

Abstract
Turnover of mitochondria by autophagy constitutes an essential quality maintenance mechanism. Recent studies have demonstrated that efficient clearance of damaged mitochondrial components depends on mitochondrial dynamics, a process characterized by frequent fusion and fission events that enable the redistribution of mitochondrial components across a population of hundreds of individual mitochondria. The presented simulation identifies kinetic parameters of fusion and fission that may influence the maintenance of mitochondrial function. The program simulated repetitive cycles of fusion and fission events in which intact and damaged mitochondrial contents were redistributed between fusion mates. Redistribution impacted mitochondrial function, thereby influencing the fate of each mitochondrion, to be either destined for a subsequent fusion or eliminated by autophagy. Our findings indicate that, when paired with fission, fusion events may serve to accelerate the removal of damaged mitochondrial components by autophagy. The model predicts the existence of an optimal frequency of fusion and fission events that can maintain respiratory function at steady-state levels amid the existence of a continuous damaging process that inactivates mitochondrial components. A further elevation of the fusion frequency can increase the clearance efficiency of damaged content. However, this requires fusion to be a selective process in which depolarized mitochondria are excluded from the fusing population. The selectivity of fusion was found to be particularly beneficial in conditions of elevated rate of damage, because it permits the increase of fusion frequency without compromising the removal of damaged content by autophagy.
Introduction
Mitochondria in living cells exist as a dynamic network of individual organelles that continuously cycle through fusion-fission events. Ample clinical and animal model data have shown that compromising either fusion or fission leads to dysfunction of neural processes and embryonic development (1–4).
Mitochondrial fusion allows for the transfer of soluble and membranous components between mitochondria (5–7), and fusion was therefore recommended as a complementation mechanism by which damaged or poorly performing mitochondria can maintain their function. A solid body of in vitro (8,9) and in vivo (10) data support this view. However, recent data have suggested that some criteria define the fusing pool of mitochondria, leaving more depolarized ones segregated. These data demonstrated that mitochondrial fusion depends on mitochondrial membrane potential (11–14) and cannot occur in depolarized mitochondria, despite the proximity of numerous polarized mitochondria to each other (15). Although depolarized mitochondria cannot fuse with other mitochondria, they are preferentially targeted by the degrading organelle, the autophagosome (16,17). The underlying mechanism for the membrane potential dependency of fusion is thought to involve proteolytic cleavage of the fusion protein, OPA1. Cleavage of OPA1 was shown to be triggered by reduction in mitochondrial ATP and by mitochondrial depolarization (18–21).
The life cycle of mitochondria involves repetitive clusters of fusion periods, each followed by fission (Fig. 1). After a fission event, mitochondria enter a solitary state that is ∼20-fold longer than the fused period and are available for subsequent fusion events (15). If a mitochondrion maintains a polarized membrane potential, it can enter a subsequent fusion event with another mitochondrion. However, if it depolarizes, it will remain solitary and eventually be targeted for autophagy, unless it repolarizes and is then recovered.
Figure 1.
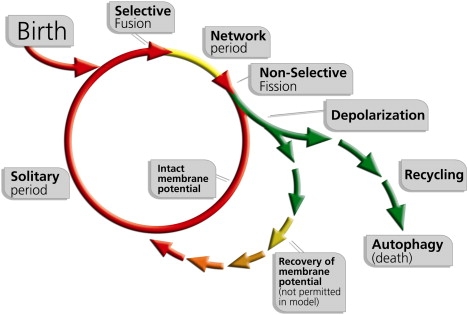
Schematic model of the mitochondrion's life cycle and the roles of mitochondrial dynamics and autophagy in the segregation of dysfunctional mitochondria. The mitochondrion shifts repeatedly between a postfusion state (Network) and a postfission state (Solitary). Fusion is brief and triggers fission. After a fission event, the daughter mitochondrion may either maintain intact membrane potential (red line), or depolarize (green line). If it depolarizes, it will unlikely reengage in further fusion events for the entire depolarization interval. In the case that mitochondrial depolarization is transient and Δψm recovers, fusion capacity is restored (green to red short arrows). Note that, whereas the scheme includes the possibility of autonomous recovery of the depolarized mitochondrion, in the current simulation mitochondria cannot be self-repaired and therefore cannot recover in a mitochondrion-autonomous fashion.
The link between mitochondrial fission and autophagy may simply be attributed to the obvious reduction in mitochondrial size resulting from fission, which makes it physically digestible for the autophagosome. Remarkably, however, a key observation has recently shown that fission generates functionally dissimilar daughter units (15,22). This observation suggests that during fusion events some functional components may be redistributing unevenly between the two fusion mates, resulting in two dissimilar daughter mitochondria generated by the subsequent fission event. The mechanism underlying this metabolic asymmetry is not clear, but it might facilitate the ability of the cell to segregate and remove damaged mitochondrial material at a much faster rate than that achieved without such reorganization.
In our investigation we presented a simulation that integrates the effect of fusion, fission, and autophagy in a large population of mitochondria over extended periods of time. The model was based on the hypothesis that, when integrated, fusion, fission, and autophagy form a quality-control mechanism (23). The goal of our simulation was to understand how factors such as the frequency, selectivity and duration of fusion events alter the efficiency of the quality-control process and, thereby, mitochondrial function. The model allowed for the extrapolation of the potential contribution of the integrated function of fusion, fission, and autophagy into quality maintenance of mitochondria over periods of time beyond those that can be tested using RNAi or knockout techniques for a variety of reasons. First, the isolated contribution of mitochondrial dynamics and autophagy cannot be tested as DNA, and protein repair cannot be silenced in living cells over days. Second, moderate changes in the rates of fusion or fission cannot be accomplished in vivo because of compensatory mechanisms.
