

Abstract
Theoretical models predict that macromolecular crowding can increase protein folding stability, but depending on details of the models (e.g., how the denatured state is represented), the level of stabilization predicted can be very different. In this study, we represented the native and denatured states atomistically, with conformations sampled from explicit-solvent molecular dynamics simulations at room temperature and high temperature, respectively. We then designed an efficient algorithm to calculate the allowed fraction, f, when the protein molecule is placed inside a box of crowders. That a fraction of placements of the protein molecule is disallowed because of volume exclusion by the crowders leads to an increase in chemical potential, given by Δμ = −kBT lnf. The difference in Δμ between the native and denatured states predicts the effect of crowding on the folding free energy. Even when the crowders occupied 35% of the solution volume, the stabilization reached only 1.5 kcal/mol for cytochrome b562. The modest stabilization predicted is consistent with experimental studies. Interestingly, a mixture of different sized crowders was found to exert a greater effect than the sum of the individual species of crowders. The stabilization of crowding on the binding stability of barnase and barstar, based on atomistic modeling of the proteins, was similarly modest. These findings have profound implications for macromolecular crowding inside cells.
Introduction
Inside cells, protein and RNA molecules occupy >30% of the volume (1). The crowded conditions are expected to have significant effects on biophysical properties of proteins. Thanks to concerted efforts based on theoretical modeling (2–7), atomistic simulations (8,9), and in vitro experiments (10–13), a qualitative understanding on the effects of macromolecular crowding on protein folding and binding stability has been reached. Crowders, because of excluded volume, raise the chemical potential of a test protein, and more so to the protein molecule when its conformation is more open (14). Thereby, crowding shifts the folding equilibrium toward the native state and the binding equilibrium toward the bound state. Here we report quantitative predictions of the effects of crowding on folding and binding stability based on atomistic modeling of protein molecules.
Simple theoretical models can be valuable in providing physical insight into how crowding may affect protein folding and binding. They are designed to capture the essential physics but lack realistic details. For examples, proteins in the native state have been modeled as spheres. Although theoretical models can be constructed to fit a specific set of experimental data, the predictive power of such models is questionable. In fact, the predicted levels of crowding-induced stabilization by different models can be significantly different (5,6). Therefore, quantitatively the value of simple theoretical models may be limited. This limitation was a main motivation for this approach of atomistic modeling.
Cheung et al. (8) and Stagg et al. (9) carried out Langevin dynamics simulations of the folding and unfolding of two different proteins, each represented by a coarse-grained Cα side-chain model, in the presence of spherical crowders. The simulations allowed them to calculate the changes in folding free energy by the crowders. Similarly, Minh et al. (15) carried out Brownian dynamics simulations of the flap motions of a protein, represented by a coarse-grained Cα-only model, in the presence of spherical crowders. The simulations showed that crowding shifts the equilibrium between open and closed conformations toward the latter.
In this study, we took a different approach to calculate the effects of crowding on thermodynamic properties. Instead of calculating the free-energy differences between two end states in the absence and presence of crowders, represented by the two horizontal legs in Fig. 1 A for protein folding and Fig. 1 B for protein binding, we calculated the transfer free energies of protein molecules from a dilute solution to a crowded solution, as represented by the vertical legs in Fig. 1, A and B. The change in folding free energy by crowding is (14)
| (1) |
where ΔμN and ΔμD, respectively, are the changes in chemical potential when a protein molecule in the native and denatured states are placed in a crowded solution. Similarly, the change in binding free energy of two proteins, A and B, by crowding is
| (2) |
where ΔμA, ΔμB, and ΔμC, respectively, are the changes in chemical potential when the two protein molecules and their complex are placed in a crowded solution.
Figure 1.
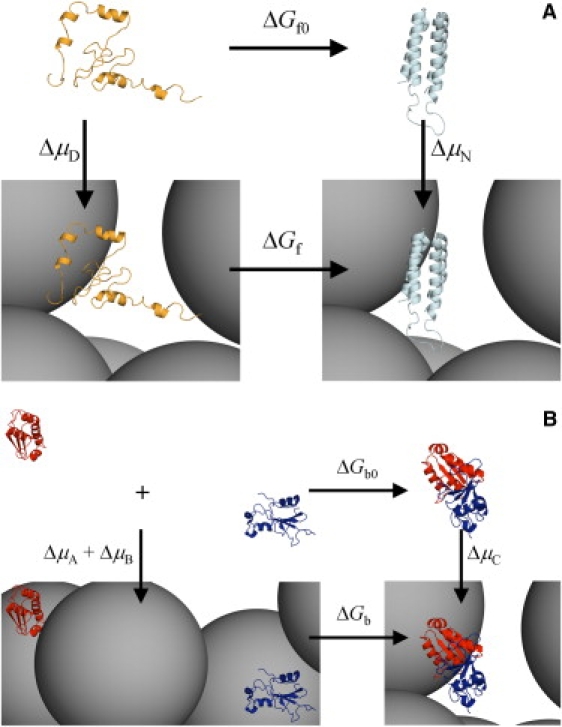
Thermodynamic cycles for calculating the effects of crowding on (A) folding free energy and (B) binding free energy. The horizontal legs are folding or binding free energies in the absence (ΔGf0 or ΔGb0) and presence (ΔGf or ΔGb) of crowders; the vertical legs are the changes in chemical potential of the denatured or unbound state (ΔμD or ΔμA + ΔμB) and native or bound state (ΔμN or ΔμC). Orange and cyan ribbons show denatured and native cytochrome b562, respectively; blue and red ribbons show barnase and barstar, respectively. Black spheres represent crowders.
