

Abstract
Fibril formation from amyloidogenic peptides is a hallmark of a wide range of diseases, including Alzheimer's disease and type II diabetes. Characterization of the aggregation process should include intrinsic factors, such as sequence variation, and extrinsic factors, such as crowding effects. To this end, we examined the interactions of dimers composed of residues 20–29 of human islet amyloid polypeptide (hIAPP), which form fibrils in vitro, and the nonamyloidogenic rat IAPP (rIAPP) using molecular dynamics simulations modeled at different peptide concentrations. There is a substantial free energy barrier to unbind the hIAPP dimer whereas no barrier exists for separating the rIAPP dimer. The profound differences in the free energy landscapes of the rIAPP and hIAPP dimers explains the lack of fibril formation in hIAPP upon substitution of the C-terminal residues by proline. Enhancing the extent of crowding has a substantial effect on both the barrier for separating a hIAPP β-sheet dimer and the formation of potential β-sheet nucleation sites. Our results show that the propensity for forming nucleation sites is dependent not only on the amino-acid sequence but also on the context in which it is found.
Introduction
Human islet amyloid polypeptide (hIAPP), or amylin, is a 37-amino-acid peptide that is co-secreted with insulin as part of the normal metabolic process (1). Fibrils formed from aggregation of hIAPP in vivo are found in 90% of patients with type II diabetes, and their presence is, in all likelihood, associated with the severity of the disease (2). Nuclear magnetic resonance (NMR) and circular dichroism measurements have confirmed that, as with most amyloidogenic peptides, the fibrils formed by amylin aggregates possess a β-sheet morphology (3). Experimental studies have shown that residues 20–29 of hIAPP are capable of forming fibrils in vitro (4,5). Proline substitution within this region has been found to inhibit, and in some cases prevent, fibril formation (6,7). The rat analog of hIAPP (rIAPP), which does not form fibrils, contains five point mutations within this region, three of which are proline substitutions. As a result, many of the studies done on hIAPP to date have focused on these residues, or subsets of them that form fibrils.
The formation of amyloid fibrils that possess a β-sheet morphology from naturally secreted peptides is a characteristic of several diverse pathologies such as Alzheimer's disease, type II diabetes, and Huntington's disease (8). Although kinetic studies of hIAPP aggregation have suggested that amyloidogenesis occurs via a nucleation-dependent polymerization mechanism (3), the driving forces behind oligomer formation remain elusive. In the case of the Aβ-peptide associated with Alzheimer's disease, studies have suggested that low molecular weight aggregates of Aβ-peptide can have a deleterious effect on cells (9–11). Probing the nature of low molecular weight aggregates of IAPP, as intermediates on the pathway to fibril formation and possibly as pathogenic disease agents, is a problem suited for molecular dynamics (12).
Here we explore the effects of peptide sequence and concentration on hIAPP and rIAPP aggregation. We have examined the effect of crowding on the stability of β-sheet dimers of hIAPP peptide fragments composed of residues 20–29. Crowding, which may be expected in a cellular environment, can have significant effects on the kinetics of formation and the stability of many biological systems (13–15). If the spatial volume accessible to the peptide is reduced by the presence of crowding agents (whether they are other macromolecules or peptides at elevated concentration), the conformational entropy of the peptide is similarly reduced. More specifically, the equilibrium of species in solution is shifted toward the state that will occupy the least volume; in the case of an amyloidogenic peptide, crowding may provide a conformational entropic driving force for aggregation.
We have examined the effect of sequence on the β-sheet stability and the overall peptide-peptide interactions by carrying out simulations for dimers composed of the hIAPP sequence as well as the analogous rIAPP sequence. The sequence for the hIAPP monomer is S20NNFGAILSS29; it contains a hydrophobic core (residues F23GAIL27) surrounded by polar, uncharged residues. The rIAPP sequence is SNNLGPVLPP; unlike the hIAPP monomer, it has a polar N-terminus and a nonpolar C-terminus. Although the characteristics of model peptides need not be reflected in the full-length peptides (16), the simulations provide insight into the biophysical basis of sequence effects on aggregation in a structurally well-characterized system.