The model simulated repetitive cycles of fusion and fission events in which mitochondrial contents were exchanged, and it monitored mitochondrial bioenergetic function over time. The model tested a spectrum of fusion frequencies, fusion-fission kinetics, and the importance of having fusion as a selective process in conditions of increased rate of damage.
Our findings indicated that the combination of fusion and fission contributes to steady-state mitochondrial activity of the cell in a frequency-dependent manner, displaying a direct relationship between mitochondrial activity and dynamics rate. Moreover, the combination of fusion and fission with the degrading function of autophagy exceeds the contribution of autophagy alone to quality maintenance. The selective character of the fusion process prevents a decline in the steady-state mitochondrial activity under increased fusion frequencies, during which, under similar conditions, nonselective fusion leads to catastrophic failure. This advantage of selective fusion is suggested to be essential for survival under an increased damage rate.
Materials and Methods
The model simulated mitochondrial fusion, fission, and autophagy in 100 cells, each containing 300 mitochondria. Mitochondrial activity per cell was determined in discrete two-minute increments (Fig. 2 a). These values were based on data obtained from INS1 and COS7 cells. These cell lines contain several hundreds of discrete mitochondrial structures per cell (24), which, on average, undergo a cycle of fusion-fission every ∼20 min per mitochondrion (15). The following describes the heuristics of the model for the three major groups of its parameters.
Figure 2.
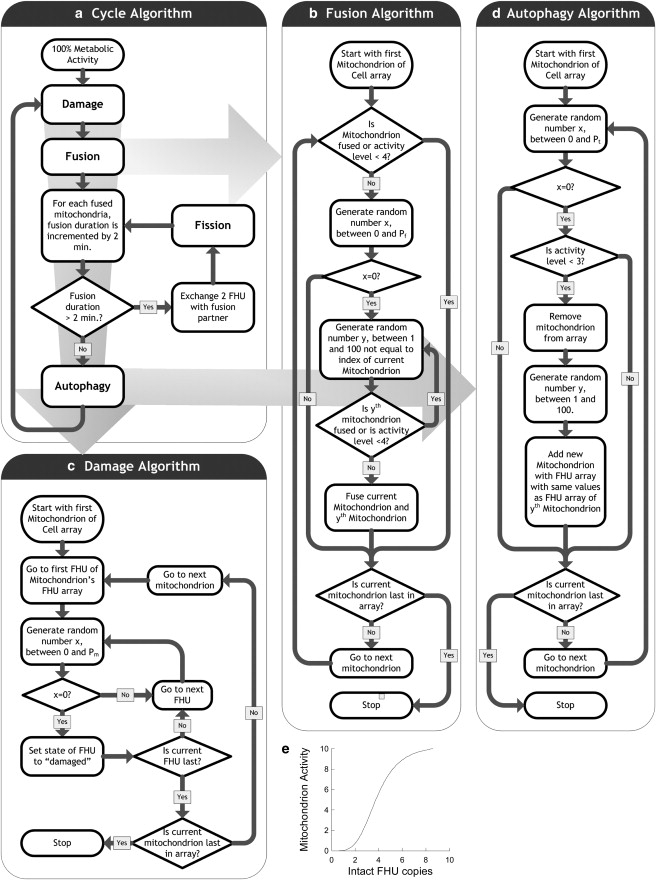
Construction of the simulation. (a) Cycle algorithm. The cycle algorithm controls the fusion, damage, and autophagy algorithms. Pf, frequency of fusion for single mitochondria; Pm, frequency of damage to FHUs of single mitochondria; Pt, frequency of autophagy for single mitochondria. (b) Fusion algorithm. Each mitochondrion is selected as a fusion candidate with a likelihood of Pf. If the candidate mitochondrion is not already fused, it is paired to another unfused mitochondrion that is randomly selected, and both are then labeled as “fused”. Two FHUs in each of the fused mitochondria are randomly selected and exchanged. A background procedure verifies that each fused mitochondrion is set to be unfused after 4 min to allow fission occur. In the case of selective fusion an additive condition is an activity level ≥ 4. (c) Damage algorithm. Each FHU is classified in a binary manner as either “intact” or “damaged”. The FHUs are subject to a fixed rate of random damage (Pm) that irreversibly flags the FHUs as damaged (nonfunctional). This value was kept constant at 1 per 100 min. (d) Autophagy algorithm. Removal of damaged mitochondria (mitochondrial activity < 3) is a 2-min event that is immediately followed by the duplication of a mitochondrion chosen randomly from within the cell's mitochondrial population. Therefore, the total number of mitochondria in a cell remains constant. For any particular mitochondrion, likelihood of autophagy is fixed at 1 per 50 min (Pt). (e) Relationship between mitochondrial activity and numbers of FHUs. The ratio between the two follows a sigmoidal relationship, with a mitochondrion activity of 50% when 5.0 intact FHUs are present.