The approach based on the vertical legs complements those based the horizontal legs. In particular, the crowding-induced changes in chemical potential of the end states (e.g., the native and denatured states in protein folding) yield valuable insight into the physical basis of crowding effects. In addition, the approach makes it possible to dissect the different contributing factors of crowding. In this study, we specifically focused on the volume exclusion of crowders. This volume exclusion precludes some of the placements when a protein molecule is introduced into a crowded solution, so that only a fraction, f, of all placements is allowed. The resulting change in chemical potential is given by
| (3) |
where kB is Boltzmann's constant and T is absolute temperature. Since f < 1, the change in chemical potential due to volume exclusion of crowders is always positive. Equation 3 can be seen as a specialization of Widom's insertion theorem (16) to hard particles.
Here we designed a very efficient algorithm for calculating the allowed fraction f of a test protein and applied it to study the effects of crowder volume exclusion on protein folding and binding stability. The approach has significant potential in other applications and in enabling more and more realistic modeling of cellular environments.
Methods
In the overall design of our approach, the test protein in a dilute solution and a collection of crowders were independently simulated. Representative conformations (totaling Mp) of the test protein and representative configurations (totaling Mc) of the crowders were saved. The protein in each representative conformation was then tested against each representative configuration of the crowders to calculate a value for the allowed fraction f. The Mp × Mc such values were averaged to give the final value for f.
Below we describe the procedures for the simulations of the test proteins, the crowders, and the algorithm for calculating f.
Conformational sampling for cytochrome b562 in native and denatured states
Cytochrome b562 is a four-helix protein with 106 residues (Fig. 1 A). In a separate study to elucidate its unfolding mechanism, molecular dynamics simulations of this protein were carried out at 300 K and 500 K in explicit solvent (H. Tjong and H.-X. Zhou, unpublished), following protocols published in a similar study (17). The total lengths of the two trajectories were 30 ns and 15 ns, respectively. Here we sampled 1000 conformations each from the room temperature and high temperature trajectories to represent cytochrome b562 in the native and denatured states, respectively. (Note that the focus in this study is on the folding stability. The effects of crowding on folding intermediates such as the transition state will be presented in a further study (H. Tjong and H.-X. Zhou, unpublished)). The average radii of gyration in the two sets of conformations were 15.8 and 17.9 Å, respectively. Representative conformations of native and denatured cytochrome b562 are displayed in Fig. 1 A.
Conformational sampling for barnase and barstar in bound and unbound states
To represent the bound state of barnase and barstar, 548 conformations of their complex were sampled from a 7.2-ns trajectory generated in a previous study (18). Each of these conformations was then separated to yield the conformations of unbound barnase and barstar. A representative conformation of the barnase-barstar complex is displayed in Fig. 1 B.
Generation of configurations for crowders
In this study, the crowders were represented by spheres. We have previously used such a hard-sphere system to study the effects of crowding on protein binding kinetics (19). Following that study, we used the hard-sphere simulation program in the book of Allen and Tildesley (20) to generate configurations of crowders with the same size. Briefly, hard sphere were initially present at the sites of a face-centered cubic (FCC) lattice and randomly assigned velocities according to the Maxwell distribution. They moved with uniform velocity until elastic collision with other spheres. The periodic boundary condition and minimum-image convention were applied during the simulations. Snapshots were then saved as representative configurations of the crowders.
The primary simulation box was a cube with side length of 103 Å. For a desired crowder radius Rc and a desired crowder volume fraction ϕ, the number, N, of crowders was determined from N(4π/3)Rc3 = ϕ × 109 Å3. The total number of sites of an FCC lattice is given by 4n3, where n is an integer. We chose the minimum n that satisfied 4n3 ≥ N. From the 4n3 sites, N was randomly selected as initial positions for the crowders. Four crowder radii, at 15, 20, 30, and 50 Å, and four volume fractions, at 5%, 15%, 25%, and 35%, were studied. The 16 combinations of Rc and ϕ, the number of crowders in the simulation box for each combination, and the size of the FCC lattice used for initial positions of the crowders, are listed in Table 1.
Table 1.