Free energy profiles for peptide association were computed, using umbrella sampling, at three different peptide concentrations (Cp values) using three different computational protocols. The changes in peptide concentration mimic the effects of crowding. In our study, crowding included nonspecific (entropic component) and specific interactions that give rise to an enthalpic contribution. In the umbrella runs, periodic boundary conditions have been employed such that, at high Cp values, it is possible for the peptide to sense its periodic image. Although direct interaction between the peptides and their periodic images is possible, especially in the most concentrated system, interactions, when they occur, are infrequent, short-lived, and statistically insignificant. The image peptides exert their effect on the surrounding environment of the primary peptides, specifically as competitors for free volume and hydrating waters. We have simulated a random dimer for ∼100 ns in both a moderately sized box of water, where we expect that the presence of the periodic images will be felt by the dimer, and a larger truncated octahedron, where image effects are expected to be negligible. The simulation results demonstrate the importance of solvation effects in the aggregation of the hIAPP peptide, as well as the significance of the central hydrophobic core in determining conformations of the dimer. Our study shows that sequence effects and the context in which nucleation sites are formed play a vital role in the aggregation process. Both crowding-induced entropic and enthalpic driving forces, which have opposing effects on hIAPP and rIAPP dimers, determine the stability of oligomers.
Methods
Sequences and initial structures
For both the hIAPP and rIAPP sequences, the N-terminus of the monomer was acetylated and the C-terminus was amidated. The sequence of the hIAPP monomer is SNNFGAILSS, whereas the rIAPP sequence is SNNLGPVLPP. The hIAPP and rIAPP β-strand monomers were generated using backbone torsional angles of (ϕ,ψ) = (–139, 135) degrees, appropriate for an antiparallel β-sheet. The initial configuration of the hIAPP and rIAPP β-sheet dimers was an antiparallel configuration that mimicked that of the fibril structure (5).
Concentration-dependent free energy profiles
All-atom molecular dynamics simulations were performed using the CHARMM 27 all-atom force field (17). All simulations were performed using the NAMD 2.5 package (18). We use three peptide concentrations (20.7 mM, 36.4 mM, and 61.3 mM) for hIAPP β-sheet dimers. The changing concentration effectively mimics crowding effects as the center-of-mass distance between the peptides RCM ∼ Cp−1/3. For comparison, the simulations for rIAPP were performed at 36.4 mM and 61.3 mM. These concentrations are at least three orders-of-magnitude greater than the conditions under which in vitro experiments have been performed.
For each simulation window, periodic boundary conditions were employed and Ewald sums were used to evaluate the electrostatics; a cutoff of 13 Å was used for the van der Waals forces. A time step of 1 fs was used and the lengths of bonds to hydrogen atoms were constrained using SHAKE. Simulations were run in the NVT ensemble at 300 K using Langevin dynamics to control the temperature. Coordinates were saved every 2000 time steps.
We determined the free energy profile for the separation of the monomers from the dimer using the umbrella-sampling method (19). The center-of-mass distance of the monomers was used as the reaction coordinate in generating the free energy profiles. The harmonic potentials used to sample the center-of-mass distance were of the form , where k = 5.0 kcal/(mol Å2), di is the position of the potential minimum of the ith window, and dt is the value of the reaction coordinate at time t. The initial value of di was chosen to be the center-of-mass distance between the monomers in the smallest water box, which was 5.3 Å. The values of di ranged from 5.3 Å to 15.3 Å in 1 Å increments. Each window of the smallest water box was simulated for 1.1 ns and the value of dt was recorded every 10 fs after the first 100 ps of equilibration. Each window of the larger water boxes was simulated for 1.2 ns and the value of dt was recorded every 10 fs after the first 200 steps of equilibration. The initial coordinates used for each window were the coordinates generated from the prior window after the equilibration time. The PMF of each system was obtained by combining the distributions using the weighted histogram analysis method (WHAM) (20,21).