Mitochondrial DNA/protein and activity
Each mitochondrion was set to include 10 copies of mitochondrial material representing both the mitochondrial DNA (mtDNA), protein, and lipids required to maintain mitochondrial respiratory function. The reasons for having multiple copies per mitochondrion were: i), to allow partial exchange of components as observed in experiments tracking membranous protein exchange during a fusion event; and ii), to allow the varied parameters of fusion and fission to generate a wide range of mitochondrial respiratory functionality. In addition, previous observations in cultured human cells have shown that a mitochondrion includes in average 2–3 nucleoids, each containing 115 copies of mtDNA (25–27). We named these copies “functional hereditary units” or FHUs. Each FHU was classified as either “intact” or “damaged” (Fig. 2, a and c). The FHUs are subject to a steady rate of random damage (Pm) that irreversibly flags them as damaged (nonfunctional). Numerous studies have shown that, under unstressed conditions, mitochondrial oxidative phosphorylation (indicated by oxygen consumption) functions at ∼30% of its maximum, a capacity that maintains both mitochondrial membrane potential and cellular functions (28). When respiration is blocked, mitochondria have the ability to maintain their membrane potential through a reversed action of ATP synthase. However, at least in the beta cell under physiological conditions, this accounts for only a negligible fraction of mitochondria (28,29). Accordingly, the number of nondamaged FHUs was used to deduce the level of mitochondrial activity (Δψm equivalent) on a 0–10 scale. The relationship between the number of undamaged FHUs and activity is sigmoidal (Fig. 2 e), with an inflection point between 20–30%. At 60%, intact FHU mitochondrial activity is near maximal (90% of the normal Δψm), and any value below 30% results in a depolarized Δψm (28). This rule follows results from previous studies showing that, under normal culture conditions, most cells utilize only ∼40% of their respiratory chain capacity and that reduction of mitochondrial cytochrome oxidase of up to ∼60–70% does not result in mitochondrial depolarization (30). Similarly, mtDNA mutations that may disable respiratory functions do not impact mitochondrial function unless they appear in 70% or more of the mtDNA copies. The plotted cellular mitochondrial activity is generated by summing the activity of 300 mitochondria in each individual cell and then averaging the 100 different cells that are simulated in parallel.
Mitochondria with activity level at or below 3 were removed by autophagy (see below), as seen in the experimental data. In some of the tests run by the model we applied “selective fusion”, in which fusion events could only occur between two mitochondria with activity level above at or above 4. The dependence of fusion on mitochondrial activity is in accordance with numerous studies showing that collapse of mitochondrial membrane potential prevents mitochondrial fusion and that partial dissipation of the membrane potential is associated with reduced probability of becoming involved in a fusion event (15).
Fusion and fission (Fig. 2b)
Fusion and fission events were reported to be paired (5,15). Therefore, in the simulation, each mitochondrion was set to be in one of two states: a solitary state (where it is available for fusion) or a fused state (which can be terminated by fission). Fusion occurs randomly, with a variable probability (Pf). The efficiency of intermitochondrial exchange of contents seems to vary between membranous proteins, matrix proteins, and intermembrane proteins, and is not uniform across cell types (8,31,32). However, for the purpose of testing the influence of fusion rate and selectivity on mitochondrial function, we chose to use a single exchange rate that represents a component that is not fully equilibrated. This is based on the observation that fusion events are followed by an asymmetric fission and the generation of functionally disparate daughter units, indicating that the exchange of functional components is not complete. In the current simulation, a fusion event was set to allow each mitochondrion to randomly exchange 2 FHUs (total of 4 out of 20). Each fusion event lasted 4 min and was followed by a fission event. The later value was set based on the observation that fusion duration varied between tens of seconds to several minutes (15).
Mitochondrial autophagy (Fig. 2d)
Removal of dysfunctional mitochondria (mitochondrial activity < 3) was a 2-min event. This was immediately followed by the duplication of a mitochondrion chosen randomly from within the cell's remaining population of “healthy” mitochondria (activity > 3), resulting in a constant number of mitochondria in a cell. We have previously shown that mitochondrial fission is a prerequisite for mitochondrial autophagy (mitophagy) (15), consistent with data showing that giant (mega) mitochondria do not undergo autophagy (33–36). In our simulation the elimination of a mitochondrion by autophagy was assumed to occur only when it was found in the solitary state.
Construction of the model (Fig. 2, a–e)
The code of the simulation (a simple Monte Carlo model) was divided into three sections, a main class that manages the whole simulation, a mitochondria class, and a cell class. The mitochondria class contained an array of 10 Boolean values representing FHU copies, each set to “damaged” or “intact”, an activity value representing its metabolic level (no activity = 0; maximal activity = 10); a Boolean fusion-state value (solitary or fused); a fusion duration value; and a fusion partner value, which is the index of the mitochondrion to which it is fused.
The cell class consisted of an array of 300 mitochondria, a fusion maintenance function, a collection of three event functions (fusion, autophagy, and damage), and a cycle function that, when run, represented 2 min in the life of the entire cell. When the simulation was begun, an array of 100 cells was generated in the main class and, for each member of the cell array, its respective cycle function was called in an iterative loop, until the final member of the array was reached. This loop itself was run 100,000 times, to represent 200,000 min of activity. The cycle function ran the maintenance function, the damage function, the fusion function, and the autophagy function, in that order.
In the fusion-maintenance function each mitochondrion was checked for its fusion state. If the fusion state was “fused”, the fusion duration value was incremented by 2. If the fusion-duration value was then 4 (i.e., 4 min in the fused state have passed), the fusion duration value was reset to 0 and the fusion state was set to “solitary”.
The damage function executed an iterative loop that proceeded from the first member of the mitochondrion array to the last. For each iteration of this loop (i.e., for each mitochondrion), another loop was executed. During each iteration of this loop, a pseudorandom number was generated between 0 and 1/Pm − 1, and, if the output was not 0, the iteration terminated and the loop proceeded to the next FHU. Otherwise, “damage” occurs to the FHU; if its state was “intact”, it was set to “damaged”, and, if the state was “damaged”, it remained in the same state.