Crowded solutions generated by crowders with a single size
| Rc (Å) | ϕ (%) | N | 4n3 |
|---|---|---|---|
| 15 | 5 | 3537 | 4 × 103 = 4000 |
| 15 | 15 | 10,610 | 4 × 143 = 10976 |
| 15 | 25 | 17,684 | 4 × 173 = 19652 |
| 15 | 35 | 24,757 | 4 × 193 = 27436 |
| 20 | 5 | 1492 | 4 × 83 = 2048 |
| 20 | 15 | 4476 | 4 × 113 = 5324 |
| 20 | 25 | 7460 | 4 × 133 = 8788 |
| 20 | 35 | 10,445 | 4 × 143 = 10976 |
| 30 | 5 | 442 | 4 × 53 = 500 |
| 30 | 15 | 1326 | 4 × 73 = 1372 |
| 30 | 25 | 2210 | 4 × 93 = 2916 |
| 30 | 35 | 3095 | 4 × 103 = 4000 |
| 50 | 5 | 95 | 4 × 33 = 108 |
| 50 | 15 | 286 | 4 × 53 = 500 |
| 50 | 25 | 477 | 4 × 53 = 500 |
| 50 | 35 | 668 | 4 × 63 = 864 |
For each combination of Rc and ϕ except for Rc = 15 Å and ϕ = 35%, a single simulation trajectory was initiated; 10 configurations of the crowders, separated by 106 collisions, were saved as representatives. The case Rc = 15 Å and ϕ = 35% yielded the lowest value for the allowed fraction (see below) and hence had the greatest demand on calculation accuracy. In this case, we started 84 independent trajectories. After 1.5 × 107 collisions, the final configurations of the 84 trajectories were saved as representatives.
Mixtures of crowders with two different sizes, at 15 and 50 Å, were also studied. With the total volume fraction kept at 35%, the two types of crowders were mixed in 18 different proportions (see Table 2). Generating representative configurations for these mixtures required modifications of the procedure just described for crowders with the same size. In preparing the initial configuration for each mixture, two FCC lattices were used. One was for the 15 Å crowders and had 4 × 193 sites; the other was for the 50 Å crowders and had 4 × 63 sites. From the second lattice, the desired number of sites was randomly selected as initial positions of the 50 Å crowders. The first lattice was then superimposed onto the second, and all sites were eliminated where the placement of a 15 Å crowder would lead to overlap with the existing 50 Å crowders. Among the remaining sites in the first lattice, the desired number was randomly selected as initial positions of the 15 Å crowders. In the propagation of the mixed crowders, the Allen-Tildesley program was also modified to account for the different radii of the crowders. For each mixture, 10 representative configurations of the crowders, separated by 106 collisions, were saved.
Table 2.
Crowded solutions generated by mixed crowders with two sizes
| ϕ1 (%) | ϕ2 (%) | N1 | N2 |
|---|---|---|---|
| 35 | 0 | 24,757 | 0 |
| 31.5 | 3.5 | 22,281 | 66 |
| 28 | 7 | 19,805 | 133 |
| 24.5 | 10.5 | 17,330 | 200 |
| 21 | 14 | 14,854 | 267 |
| 17.5 | 17.5 | 12,378 | 334 |
| 14 | 21 | 9902 | 401 |
| 10.5 | 24.5 | 7427 | 467 |
| 7 | 28 | 4951 | 534 |
| 6.125 | 28.875 | 4332 | 551 |
| 5.25 | 29.75 | 3713 | 568 |
| 4.375 | 30.625 | 3094 | 584 |
| 3.5 | 31.5 | 2475 | 601 |
| 2.8 | 32.2 | 1980 | 614 |
| 1.75 | 33.25 | 1237 | 635 |
| 0.7 | 34.3 | 495 | 655 |
| 0.35 | 34.65 | 247 | 661 |
| 0 | 35 | 0 | 668 |
Algorithm for calculating the fraction of allowed placements
In calculating the allowed fraction f, we treated the test protein molecule in each conformation and the crowders in each configuration as hard particles. The protein molecule was modeled as a collection of van der Waals spheres with Bondi radii (21). In principle, f can be calculated by randomly placing the protein molecule, with a given conformation, inside a given configuration of the crowders (the system was kept at constant volume). If out of P random placements, Pa placements were successful in that they do not lead to overlap between the test protein and the crowders, then f = Pa/P.