Dimer evolution
Using the same sequence as described above, the initial configuration for the monomers composing the hIAPP dimer was randomly chosen from the set of NMR structures determined by Mascioni et al. (22). The initial configuration of the rIAPP monomers was chosen from among the final configurations of a series of simulated annealing runs. The structure with the smallest root mean-square deviation as compared to the hIAPP monomer was selected. For each sequence, the two monomers were minimized with the initial RCM fixed at 6 Å, so as to be in close contact at the start of the simulation. The dimers of the two sequences were solvated in two different systems: the first was an orthorhombic box with a peptide concentration of 36 mM; the second was a truncated octahedron with a peptide concentration of 28 mM. The simulations were performed using the NAMD 2.5 package. The systems were allowed to evolve freely for 100 ns. Table S1 of the Supporting Material provides a summary of the parameters for the systems simulated in the extended dynamics runs.
Each simulation was performed under conditions similar to those of the umbrella simulations. Periodic boundary conditions were employed and Ewald sums were used to evaluate the electrostatics; a cutoff of 12 Å was used for the van der Waals forces. A time step of 1 fs was used and the lengths of bonds to hydrogen atoms were constrained using SHAKE. Simulations were run in the NVT ensemble at 300 K using Langevin dynamics to control the temperature. Coordinates were saved every 10,000 time steps.
Analysis
We used principal coordinate analysis (23) to infer collective fluctuations of the peptides. The covariance matrix for each trajectory was calculated using
| (1) |
where the xi values represent the 3N Cartesian coordinates and 〈…〉 denotes the average over the conformations sampled in the trajectory. Since C is a symmetric matrix, it can be diagonalized as C = BΛBT, where B is composed of eigenvectors of C and Λ is a diagonal matrix whose elements are the eigenvalues of C. These eigenvalues represent the variation along the axis defined by the corresponding eigenvector. The largest eigenvalues correspond to the axes along which the largest motions of the system occur. Projecting each frame of the trajectory onto the first few principal axes allows one to effectively visualize the dynamics in the multidimensional energy landscape using a reduced representation.
Results
Monomers form kinked β-strand-like structures
To validate our simulations, we compared the calculated monomer structures with the amyloid microcrystal of hIAPP residues 21–27. The distinctive feature of the ordered monomer in the crystal is a bend in the backbone that results in a kink between the N- and C-terminal residues centered at G24. In our simulations the kink angle (θ ≈ 109° in (24)), which is formed between the α-carbon atoms of N21, G24, and L27, is broadly distributed (Fig. 1 a and Fig. S7 a in the Supporting Material) with a mean of 128° and a standard deviation of 32° in the 36-mM simulation. Considering that the structural fluctuations in the simulated systems are much larger than in the crystal, the computed mean θ and the experimental value are in good agreement. The 28-mM system has a bimodal distribution in θ (Fig. S7 a) with a second peak at ≈75°, which shows that the monomers adopt collapsed structures at low concentrations.
Figure 1.
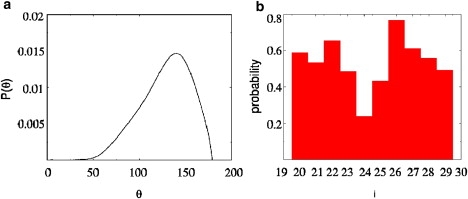
(a) Probability distribution of θ values for the two monomers in the 36-mM simulation. (b) Probability of finding each residue of the hIAPP sequence in a β-strand configuration for the 36-mM simulation. The results are averaged over both monomers.
