedures differed slightly between these sets of studies. For the K562 cells, immunogold labeling of PSGL-1 was performed before cell fixation and was visualized by scanning electron microscopy (25,26), whereas for neutrophils, partial fixation was performed before immunogold labeling of PSGL-1 which was visualized by transmission electron microscopy (6,8). Based on our present findings, we propose that the latter approach resulted in an artifactual redistribution of PSGL-1 and that the conclusion that it is confined to microvillar tips was erroneous. The fluorescence data presented herein shows that, in living cells, PSGL-1 distribution far more resembles that of Mac-1 than L-selectin.
The idea that PSGL-1 appeared on the tips of microvilli-like L-selectin was appealing in a teleological sense, because the distribution of molecules seemed to segregate with their functional roles (27,28). Along a similar line, we have shown recently that the lateral mobility of PSGL-1 is similar to that of L-selectin and substantially slower than that of the β2 integrins (29). Thus, the finding that PSGL-1 distribution resembles that of the β2 integrins was a surprise. L-selectin has a very short extracellular length compared to PSGL-1 which extends at least four times farther (∼60 nm) from the membrane (30–32). Furthermore, the critical recognition site for P-selectin on PSGL-1 lies relatively far from the membrane (31). Similarly, the carbohydrate-binding lectin domains of L-, E-, and P-selectins are all located at their NH2 terminus, the farthest from the cell surface (30), and human P-selectin (at ∼40 nm) is at least three times longer than L-selectin. (P-selectin has nine short consensus repeats instead of two for L-selectin.) Thus, PSGL-1 and P-selectin extend well above the membrane surface, and this might help to overcome a seemingly less-favorable distribution of PSGL-1 over the cell topography.
Conclusion
A novel experimental approach for assessing the relative accessibility of adhesion molecules at an interface has been applied to examine the localization of the principal adhesion molecules on living human neutrophils relative to the surface topography. Consistent with prior ultrastructural evidence, L-selectin is localized to the tips of microvilli and the β2 integrins are sequestered away from the microvillus tips. PSGL-1, previously thought to be localized at microvilli tips, was found to be markedly dissimilar to L-selectin but to match the distribution of Mac-1 on the cell surface. These findings overturn existing paradigms for PSGL-1 localization, but agree with other studies showing widespread distribution of PSGL-1 when it is transfected into model cells.
Acknowledgments
The authors thank Thomas Gaborski, Elena Lomakina, Richard Bauserman, Margaret Youngman, Joanne Schultz, and Nathan Clark for technical support and scientific discussions.
This work was supported by National Institutes of Health grant No. PO1-HL18208 (R.W. and S.H.) and RO1-CA134839 (O.H.).
References
- 1.Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 2.Lawrence M.B., Springer T.A. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell. 1991;65:859–873. doi: 10.1016/0092-8674(91)90393-d. [DOI] [PubMed] [Google Scholar]
- 3.Majstoravich S., Zhang J., Nicholson-Dykstra S., Linder S., Friedrich W. Lymphocyte microvilli are dynamic, actin-dependent structures that do not require Wiskott-Aldrich syndrome protein (WASp) for their morphology. Blood. 2004;104:1396–1403. doi: 10.1182/blood-2004-02-0437. [DOI] [PubMed] [Google Scholar]
- 4.Williams T.E., Nagarajan S., Selvaraj P., Zhu C. Quantifying the impact of membrane microtopology on effective two-dimensional affinity. J. Biol. Chem. 2001;276:13283–13288. doi: 10.1074/jbc.M010427200. [DOI] [PubMed] [Google Scholar]
- 5.Erlandsen S.L., Hasslen S.R., Nelson R.D. Detection and spatial distribution of the β2 integrin (Mac-1) and L-selectin (LECAM-1) adherence receptors on human neutrophils by high-resolution field emission SEM. J. Histochem. Cytochem. 1993;41:327–333. doi: 10.1177/41.3.7679125. [DOI] [PubMed] [Google Scholar]
- 6.Bruehl R.E., Moore K.L., Lorant D.E., Borregaard N., Zimmerman G.A. Leukocyte activation induces surface redistribution of P-selectin glycoprotein ligand-1. J. Leukoc. Biol. 1997;61:489–499. doi: 10.1002/jlb.61.4.489. [DOI] [PubMed] [Google Scholar]
- 7.Bruehl R.E., Springer T.A., Bainton D.F. Quantitation of L-selectin distribution on human leukocyte microvilli by immunogold labeling and electron microscopy. J. Histochem. Cytochem. 1996;44:835–844. doi: 10.1177/44.8.8756756. [DOI] [PubMed] [Google Scholar]
- 8.Moore K.L., Patel K.D., Bruehl R.E., Li F.G., Johnson D.A. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 1995;128:661–671. doi: 10.1083/jcb.128.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fernandez-Segura E., Garcia J.M., Campos A. Topographic distribution of CD18 integrin on human neutrophils as related to shape changes and movement induced by chemotactic peptide and phorbol esters 1. Cell. Immunol. 1996;171:120–125. doi: 10.1006/cimm.1996.0181. [DOI] [PubMed] [Google Scholar]