We developed a far more efficient algorithm for calculating the allowed fraction when a protein, represented with atomic details, is placed into a random distribution of crowders. The basic idea of the algorithm is to first expand each crowder to the largest distances at which the test protein can still overlap with the crowder and then calculate f as the fraction of space not filled by the expanded crowders (Fig. 2). In our calculation, the center of the test protein was placed on a three-dimensional grid (and the center of each crowder is moved to the nearest grid as an approximation). To find the required expansion of a crowder, the test protein was placed at grid points around the crowder. At each grid point, overlap between the test protein and the crowder was checked by calculating the distance between each protein atom and the crowder. The expansion was comprised of all the grid points that led to overlap. This part of the calculation was done only once and then by translation mapped to all the crowders in the simulation box. After the mapping, f was obtained by counting the number of grid points not covered by any expanded crowder. The value of f was then averaged over crowder configurations and over conformations of the test protein. Finally, the change in chemical potential by crowding was calculated by Eq. 3. For protein folding, the calculations were done separately for the native protein and for the denatured protein, yielding the effect of crowding on the folding stability (Eq. 1). Similarly, for protein binding, the calculations were done separately for the complex and for the two subunits, yielding the effect of crowding on the binding stability (Eq. 2).
Figure 2.
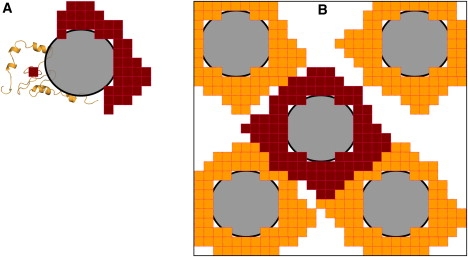
Illustration of the algorithm for calculating the allowed fraction. (A) The region around a crowder is filled by grids, each representing a location where the test protein will overlap with the crowder. (B) The filled region is mapped to each crowder in the simulation box. The total number of unfilled grids divided by the total number of grids in the box gives the allowed fraction.
Test of the algorithm
This algorithm was exhaustively tested. Because the test protein was placed on a grid, a sufficiently fine grid spacing must be used. To illustrate the effect of the grid spacing, in Fig. 3 we display the volume of the expanded crowder calculated at grid spacings from 0.14 to 3.0 Å. The relative error decreased to <0.1% as the grid spacing was reduced to 1.5 Å and further decreased to 0.01% at a grid spacing of 1 Å. We used a 1 Å grid spacing in all subsequently calculations.
Figure 3.
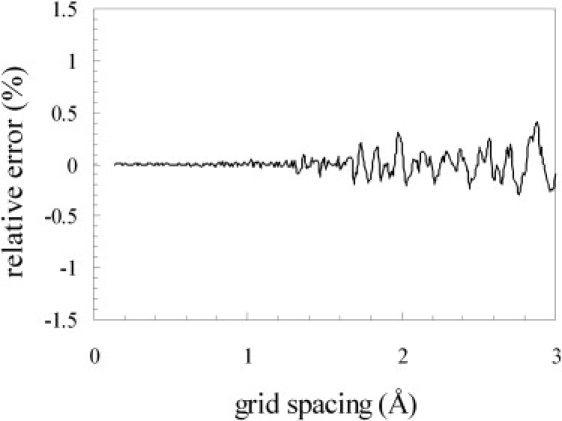
Effect of grid spacing on the calculated volume of an expanded crowder. The radius of the bare crowder was 15 Å. At a given grid spacing, the relative error was calculated by comparing the calculated volume against the exact result; the latter was taken as the average of the values calculated at six fine grid spacings: 0.14–0.19 Å with an increment of 0.01 Å.
By comparing the results for ΔΔGf calculated on different configurations of the crowders, the calculation errors were found to be under 0.05 kBT except for the case Rc = 15 Å and ϕ = 35%; in that case, the error was 0.2 kBT.
The problem of placing a spherical test protein into a box of spherical crowders has an approximate analytical solution, provided by the scaled particle theory (SPT) (22). The theory, which has been used to model macromolecular crowding (2,3,5–7), predicts the allowed fraction according to
| (4) |
where a, S, and V are the radius, surface area, and volume, respectively, of the test protein, Sc = 4πRc2 is the surface area of the crowders, and C is their number density. For a = 20 Å and Rc = 20 Å, the f values obtained from our calculations were 0.644 ± 0.001, 0.194 ± 0.001, 0.0296 ± 0.0005, and 0.00130 ± 0.00009, respectively, at ϕ = 5%, 15%, 25%, and 35%. The values predicted by the SPT are 0.643, 0.192, 0.0283, and 0.00107, respectively. Overall, there is good agreement between calculation and theory, but there are systematic discrepancies at high ϕ-values. The discrepancies increase with decreasing Rc. We attributed the discrepancies to underestimation by the SPT. The severity of the approximation made in the SPT is known to increase both with increasing ϕ and with decreasing Rc.
Results
Folding stability in the presence of a single species of crowders
The change in folding stability of cytochrome b562 by the addition of 85 g/L of PEG 20 K has been measured by Ai et al. (12). We calculated ΔμN and ΔμD, the crowding-induced increases in chemical potential of this protein in the native and denatured states. The resulting change in the folding free energy is displayed in Fig. 4 for four crowder sizes (Rc from 15 to 50 Å) and four crowder volume fractions (ϕ from 5% to 35%). At a given crowder size, the level of stabilization increases with increasing volume fraction occupied by the crowders, consistent with expectation.