We also computed the residue-dependent β-strand propensity using the (Φ, Ψ) angles (see Supporting Material for details). In the crystal structure N21, N22, and I26 adopt β-strand conformations, whereas the residues in the kink region (F23, G24, and A25) do not. These findings are in excellent agreement with our simulations (Fig. 1 b and Fig. S7 b), which show that the propensity to be in a β-strand configuration is lowest for the residues in the kink region. The correspondence between the calculated values and the crystal structure using the measures in Fig. 1 and the root mean-square deviation (Fig. S7 c) validates the simulations. Despite the kink in the monomer structure, for simplicity, we will refer to the ordered monomer as β-strand in the rest of the article.
Our simulations can be directly compared with an illuminating solid-state NMR study by Madine et al. (25), who have shown that hIAPP forms two distinct amyloidlike fibrils. In one of the structures the peptides form parallel fibrils, as seen in our simulations, whereas in the other they are found in an antiparallel arrangement. However, when seeded with hIAPP8–37 fibrils, it appears that only the parallel structure is formed. Most importantly, their analysis suggests that all of the residues are in the β-strand conformation. Our simulations, although in broad agreement with their experiments, also differ in one respect. We find that the probability of forming a β-strand at position G25 is the lowest, which is in part due to the observed bend at that position. This discrepancy could be due to finite size effects. Our simulations do not mimic the fibril structure, and hence the peptides are subject to larger conformational fluctuations. Clarification of the detailed structural differences and the effects of point mutations on the ability to form fibrils require additional studies.
The level of confinement affects both the mechanism and energetics of hIAPP β-sheet separation
Fig. 2 a shows the PMFs representing the system free energy as a function of RCM at three different effective hIAPP concentrations. The starting structures for the umbrella simulations are displayed in Fig. S8. The free energy cost for dissociating the dimer increases significantly with the concentration of the peptide, with the lowest concentration having essentially no barrier for separation.
Figure 2.
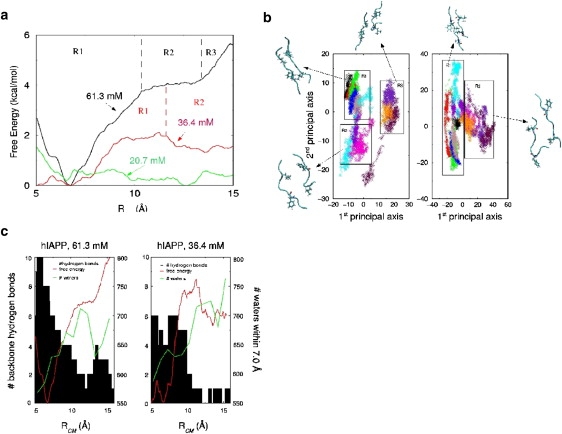
(a) Free energy profiles as a function of the center-of-mass distance, RCM, among the hIAPP β-sheet dimer at concentrations of 61.3 mM (black), 36.4 mM (red), and 20.7 M (green). The black dashed lines define the three regions for Cp = 61.3 mM and the red dashed line defines the two regions for Cp = 36.4 mM. (b) Projection of the hIAPP dimer onto the first and second principal component axes at 61.3 mM (left) and 36.4 mM (right). Boxes have been drawn to cluster structures with similar features. (c) The number of backbone hydrogen bonds between the two hIAPP monomers at a particular RCM for Cp = 61.3 mM (left) and Cp = 36.4 mM (right). The free energy profile (red) and the average number of waters within 7 Å of the monomers (blue) has been overlaid on both plots.