For each of the fusion and autophagy functions, an iterative loop proceeded from each mitochondrion to the next and a pseudorandom integer between 0 and 1/Pf − 1 (for fusion) and between 0 and 1/Pt − 1 (for autophagy) were generated. If the output was not 0, the current iteration of the loop terminated and the function proceeded to the next member of the array. Otherwise, the fusion, or autophagy, process occurred before proceeding to the next array member.
For the fusion process, the iteration terminated if the mitochondrion was already in the fused state. Additionally, when the model was set to simulate selective fusions, the iteration also terminated if the activity level of the mitochondrion was <4. Otherwise (i.e., if the mitochondrion was at the “solitary” state), pseudorandom integers between 0 and 300 were iteratively generated until the output number equaled the index of a member of the mitochondrion array whose state was also “solitary”, so that an “available” fusion partner was obtained. (Note that solitary mitochondria were the only ones available for fusion, because of the provision that mitochondria may only fuse in pairs.)
Next, the fusion-state property for both mitochondria was set to “fused”, and the fusion partner value of each mitochondrion was set to the index of the other mitochondrion. At this point, two pairs of distinct integers between 0 and 9 were psuedorandomly generated (representing the indices of two pairs of FHUs from the FHU array of the original mitochondrion and its fusion partner, respectively).
The damage state values of the FHU (in the original mitochondrion) at an index of the first integer in the first integer pair and FHU (in the fusion partner) with an index of the first integer in the second pair were then swapped. This was repeated for the two FHUs, with indices of the second integer of the first pair and the second integer of the second pair. In this way, two distinct FHUs in the first mitochondrion were “swapped” with two distinct FHUs from its fusion partner. If, for example, the first pair of FHU (A, an FHU in the first mitochondrion, and B, one in its partner), were “damaged” and “intact”, respectively, A was set to “intact” and B was set to “damaged”.
For the autophagy process, if the activity level of a mitochondrion was ≥3, the iteration terminated. If the activity was < 3, the mitochondrion was eliminated and replaced by a duplicate of another mitochondrion in the cell. To select the parent (template) of the duplicate, pseudorandom integers between 0 and 100 were generated until one was output not equal to the index of the current mitochondrion (i.e., the index of a distinct mitochondrion). Each value in the FHU array of the current mitochondrion was then set to the value of the corresponding member of the FHU array in the mitochondrion whose index was equal to the aforementioned pseudorandom integer (i.e., the current mitochondrion is eliminated from the population and replaced by a duplicate of another mitochondrion).
Finally, the main class consisted of an array of 100 cells, and a loop, which iteratively executes the cycle function of each cell class and that is executed 100,000 times (i.e., 200,000 min). Note that all pseudorandom number-generating functions are seeded against time, ensuring that the behavior of each cell, and each mitochondrion in those cells, is different.
The values used in the model are given in Table 1. The values for Pt and Pm were set to 1/25, and 1/50 events per cycle, respectively, so that autophagy occurred with a probability of once every 50 min and damage once every 100 min unless mentioned otherwise. Fusion occurred every 2 min to once every 18,000 min (i.e., Pf ranged from 1 to 1/9500).
Table 1.
Simulation parameters and their values
| Parameter symbol | Description | Value |
|---|---|---|
| Pf | Frequency of fusion and fission events for single mitochondria per min | Variable (1/2–1/9500 min−1) |
| Pm | Rate of damage to FHUs per single mitochondria (activity/ min) | Variable (1/50, 1/100, or 1/200 min−1) |
| Pt | Rate of autophagy for single mitochondria | Constant (1/100 min−1) |
| Threshold for fusion (fusion was either selective or nonselective in any given simulation) | Selective fusion: 40% of maximum Δψm (equivalent to mitochondrial activity = 4); Nonselective fusion: 0% of the maximum Δψm (mitochondrial activity = 0). | |
| Threshold for autophagy | 30% of maximum Δψm (mitochondrial activity = 3) |
The simulation was also run for the two extreme cases in which either fusion or fission were silenced. To silence fusion, the fusion-maintenance and fusion function were omitted from the cycle function of the cell class, so that a cycle would proceed from “damage” to “autophagy” only. To silence fission, the fusion-maintenance function was omitted, so that no process would unfuse the mitochondria after a certain amount of time had passed.
The code was written in C# and executed on a Pentium D 830 computer, using the open-source Mono framework.
Results
Contribution of the combination of fusion and fission to quality maintenance
To determine the contribution of fusion and fission to the steady-state mitochondrial activity, we simulated mitochondrial activity of a cell with 300 mitochondria over time. These mitochondria underwent fusion-fission events at variable rates, exchanged their FHU content, and were subject to variable damage rate and autophagy (see Methods section for details). In this model, autophagy acted solely as a removal mechanism of low-activity mitochondria (30% of maximum and lower) that are generated by a random damage algorithm.
Running the model at any given rate of fusion-fission resulted in a drop in the level of mitochondrial activity from an initial level (maximal, where none of the FHUs are damaged) to a certain plateau, as shown in Fig. 3 a. Reducing the fusion rate to 0 tested a condition in which all mitochondria were constantly solitary and resulted in a steady-state activity of 32% of its initial value (Fig. 3 a, No Fusion). Under this condition, autophagy serves as a threshold mechanism that removes mitochondria once their activity drops to 30% or below. When a mitochondrion is removed, a random mitochondrion within the population is duplicated. As expected, the system stabilizes just above the autophagy threshold.
Figure 3.