Figure 4.
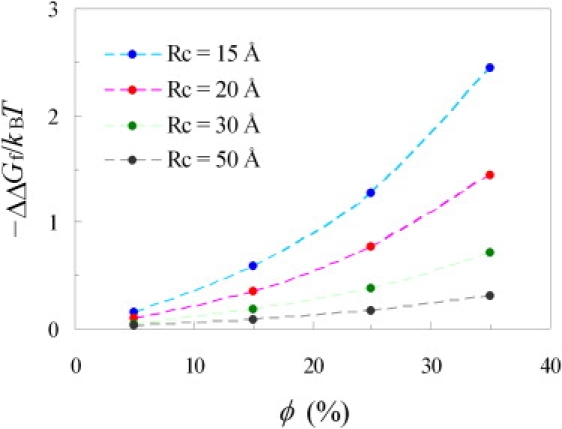
Dependence of ΔΔGf on crowder radius Rc and volume fraction ϕ. Circles show calculated results; curves show results from fitting to Eq. 4.
At a given crowder volume fraction, the level of stabilization decreases as the crowder size increases from 15 to 50 Å. This result can be understood as follows. Only a small number of large crowders is needed to produce the same volume fraction as a large number of small crowders. The small number of large crowders leaves large voids in which the more open denatured state can be accommodated nearly as well as the compact native state, leading to a diminishing ΔΔGf. In contrast, a large number of small crowders would lead to small voids, which become much more discriminating against the more open denatured state, leading to a relatively large magnitude in ΔΔGf.
The dependences of ΔμN and ΔμD on Rc and ϕ can be fitted very well to Eq. 4 when a, S, and V were treated as adjustable parameters. The fitted values of these parameters were 20.6 Å, 5165 Å2, and 16,569 Å3, respectively, for native cytochrome b562 and 24.9 Å, 6873 Å2, and 18,154 Å3, respectively, for the denatured protein. Fig. 3 shows that the results for ΔΔGf calculated from the fitting of ΔμN and ΔμD and from the actual values of ΔμN and ΔμD are indistinguishable.
Experimentally, Ai et al. (12) observed a level of stabilization at 0.4 kBT by the addition of 85 g/L of PEG 20 K. In our atomistic modeling, this level of stabilization could be produced by crowders with a radius of 20 Å and occupying 15% of volume. The highest level obtained in our study, produced by crowders with a radius of 15 Å and occupying 35% of volume, was 2.5 kBT, or 1.5 kcal/mol at room temperature.
Folding stability in the presence of mixed crowders
The cytosol is crowded by many species of macromolecules. To make progress toward mimicking cellular environments, we studied the effects of mixtures of crowders with two different sizes on folding stability. Crowders with radii of 15 and 50 Å were mixed to yield a fixed total volume fraction of 35%. Fig. 5 displays the results for ΔΔGf as a function of ϕ2/(ϕ1 + ϕ2), which represents the percentage of the total volume fraction occupied by the larger crowders. The level of stabilization decreases as the content of the larger crowders increases, echoing the finding in Fig. 4 that the larger crowders are less effective in stabilization than the smaller crowders.
Figure 5.
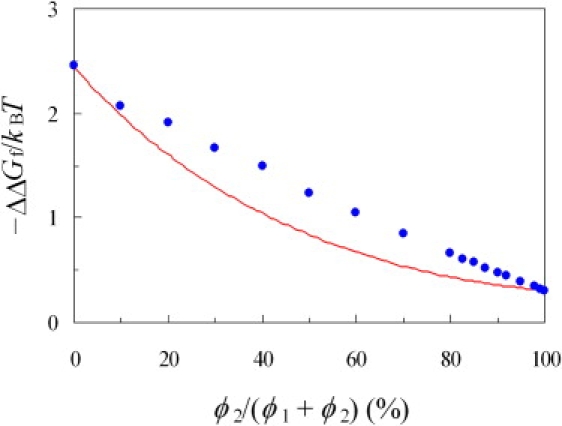
Increase in folding stability of cytochrome b562 by mixtures of crowders with 15 and 50 Å radii. Circles show the actual results for ΔΔGf obtained from atomistic modeling; the curve shows additive prediction.
An important question is whether the stabilization effect of the mixture is additive of the separate effects of the constituents. As an example, let ΔΔGm be the change in folding stability by a mixture of the smaller and larger crowders, respectively occupying 15% and 20% of the total volume. Now let ΔΔG1 be the change in folding stability by the smaller crowders alone at a volume fraction of 15%, and ΔΔG2 be the counterpart for the larger crowders alone at a volume fraction of 20%. Is ΔΔGm predicted well by adding ΔΔG1 and ΔΔG2? Because the fit to Eq. 4 worked so well for crowders with a single size, we simply used the fit to generate ΔΔGf for the smaller or the larger crowders alone at desired volume fractions. Upon pairing the respective volume fractions to yield a total of 35%, the levels of stabilization due to the two types of crowders alone were added and the results are displayed in Fig. 5. It can be seen that the levels of stabilization produced by mixtures of the two types of crowders are greater than predicted by additivity.