The projection of the umbrella trajectories onto their first and second principal axes, at the higher concentrations, in Fig. 2 b shows that two different mechanisms are responsible for the dimer separation. At Cp = 61.3 mM, the free energy landscape can be divided into three regions that correspond to distinct features of the PMF, suggesting a three-state unbinding mechanism. The first region, R1, encompasses the basin containing the minimum of the PMF (RCM = 5.0–10 Å); R2 refers to the flat region of the PMF (RCM = 10.0–13.0 Å); and in R3, the PMF begins to rise again (RCM = 13.0–15.5 Å). The projection of the moderately concentrated system (Cp = 36.4 mM) has only two observable regions corresponding to motion along the first principal axis, though the distinction is not as well defined as in the concentrated system (Fig. 2 b). The R1 region encompasses the PMF minimum, as well as the flat region of high energy (RCM = 5.0–11.3 Å) and the R2 region consists of those windows for which the PMF begins to decline (RCM = 11.3–15.5 Å in Fig. 2 a). Thus, self-crowding (15) by other polypeptide chains alters the energy landscape of hIAPP.
Fig. 2 c shows the number of backbone hydrogen bonds in the dimer at a given RCM. At Cp = 61.3 mM there is a gradual decrease in the backbone hydrogen bonding up to 12 Å, where it goes to zero. As the PMF begins to rise again near 13 Å, there is an increase in hydrogen bonding due to the formation of a small section of β-sheet formed between the dimers that is off-register in comparison to the initial state. In contrast, under moderately concentrated conditions, there is a sharp drop in the number of backbone hydrogen bonds when the PMF is at its peak (9.6–11.0 Å). After this point, only transient hydrogen bonds form along the backbone. From these observations, we infer that the three states of the separation mechanism in the concentrated system are the initial sheet as it is dissociating, the separated monomer pair, and second off-register β-sheet. In the less concentrated system, the two observed states are simply the initial sheet and the separated monomers. The inference from the decrease in hydrogen bonds is consistent with the projections onto the principal component axes in Fig. 2 b.
The origin of the two different mechanisms of separation are also reflected in Fig. 2 c, which depicts the average number of water molecules within 7.0 Å of the monomers for each window of the umbrella run. The averages are plotted at the constraint distance for the window. The most concentrated system attains the maximum in its solvation shell near 11.3 Å. As the constraint distance is further increased, the images of the monomers and their surrounding solvation shells become too close to allow the original monomers to add waters to their own solvation shells. This solvation constraint forces the monomers to collapse into a second β-sheet to minimize the excluded volume. The moderately concentrated system shows very minute changes in solvation number until a sharp increase occurs near 11.2 Å. Beyond RCM > 11.2 Å, the number of waters in the second solvation shell is approximately the same as in the more concentrated system. However at RCM = 13.3 Å there is enough space between the monomers and their images to allow for the addition of waters to the second solvation shell. With this addition, the PMF begins to decrease. At the lower concentration (Cp = 36.4 mM), the peptides maintain their separation from each other.
The rIAPP β-sheet is not stable, whereas interactions between the hydrophobic core residues stabilize the hIAPP β-sheet
The PMF for the unbinding of the rIAPP β-sheet dimers, in contrast to the hIAPP analog, shows (Fig. 3 a) that there is virtually no free energy barrier to peptide separation, even at high Cp. No free energy minimum stabilizing the rIAPP dimer is observed. Structural details of the differences between the two monomers in the hIAPP and rIAPP simulations are revealed in the contact maps (Fig. 3 b). At both high and low Cp, most contacts in the hIAPP dimer occur between residues of the hydrophobic core, indicating that association of the hydrophobic residues is responsible for stabilizing the β-sheet. The rIAPP residues at Cp = 61.3 mM also have the highest degree of association between the central hydrophobic residues. However, they are fewer than those of the hIAPP system and insufficiently strong to impose a barrier to separation. In rIAPP at Cp = 36.4 mM, contact occurs primarily between the N-terminus of one peptide and the C-terminus of the other. However, strong association between these residues does not impose a significant barrier to peptide separation.
Figure 3.