Dependence of mitochondrial activity on the frequency of fusion and fission. The integrated mitochondrial activity (Δψm relative to maximum) of 100 cells, each containing 300 mitochondria, is shown. The duration of each fusion event is 4 min; Pt = 1/50, Pm = 1/100 events per minute. (a) Changes of mitochondrial activity over time with nonselective fusion. At t = 0 all mitochondria start at maximal activity, which gradually reduces because of the accumulation of damage. Mitochondrial activity is shown for conditions of no fusion (0), no fission (permanent fused state, ∞), and two frequencies of fusion events: 55 events/mitochondrion/day and 240 events/mitochondrion/day. (b) Effect of the selectivity of fusion on the steady-state mitochondrial activity. Selective (black) and nonselective (white) fusion are shown.
A condition in which mitochondria are constantly in the fused state (Fig. 3 a, Permanent fusion) was characterized by a fast drop of mitochondrial activity to zero. In the constant fused state mitochondria are unavailable for autophagy, which can occur only when mitochondria are in a solitary state. Because, in the simulation, mitochondria were being subjected to a baseline damage rate, the prevention of autophagy resulted in the accumulation of damaged mitochondrial components and a drop in activity. Experimental support for this link is based on the observation that increased fission is accompanied with an increase in mitochondrial autophagy (22), whereas silencing of the fission machinery prolongs fusion interval, inhibits selectively mitochondrial autophagy, and leads to accumulation of damaged units, despite lower reactive oxygen species production rate (15). Accumulation of damaged (low-activity) mitochondria results in a decline of cellular mitochondrial activity, eventually reaching a steady state of zero activity.
When 4-min fusion events were allowed to occur nonselectively at a rate of 55 events per mitochondrion per day, mitochondrial activity stabilized at 75% of its initial level. Further increase in the frequency diminished the beneficial effect of fusion and fission. The dependency of the steady-state activity values on the frequency of fusion-fission clusters is shown in Fig. 3 b.
Selective fusion benefits from higher fusion-fission frequencies
Fig. 3 b compares the steady-state values of mitochondrial activity when fusion is selective (only mitochondria with activity ≥ 40% are allowed to fuse) or nonselective (fusion occurs independent of mitochondrial activity). In the nonselective fusion group, steady-state activity peaks at 75%, and steeply drops to zero for frequencies > 100 events/mitochondrion/day. In the selective fusion group, however, steady-state activity rises for any increments in frequency. The peak activity of selective fusion reached 94% of maximum at 720 fusion-fission events/day (the highest frequency tested). At a fusion-fission frequency of ∼70 events/mitochondrion/day, the frequency reported in INS1 and COS7 cells, mitochondrial activity was 86% and 70% for the selective and nonselective fusion groups, respectively. These data indicate an additive positive metabolic contribution by selectivity of the fusion machinery.
Stabilization of mitochondrial activity performance under increased damage rate with selective fusion
Overproduction of free radicals, a decrease in the activity of DNA and protein repair mechanisms, or a decrease in radical scavenging capacity can increase mitochondrial baseline damage rate. Here we tested whether increment frequencies of selective or nonselective fusion facilitate maintenance of mitochondria activity when the damage rate is doubled or halved (Pm = 1/50 min and 1/200 min, respectively) (Fig. 4). When the damage rate is doubled, mitochondrial activity is attenuated for any fusion-fission frequency in a frequency-dependent manner. Increasing fusion frequency can restore mitochondrial activity as long as fusion is selective. As shown in Fig. 4, when fusion is nonselective, the optimal frequency (the one that yields highest steady-state activity) depends on the rate of damage; optimal fusion frequency is decreased when damage rate is increased. Even at optimal fusion frequency, mitochondria undergoing nonselective fusion are severely impacted by the increased damage rate. Selective fusion, on the other hand, benefited from very high fusion event frequencies, resulting in a near complete restoration of mitochondrial activity under increased damage rate. At high damage rate and low fusion frequency, mitochondrial activity of both selective and nonselective fusion stabilizes at zero. At low fusion rates, redistribution of damaged FHUs does not occur, and, therefore, the activity is derived from the balance between damage infliction and damage removal rates. Whereas, at low damage rate, the frequency of damage equals that of autophagy, in the case of increased damage rate, the frequency of damage is fivefold the frequency of the autophagy module.
Figure 4.

Effect of increased damage rate on the steady-state mitochondrial activity at different frequencies of fusion events. Selective and nonselective fusion are compared under (a) high (Pm = 1/50) and (b) low (Pm = 1/200) damage rates. The values of Pt, Pf, and duration of fusion event are identical to those used in Fig. 3. Note that, under increased levels of damage, mitochondrial activity is severely reduced when fusion is nonselective. Dotted lines demonstrate that, under increased damage rate, increased frequency of fusion-fission clustering is needed to reach a certain steady-state mitochondrial activity.
Duration of fusion events
Inhibitors of mitochondrial fission are commonly used in the study of mitochondrial dynamics and metabolism. Here we simulated a condition in which the probability of fission is diminished, resulting in extended duration of the fused state. Fig. 5 shows the consequences of increasing the duration of the fused state from 4 to 120 min. This prolongation resulted in a decrease of mitochondrial activity for mid- and high-range fusion frequencies, regardless of fusion selectivity. Extending fusion duration to 4 h caused mitochondrial activity to slowly decline to zero (data not shown). These data indicate that extension of mitochondrial fusion interval results in a drop in mitochondrial activity similar to results reported under silencing of Fis-1 and Drp-1 (3,15,35–38).
Figure 5.