Binding stability under crowding
Because two protein molecules become more compact when they form a complex, crowding is expected to stabilize the bound state (14). In this study, we used barnase and barstar as a test system. Fig. 6 displays the changes in binding free energy by crowders at four sizes (Rc from 15 to 50 Å) and at four volume fractions (ϕ from 5% to 35%). The trends observed here are very similar to those displayed in Fig. 3 for protein folding stability. The maximal increase in binding free energy, produced by crowders with a radius of 15 Å and occupying 35% of volume, was 3.2 kBT, or 1.9 kcal/mol at room temperature.
Figure 6.
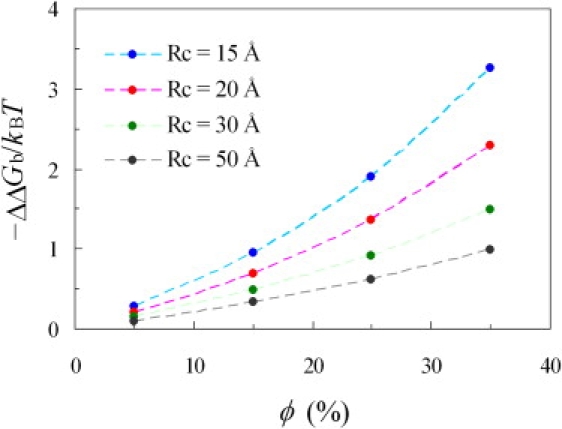
Dependence of ΔΔGb on crowder radius Rc and volume fraction ϕ.
Discussion
The magnitude of crowding-induced stabilization
The modest stabilization obtained in this study, based on atomistic modeling of macromolecular crowding, is in line with experimental measurements of crowding effects on protein folding stability (10–13). The maximal levels of stabilization observed in these experimental studies, using dextran, Ficoll, or polyethylene glycol as a crowding agent, were ∼1 kcal/mol. Interestingly, simulations based on a coarse-grained model also yielded similarly modest stabilization by crowding (8,9). On the other hand, simple theoretical models, depending on how the denatured state is represented, have either predicted modest (5) or substantial (6) stabilization, indicating that these models are of limited value at a quantitative level.
The modest level of stabilization, of ∼1 kcal/mol, can nevertheless be of physiological importance. In particular, in protein aggregation the modest stabilization of individual folding and binding equilibria can accumulate into significant stabilization of the nucleus for aggregation, resulting in considerable reduction in the lag time of aggregation. As illustrated in Fig. 7, a nucleus consisting of four monomers involve four folding events and three bimolecular binding events. If crowding favors each equilibrium in the forward direction by 1 kcal/mol, cumulatively, the stability of the nucleus (relative to the unfolded and unbound monomers) will be enhanced by 7 kcal/mol. This stabilization may explain the shortening of aggregation lag time from months to days observed for α-synuclein by the addition of crowding agents (23,24). Aggregation of this natively unfolded protein is implicated in Parkinson's disease.
Figure 7.

Illustration of a protein aggregation process. (Top row) Four unfolded monomers slowly form a nucleus; thereafter aggregation proceeds quickly. (Bottom row) The free-energy difference between the nucleus and the four unfolded monomers can be reconstructed sequentially, first forming four folded monomers and then adding monomers one at a time.
The influence of crowder size on the level of stabilization
Our calculation results show that, when present at the same volume fraction, crowders with different sizes can lead to different levels of increase in protein folding and binding stability. This finding suggests that different crowding agents, when present at the same concentration, will produce different levels of stabilization.
This study only explored the variation in crowder size. Another important variable is crowder shape. When crowders differing in both shape and size are compared, one can only expect greater variation in the level of stabilization. Such variations in level of stabilization have been observed in an experimental study in which the effects of dextran species with different sizes and Ficoll were compared (25).
Nonadditive effects of mixed crowding
We found that the effect of a mixture of two types of crowders on protein folding stability is greater than the sum of the effects of the constituents. This nonadditive behavior has also been observed in an experimental study using dextran and Ficoll as mixed crowding agents (25). The nonadditivity can be viewed as a manifestation of the nonlinear dependence of the crowding-induced change in chemical potential on crowder concentrations (see Eq. 4 and Fig. 4). In particular, Fig. 4 shows that the –ΔΔGf function curves upward as ϕ increases, The upward curving explains why the effect of mixed crowding, insofar as volume exclusion is concerned, is greater than predicted by additivity, The fact that this nonadditive behavior is precisely what is observed on mixtures of dextran and Ficoll (25) is strong evidence that volume exclusion plays an important role in the effects of these crowding agents on folding stability.