- 10.Lomakina E.B., Spillmann C.M., King M.R., Waugh R.E. Rheological analysis and measurement of neutrophil indentation. Biophys. J. 2004;87:4246–4258. doi: 10.1529/biophysj.103.031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spillmann C.M., Lomakina E., Waugh R.E. Neutrophil adhesive contact dependence on impingement force. Biophys. J. 2004;87:4237–4245. doi: 10.1529/biophysj.103.031773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 13.Shao J.Y., Ting-Beall H.P., Hochmuth R.M. Static and dynamic lengths of neutrophil microvilli. Proc. Natl. Acad. Sci. USA. 1998;95:6797–6802. doi: 10.1073/pnas.95.12.6797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hocdé S.A., Hyrien O., Waugh R.E. Molecular accessibility in relation to cell surface topography and compression against a flat substrate. Biophys. J. 2009;97:369–378. doi: 10.1016/j.bpj.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Searle S.R., Casella G., MacCulloch C.E. John Wiley & Sons; Hoboken, NJ: 1992. Variance Components. [Google Scholar]
- 16.Nelder J.A., Mead M.R. A simplex method for function minimization. Comput. J. 1965;7:308–313. [Google Scholar]
- 17.Waugh R.E., Lomakina E.B. Active site formation, not bond kinetics, limits adhesion rate between human neutrophils and immobilized vascular cell adhesion molecule 1. Biophys. J. 2009;96 doi: 10.1016/j.bpj.2008.09.009. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Andrian U.H., Hasslen S.R., Nelson R.D., Erlandsen S.L., Butcher E.C. A central role for microvillus receptor presentation in leukocyte adhesion under flow. Cell. 1995;82:989–999. doi: 10.1016/0092-8674(95)90278-3. [DOI] [PubMed] [Google Scholar]
- 19.Hasslen S.R., von Andrian U.H., Butcher E.C., Nelson R.D., Erlandsen S.L. Spatial distribution of L-selectin (CD62L) on human lymphocytes and transfected murine L1–2 cells. Histochem. J. 1995;27:547–554. [PubMed] [Google Scholar]
- 20.Pavalko F.M., Walker D.M., Graham L., Goheen M., Doerschuk C.M. The cytoplasmic domain of L-selectin interacts with cytoskeletal proteins via α-actinin: receptor positioning in microvilli does not require interaction with α-actinin. J. Cell Biol. 1995;129:1155–1164. doi: 10.1083/jcb.129.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berlin C., Bargatze R.F., Campbell J.J., von Andrian U.H., Szabo M.C. α4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. 1995;80:413–422. doi: 10.1016/0092-8674(95)90491-3. [DOI] [PubMed] [Google Scholar]
- 22.Abitorabi M.A., Pachynski R.K., Ferrando R.E., Tidswell M., Erle D.J. Presentation of integrins on leukocyte microvilli: a role for the extracellular domain in determining membrane localization. J. Cell Biol. 1997;139:563–571. doi: 10.1083/jcb.139.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tandon R., Sha'afi R.I., Thrall R.S. Neutrophil β2-integrin upregulation is blocked by a p38 MAP kinase inhibitor. Biochem. Biophys. Res. Commun. 2000;270:858–862. doi: 10.1006/bbrc.2000.2540. [DOI] [PubMed] [Google Scholar]
- 24.Lomakina E.B., Waugh R.E. Dynamics of increased neutrophil adhesion to ICAM-1 after contacting immobilized IL-8. Ann. Biomed. Eng. 2006;34:1553–1563. doi: 10.1007/s10439-006-9172-y. [DOI] [PubMed] [Google Scholar]
- 25.Snapp K.R., Craig R., Herron M., Nelson R.D., Stoolman L.M. Dimerization of P-selectin glycoprotein ligand-1 (PSGL-1) required for optimal recognition of P-selectin. J. Cell Biol. 1998;142:263–270. doi: 10.1083/jcb.142.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snapp K.R., Heitzig C.E., Kansas G.S. Attachment of the PSGL-1 cytoplasmic domain to the actin cytoskeleton is essential for leukocyte rolling on P-selectin. Blood. 2002;99:4494–4502. doi: 10.1182/blood.v99.12.4494. [DOI] [PubMed] [Google Scholar]
- 27.Dore M., Korthuis R.J., Granger D.N., Entman M.L., Smith C.W. P-selectin mediates spontaneous leukocyte rolling in vivo. Blood. 1993;82:1308–1316. [PubMed] [Google Scholar]
- 28.Mayadas T.N., Johnson R.C., Rayburn H., Hynes R.O., Wagner D.D. Leukocyte rolling and extravasation are severely compromised in P-selectin-deficient mice. Cell. 1993;74:541–554. doi: 10.1016/0092-8674(93)80055-j. [DOI] [PubMed] [Google Scholar]
- 29.Gaborski T.R., Clark A., Waugh R.E., McGrath J.L. Membrane mobility of β2 integrins and rolling associated adhesion molecules in resting neutrophils. Biophys. J. 2008;95:4934–4937. doi: 10.1529/biophysj.108.132886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bevilacqua M.P., Nelson R.M. Selectins. J. Clin. Invest. 1993;91:379–387. doi: 10.1172/JCI116210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li F., Erickson H.P., James J.A., Moore K.L., Cummings R.D. Visualization of P-selectin glycoprotein ligand-1 as a highly extended molecule and mapping of protein epitopes for monoclonal antibodies. J. Biol. Chem. 1996;271:6342–6348. doi: 10.1074/jbc.271.11.6342. [DOI] [PubMed] [Google Scholar]
- 32.Ushiyama S., Laue T.M., Moore K.L., Erickson H.P., McEver R.P. Structural and functional characterization of monomeric soluble P-selectin and comparison with membrane P-selectin. J. Biol. Chem. 1993;268:15229–15237. [PubMed] [Google Scholar]