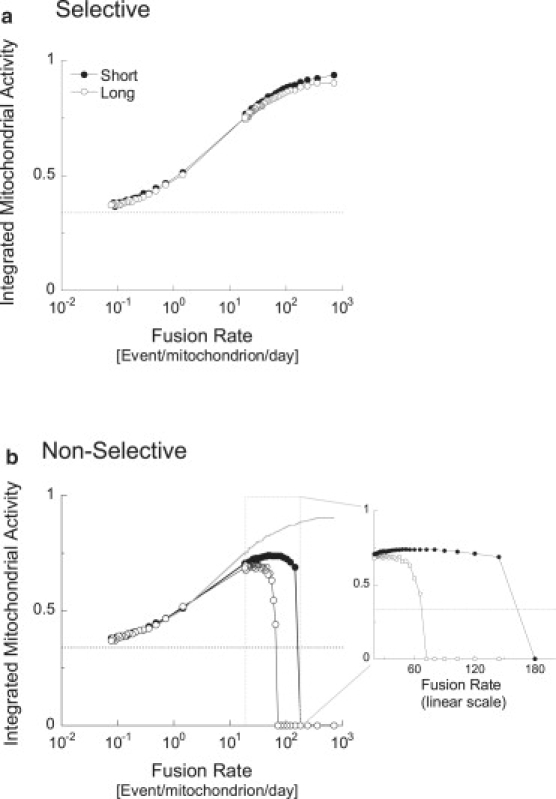
Effect of duration of mitochondrial fusion on steady- state mitochondrial activity at different frequencies of fusion events. Simulations of (a) selective and (b) nonselective fusion are compared. Short (4 min) and long (120 min) fusion events were simulated. Pt = 1/50, Pm = 1/100 events per minute. For comparison, a smooth continuous line in part b marks the curve of “Long Selective Fusion” from part a of this figure. The x axis expresses the maximal frequency of fusion events permitted. In the condition of long fusion, the actual frequency of fusion events is lower, because most mitochondria are occupied. Above 10 events per min, mitochondria in the long-fusion condition spend virtually no time in solitary state. Note that long-fusion events affect mitochondrial activity if fusions are nonselective and more so in higher and physiological fusion frequencies (∼70–100 events/mitochondrion/day).
Discussion
A vast amount of clinical and experimental evidence has demonstrated that both mitochondrial fusion and mitochondrial fission are essential for mitochondrial function (1,2). However, the benefit of having repetitive and frequent clusters of fusion-fission events has not been addressed thus far. The goal of the current simulation was to dissect the contribution of kinetic parameters of mitochondrial dynamics to the maintenance of mitochondrial function.
Contribution of fusion to quality maintenance
Our model took into account recent results demonstrating that, whereas fusion events allow for exchange of mitochondrial membranous and matrix material (6,39), it also results in the generation of dissimilar daughter units (15,22). This indicated that either not all of the material is equally distributed between the two fusion mates or that an active process of redistribution segregates inactive components into one of the daughters. In the presented study, this was simulated by a random exchange of 2 random FHUs out of the total 10 FHUs, resulting in random occurrence of uneven daughters.
Using these features, we tested the possibility that, when combined, fusion, fission, and autophagy may be able to maintain high levels of steady- state mitochondrial activity without repair. Remarkably, the simulation showed that placing fusion and fission clusters upstream of autophagy results in a net benefit in mitochondrial activity, even when fusion is allowed to occur nonselectively. This benefit exceeds the outcome of autophagy alone that functions as a threshold-based quality-control mechanism. Furthermore, it indicated that, in the absence of repair mechanisms, fusion, fission, and autophagy may be sufficient for the restoration of mitochondrial function. This is of importance for cases in which the damage is of a permanent nature (mutation/deletion) and cannot be repaired.
The simulation demonstrated that fusion allows for faster removal of damaged FHUs because of the redistribution feature. For example, in a single iteration, two mitochondria with 40% activity may go through a fusion-fission cluster and randomly generate two daughters of 30% and 50% activity. The one with 30% activity will be available for autophagy. This will remove 7 damaged FHUs from the system and will create one mitochondrion that has higher activity compared to that of each of the original two.
Simulation of increased damage rate demonstrated that the frequency might be an essential variable determining the ability of the system to cope with increased damage. Faster damage requires faster iterations that remove the damaged material in a faster rate than its generation. This finding stresses the importance of the absolute frequency of fusion and fission, rather than the sole balance between them.
Mitochondrial fusion as a recombination-like mechanism
The results obtained from the model demonstrated the advantage of redistribution of FHUs at each fusion event. The effect of redistribution on the increased ability to remove damaged FHUs from the population is very similar to the effect of genomic recombination combined with positive Darwinian selection. DNA recombination that occurs during sexual proliferation is characterized by the assembly of a new genomic material made by the contribution of both parents. Both processes, DNA recombination and mitochondrial fusion, involve the engagement of two parental units that will go through a step of redistribution of their components. Both fusion and recombination allow functional and damaged components that coexisted in one parental unit (chromosome or mitochondrion) to disengage and reorganize in a way that forms an offspring that is better than either parent. Whereas the process of enrichment through fusion and fission has parallels to the effect of recombination, it does not represent a form of mtDNA recombination. This would require a biochemical mechanism that reorganizes DNA at the level of the single nucleoid, a process that was not yet identified. Therefore, mutations are expected to accumulate in mitochondrial genomes through the random loss of less-mutated genomes (a process also referred to as Muller's ratchet) (40). The beneficial effect of fusion on removal of FHUs may therefore represent the removal of mitochondrial genomes, which appear at 1–10 copies per mitochondrion.
Selectivity of fusion
Whereas fusion may recruit dysfunctional mitochondria into the active pool, autophagy targets depolarized mitochondria for digestion and elimination. This places autophagy and fusion as competing fates of the depolarized mitochondria. However, the numerous reports indicating that mitochondrial fusion is dependent on membrane potential or respiratory function make fusion a selective process (11–13,41–43). A principal conclusion of our simulation is that selectivity of the fusion machinery makes fusion and autophagy complementary processes rather than competing ones.