Since the cytosol is crowded by many species of macromolecules, the nonadditive effects of mixed crowding have important implications. First, they suggest that future studies should be directed at more-complex mixtures of crowding agents to realistically mimic cellular environments. Second, they identify composition as another important variable in controlling the effects of crowding. Together, crowder shape, size, composition, and concentration may inject significant variability in the effects of crowding on biophysical properties of proteins. It is known that abnormal (e.g., misfolded) proteins accumulate with age inside cells because of impairment of the cellular degradation machinery. The accumulation may lead to enhanced crowding effects, which in turn may explain why susceptibility to aggregation-related diseases such as Parkinson's disease increases with age.
Extensions and other applications
The subtle dependences on crowder species, concentration, and concentration make quantitative prediction of crowding effects a formidable challenge. The approach introduced in this study is well suited to meet this challenge. In our approach, simulations of test proteins and crowders are separated. The tasks for test proteins and for crowders are very different. For test proteins, the main interest is in sampling internal conformations; for crowders, the main interest is in generating an equilibrium distribution of crowder positions and orientations. The protein simulations require short time steps; without the encumbrance of crowders, relatively long simulations can be afforded. The crowder simulations can be accomplished by running Brownian dynamics simulations with relatively large time steps (26).
We have developed an efficient algorithm for calculating the increase in chemical potential of a test protein due to volume exclusion by crowders. Like other simulation studies of crowding and confinement (8,9,15,27,28), here we only considered a simple, spherical representation of crowders. In future work we will extend the approach to model crowders with atomic details and to account for other types of interactions between a test protein and crowders. These extensions will allow us to directly compare calculation results with experimental data.
Here our approach to the modeling of macromolecular crowding was applied to protein folding and binding equilibria. There are many other potential applications. For example, the biological functions of many proteins (or protein complexes) involve different conformational states; crowding may affect their equilibria. Minh et al. (15) have simulated the opening and closing of the flaps of an HIV-1 protease dimer in the presence of crowders. Our approach can be applied to reanalyze the many published simulations of conformational transitions carried out in the absence of crowding to find how crowding affects the results.
The approach works not only for equilibrium but also for kinetic properties. For example, when applied to the reactant and transition states, the effect of crowding on the activation barrier can be obtained. For the binding of proteins, we have developed a transient-complex theory, which predicts the rate constant as (29,30)
| (5) |
where Ul-r∗ is the interaction energy, due to long-range forces, in the transient complex for protein binding, and ka0 is the binding rate constant in the absence of any force. In the absence of crowding, Ul-r∗ comes from electrostatic interactions between the binding molecules. Crowding induces an effective interaction potential; its effect on ka depends on whether it is long-ranged, i.e., with magnitude decreasing slowly as the binding molecules are moved apart. If the effective potential is short-ranged, then its effect on ka is minimal (31). On the other hand, if the effective potential turns out to be long-ranged, its effect on the binding rate can be accounted for by including it as an additive contribution in calculating Ul-r∗ (5).
Acknowledgments
This work was supported in part by National Institutes of Health grant No. GM058187.
References
- 1.Zimmerman S.B., Trach S.O. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J. Mol. Biol. 1991;222:599–620. doi: 10.1016/0022-2836(91)90499-v. [DOI] [PubMed] [Google Scholar]
- 2.Minton A.P. The effect of volume occupancy upon the thermodynamic activity of proteins: some biochemical consequences. Mol. Cell. Biochem. 1983;55:119–140. doi: 10.1007/BF00673707. [DOI] [PubMed] [Google Scholar]
- 3.Minton A.P. Effect of a concentrated “inert” macromolecular cosolute on the stability of a globular protein with respect to denaturation by heat and by chaotropes: a statistical-thermodynamic model. Biophys. J. 2000;78:101–109. doi: 10.1016/S0006-3495(00)76576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou H.-X., Dill K.A. Stabilization of proteins in confined spaces. Biochemistry. 2001;40:11289–11293. doi: 10.1021/bi0155504. [DOI] [PubMed] [Google Scholar]
- 5.Zhou H.-X. Protein folding and binding in confined spaces and in crowded solutions. J. Mol. Recognit. 2004;17:368–375. doi: 10.1002/jmr.711. [DOI] [PubMed] [Google Scholar]
- 6.Minton A.P. Models for excluded volume interaction between an unfolded protein and rigid macromolecular cosolutes: macromolecular crowding and protein stability revisited. Biophys. J. 2005;88:971–985. doi: 10.1529/biophysj.104.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou H.-X. Effect of mixed macromolecular crowding agents on protein folding. Proteins. 2008;72:1109–1113. doi: 10.1002/prot.22111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheung M.S., Klimov D., Thirumalai D. Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc. Natl. Acad. Sci. USA. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stagg L., Zhang S.Q., Cheung M.S., Wittung-Stafshede P. Molecular crowding enhances native structure and stability of α/β protein flavodoxin. Proc. Natl. Acad. Sci. USA. 2007;104:18976–18981. doi: 10.1073/pnas.0705127104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qu Y., Bolen D.W. Efficacy of macromolecular crowding in forcing proteins to fold. Biophys. Chem. 2002;101–102:155–165. doi: 10.1016/s0301-4622(02)00148-5. [DOI] [PubMed] [Google Scholar]
- 11.Spencer D.S., Xu K., Logan T.M., Zhou H.-X. Effects of pH, salt, and macromolecular crowding on the stability of FK506-binding protein: an integrated experimental and theoretical study. J. Mol. Biol. 2005;351:219–232. doi: 10.1016/j.jmb.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 12.Ai X., Zhou Z., Bai Y., Choy W.Y. 15N NMR spin relaxation dispersion study of the molecular crowding effects on protein folding under native conditions. J. Am. Chem. Soc. 2006;128:3916–3917. doi: 10.1021/ja057832n. [DOI] [PubMed] [Google Scholar]
- 13.Roberts A., Jackson S.E. Destabilized mutants of ubiquitin gain equal stability in crowded solutions. Biophys. Chem. 2007;128:140–149. doi: 10.1016/j.bpc.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 14.Zhou H.-X., Rivas G., Minton A.P. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Minh D.D., Chang C.E., Trylska J., Tozzini V., McCammon J.A. The influence of macromolecular crowding on HIV-1 protease internal dynamics. J. Am. Chem. Soc. 2006;128:6006–6007. doi: 10.1021/ja060483s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Widom B. Some topics in the theory of fluids. J. Chem. Phys. 1963;39:2802–2812. [Google Scholar]
- 17.Huang X., Zhou H.-X. Similarity and difference in the unfolding of thermophilic and mesophilic cold shock proteins studied by molecular dynamics simulations. Biophys. J. 2006;91:2451–2463. doi: 10.1529/biophysj.106.082891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tjong H., Zhou H.-X. Accurate calculations of binding, folding, and transfer free energies by a scaled generalized Born method. J. Chem. Theory Comput. 2008;4:1733–1744. doi: 10.1021/ct8001656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou H.-X., Szabo A. Comparison between molecular dynamics simulations and the Smoluchowski theory of reactions in a hard sphere liquid. J. Chem. Phys. 1991;95:5948–5952. [Google Scholar]
- 20.Allen M.P., Tildesley D.J. Clarendon Press; Oxford, UK: 1987. Computer Simulation of Liquids. [Google Scholar]
- 21.Bondi A. van der Waals volumes and radii. J. Phys. Chem. B. 1964;68:441–451. [Google Scholar]
- 22.Lebowitz J.L., Rowlinson J.S. Thermodynamic properties of mixtures of hard spheres. J. Chem. Phys. 1964;41:133–138. [Google Scholar]
- 23.Shtilerman M.D., Ding T.T., Lansbury P.T. Molecular crowding accelerates fibrilization of α-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson's disease? Biochemistry. 2002;41:3855–3860. doi: 10.1021/bi0120906. [DOI] [PubMed] [Google Scholar]
- 24.Uversky V.N., Cooper M., Bower K.S., Li J., Fink A.L. Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett. 2002;515:99–103. doi: 10.1016/s0014-5793(02)02446-8. [DOI] [PubMed] [Google Scholar]
- 25.Batra J., Xu K., Zhou H.-X. Nonaddtive effects of mixed crowding on protein stability. Proteins. 2009 doi: 10.1002/prot.22425. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGuffee S.R., Elcock A.H. Atomically detailed simulations of concentrated protein solutions: the effects of salt, pH, point mutations, and protein concentration in simulations of 1000-molecule systems. J. Am. Chem. Soc. 2006;128:12098–12110. doi: 10.1021/ja0614058. [DOI] [PubMed] [Google Scholar]
- 27.Takagi F., Koga N., Takada S. How protein thermodynamics and folding mechanisms are altered by the chaperonin cage: molecular simulations. Proc. Natl. Acad. Sci. USA. 2003;100:11367–11372. doi: 10.1073/pnas.1831920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mittal J., Best R.B. Thermodynamics and kinetics of protein folding under confinement. Proc. Natl. Acad. Sci. USA. 2008;105:20233–20238. doi: 10.1073/pnas.0807742105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alsallaq R., Zhou H.-X. Energy landscape and transition state of protein-protein association. Biophys. J. 2007;92:1486–1502. doi: 10.1529/biophysj.106.096024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alsallaq R., Zhou H.-X. Electrostatic rate enhancement and transient complex of protein-protein association. Proteins. 2008;71:320–335. doi: 10.1002/prot.21679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schreiber G., Haran G., Zhou H.-X. Fundamental aspects of protein-protein association kinetics. Chem. Rev. 2009;109:839–860. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]