Segregation of the damaged units from the fusing population prevents the mixing of damaged mitochondria with more active mitochondria, but also, and equally important, leaves them available to autophagy. This property becomes more apparent under high fusion rate or prolonged fusion time. Under these conditions, the duration spent in the solitary state is shortened, and, thereby, the probability of autophagy of depolarized mitochondria is reduced, leading to the collapse in mitochondrial activity observed at high fusion frequencies or durations. When fusion is selective, prolonged fusion durations or high fusion frequencies do not affect the removal of damaged mitochondria, because these mitochondria avoid fusion and are therefore available for autophagy at all times. However, when fusion is nonselective, autophagy is indirectly inhibited by the occurrence of most units in the fused state and results in the accumulation of low-activity units. Selectivity of fusion appears to strongly impact mitochondrial activity when high rates of damage are tested. Here, selective fusion permits the increase of fusion frequency without compromising autophagy, thereby allowing faster removal of damaged material. Therefore, selectivity of mitochondrial fusion is important, not only as an intramitochondrial complementary route (9,10), but also as an isolation step preceding autophagy.
To conclude, the presented simulation stresses the importance of the frequency and selectivity of mitochondrial fusion for mitochondrial activity. The combination of the two suggests that mitochondrial fusion is a relevant component of a quality maintenance mechanism under conditions of increased damage, in addition to its benefit as intermitochondrial complementation route. Our findings emphasized that fusion selectivity is a property that indirectly speeds the removal of damaged mitochondria by autophagy and also as a property that allows for increased fusion frequency without compromising the removal rate of dysfunctional mitochondria by autophagy.
Considerations that may benefit future development of the model
Whereas the values used in the simulation were based on experimental findings, the output of the model was expressed in arbitrary units and could only be used to assess the relative contribution of the parameters tested. Those included the mitochondrial activity, the number of copies of mitochondrial material, the extent of damage, the site of damage and the Δψm drop required for triggering autophagy. The model was therefore qualitatively testing the potential capacity of fusion, fission, and autophagy to generate a quality-control mechanism and maintain metabolic function under continuous damage of mitochondrial material. For that reason, we chose to ignore that autophagy and mitogenesis occur as independent events that can further change under various damage and possibly dynamics rates. The simulation did not take into account the dependence between rate of damage and metabolic activity. As it is generally understood, increased mitochondrial function can lead to breakdown of the electron transport chain, and, thereby, higher abundance of reactive oxygen species, which is known to cause mtDNA mutations (28). In our model, this interplay manifested solely as the damage rate.
The correlation between likelihood of selective fusion and metabolic activity and likelihood of autophagy and metabolic activity were represented as strict thresholds. These would be better represented by a more continuous correlation, but more experimental data are needed to develop the necessary equations. Finally, the behavior of mitochondrial fusion used in the presented model represented its occurrence in the most frequent forms. For simplicity of the model, we chose to ignore rare dynamics events such as a repetitive fusion event (without any fission event between them) (15) or a fusion event that involves more than two mitochondria (O. Shirihai, unpublished data).
Acknowledgments
We thank Drs. Dani Dagan, Leslie Satin, Solomon Graf, David Nicholls, Gyorgy Hajnoczky, Vipul Periwal, and Barbara Corkey for helpful discussions and comments.
This work was supported by National Institutes of Health grants 5R01HL071629-03 and 5R01DK074778.
Footnotes
Pradeep K. Mouli and Gilad Twig contributed equally to this work.
References
- 1.Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 3.Li Z., Okamoto K., Hayashi Y., Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Waterham H.R., Koster J., van Roermund C.W., Mooyer P.A., Wanders R.J. A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 2007;356:1736–1741. doi: 10.1056/NEJMoa064436. [DOI] [PubMed] [Google Scholar]
- 5.Arimura S., Yamamoto J., Aida G.P., Nakazono M., Tsutsumi N. Frequent fusion and fission of plant mitochondria with unequal nucleoid distribution. Proc. Natl. Acad. Sci. USA. 2004;101:7805–7808. doi: 10.1073/pnas.0401077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busch K.B., Bereiter-Hahn J., Wittig I., Schagger H., Jendrach M. Mitochondrial dynamics generate equal distribution but patchwork localization of respiratory complex I. Mol. Membr. Biol. 2006;23:509–520. doi: 10.1080/09687860600877292. [DOI] [PubMed] [Google Scholar]
- 7.Karbowski M., Youle R.J. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- 8.Ono T., Isobe K., Nakada K., Hayashi J.I. Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat. Genet. 2001;28:272–275. doi: 10.1038/90116. [DOI] [PubMed] [Google Scholar]
- 9.Sato A., Nakada K., Hayashi J. Mitochondrial dynamics and aging: Mitochondrial interaction preventing individuals from expression of respiratory deficiency caused by mutant mtDNA. Biochim. Biophys. Acta. 2006;1763:473–481. doi: 10.1016/j.bbamcr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Nakada K., Inoue K., Ono T., Isobe K., Ogura A. Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 2001;7:934–940. doi: 10.1038/90976. [DOI] [PubMed] [Google Scholar]
- 11.Legros F., Lombes A., Frachon P., Rojo M. Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol. Biol. Cell. 2002;13:4343–4354. doi: 10.1091/mbc.E02-06-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malka F., Guillery O., Cifuentes-Diaz C., Guillou E., Belenguer P. Separate fusion of outer and inner mitochondrial membranes. EMBO Rep. 2005;6:853–859. doi: 10.1038/sj.embor.7400488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mattenberger Y., James D.I., Martinou J.C. Fusion of mitochondria in mammalian cells is dependent on the mitochondrial inner membrane potential and independent of microtubules or actin. FEBS Lett. 2003;538:53–59. doi: 10.1016/s0014-5793(03)00124-8. [DOI] [PubMed] [Google Scholar]
- 14.Meeusen S., McCaffery J.M., Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–1752. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- 15.Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elmore S.P., Qian T., Grissom S.F., Lemasters J.J. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J. 2001;15:2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 17.Priault M., Salin B., Schaeffer J., Vallette F.M., di Rago J.P. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 2005;12:1613–1621. doi: 10.1038/sj.cdd.4401697. [DOI] [PubMed] [Google Scholar]
- 18.Duvezin-Caubet S., Koppen M., Wagener J., Zick M., Israel L. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol. Biol. Cell. 2007;18:3582–3590. doi: 10.1091/mbc.E07-02-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griparic L., Kanazawa T., van der Bliek A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishihara N., Fujita Y., Oka T., Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song Z., Chen H., Fiket M., Alexander C., Chan D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barsoum M.J., Yuan H., Gerencser A.A., Liot G., Kushnareva Y. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Twig G., Hyde B., Shirihai O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim. Biophys. Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Twig G., Graf S.A., Wikstrom J.D., Mohamed H., Haigh S.E. Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am. J. Physiol. Cell Physiol. 2006;291:C176–C184. doi: 10.1152/ajpcell.00348.2005. [DOI] [PubMed] [Google Scholar]
- 25.Legros F., Malka F., Frachon P., Lombes A., Rojo M. Organization and dynamics of human mitochondrial DNA. J. Cell Sci. 2004;117:2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- 26.Iborra F.J., Kimura H., Cook P.R. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004;2:9. doi: 10.1186/1741-7007-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Satoh M., Kuroiwa T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp. Cell Res. 1991;196:137–140. doi: 10.1016/0014-4827(91)90467-9. [DOI] [PubMed] [Google Scholar]
- 28.Nicholls D.G., Budd S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- 29.Wikstrom J.D., Katzman S.M., Mohamed H., Twig G., Graf S.A. β-Cell mitochondria exhibit membrane potential heterogeneity that can be altered by stimulatory or toxic fuel levels. Diabetes. 2007;56:2569–2578. doi: 10.2337/db06-0757. [DOI] [PubMed] [Google Scholar]
- 30.Scott I.D., Nicholls D.G. Energy transduction in intact synaptosomes: influence of plasma-membrane polarization on respiration and membrane potential of internal mitochondria determined in situ. Biochem. J. 1980;186:21–33. doi: 10.1042/bj1860021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Enriquez J.A., Cabezas-Herrera J., Bayona-Bafaluy M.P., Attardi G. Very rare complementation between mitochondria carrying different mitochondrial DNA mutations points to intrinsic genetic autonomy of the organelles in cultured human cells. J. Biol. Chem. 2000;275:11207–11215. doi: 10.1074/jbc.275.15.11207. [DOI] [PubMed] [Google Scholar]
- 32.Malka F., Lombes A., Rojo M. Organization, dynamics and transmission of mitochondrial DNA: focus on vertebrate nucleoids. Biochim. Biophys. Acta. 2006;1763:463–472. doi: 10.1016/j.bbamcr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Coleman R., Silbermann M., Gershon D., Reznick A.Z. Giant mitochondria in the myocardium of aging and endurance-trained mice. Gerontology. 1987;33:34–39. doi: 10.1159/000212851. [DOI] [PubMed] [Google Scholar]
- 34.Terman A., Dalen H., Eaton J.W., Neuzil J., Brunk U.T. Aging of cardiac myocytes in culture: oxidative stress, lipofuscin accumulation, and mitochondrial turnover. Ann. N. Y. Acad. Sci. 2004;1019:70–77. doi: 10.1196/annals.1297.015. [DOI] [PubMed] [Google Scholar]
- 35.Beregi E., Regius O., Huttl T., Gobl Z. Age-related changes in the skeletal muscle cells. Z. Gerontol. 1988;21:83–86. [PubMed] [Google Scholar]
- 36.Yoon Y.S., Yoon D.S., Lim I.K., Yoon S.H., Chung H.Y. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006;209:468–480. doi: 10.1002/jcp.20753. [DOI] [PubMed] [Google Scholar]
- 37.Lee S., Jeong S.Y., Lim W.C., Kim S., Park Y.Y. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 2007;282:22977–22983. doi: 10.1074/jbc.M700679200. [DOI] [PubMed] [Google Scholar]
- 38.Gomes L.C., Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim. Biophys. Acta. 2008;1777:860–866. doi: 10.1016/j.bbabio.2008.05.442. [DOI] [PubMed] [Google Scholar]
- 39.Karbowski M., Arnoult D., Chen H., Chan D.C., Smith C.L. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J. Cell Biol. 2004;164:493–499. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rand D.M. Mitigating mutational meltdown in mammalian mitochondria. PLoS Biol. 2008;6:e35. doi: 10.1371/journal.pbio.0060035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishihara N., Jofuku A., Eura Y., Mihara K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem. Biophys. Res. Commun. 2003;301:891–898. doi: 10.1016/s0006-291x(03)00050-0. [DOI] [PubMed] [Google Scholar]
- 42.Meeusen S., DeVay R., Block J., Cassidy-Stone A., Wayson S. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell. 2006;127:383–395. doi: 10.1016/j.cell.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 43.Meeusen S.L., Nunnari J. How mitochondria fuse. Curr. Opin. Cell Biol. 2005;17:389–394. doi: 10.1016/j.ceb.2005.06.014. [DOI] [PubMed] [Google Scholar]