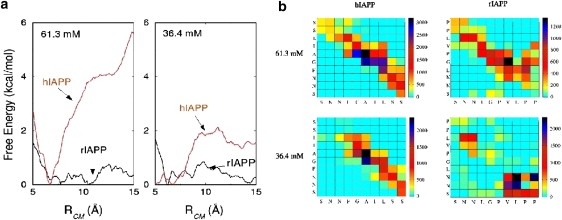
(a) Free energy profiles for separating the hIAPP (red) and rIAPP (black) β-sheet dimers in a 61.3 mM solution (left) and a 36.4 mM solution (right). (b) Contact maps for the hIAPP and rIAPP simulations Cp = 61.3 mM and Cp = 36.4 mM. The x axis represents the residues of the A monomer and the y axis represents residues of the B monomer. Two residues were considered to be in contact if their centers-of-mass were within 6 Å. The scale on the right gives the number of times a particular residue pair was found to be in contact, averaged over 5500 structures.
Under confined conditions, the hIAPP monomers are more likely to form β-sheet nucleation sites than rIAPP monomers
The top and middle graphs in Fig. 4 present a comparison between general structural features of System I = ((hIAPP)2; 36 mM) and System II = ((rIAPP)2; 36 mM), where finite size effects are expected to be important. The starting structures for the 100-ns simulations can be seen in Fig. S9. Hydrogen bonding between backbone atoms in System I is significantly more prominent than for System II. For System I, the peptide strands display a greater tendency to remain in close contact and the preferred RCM is closer than for System II (7.0 Å vs. 10 Å). The radius-of-gyration distributions for the two systems suggests that the monomers of System I show a slight preference to be elongated when compared to the monomers of System II. In general, (hIAPP)2 displays a greater propensity for multiple residue contacts between the two monomers (i.e., 5–7) than (rIAPP)2 (i.e., 1–4). Taken together, these results indicate that when monomer fluctuations are restricted, as happens upon crowding, the hIAPP peptide is more likely than the rIAPP peptide to form semistable nucleation sites for the formation and growth of β-sheets.
Figure 4.
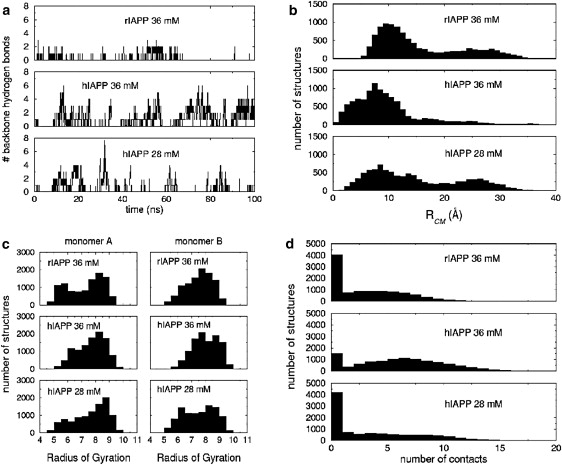
Comparison of the structural characteristics for the hIAPP monomers at Cp = 28 mM (bottom), the hIAPP monomers at Cp = 36 mM (middle), and the rIAPP monomers at Cp = 36 mM (top). (a) Number of backbone hydrogen bonds present between the monomers; (b) histogram of the sampled center-of-mass distances for the monomers where the bin size is 1 Å; (c) histogram of the sampled radii of gyration for the monomers where the bin size is 0.5 Å; and (d) the number of contacts between residues of the monomers. Two residues were considered to be in contact if their centers-of-mass were within 6 Å.
The middle and lower graphs in Fig. 4 compare structural features of System I and System III = ((hIAPP)2; 28 mM). Comparing the number of backbone hydrogen bonds between the two hIAPP runs, it is clear that the frequency of hydrogen bonding is reduced in System III. System III (weak crowding) is more likely to dissociate than System I (strong crowding), as it is in fact almost equally probable to find the monomers in System III in close contact as it is to observe them separated. The hIAPP monomers in System III are also less likely to be elongated than in System I. The number of residue-residue contacts has also been reduced. Although there is some appearance of β-sheet-like structures that could serve as nucleation sites, the frequency of such structures is noticeably reduced in the lower concentration conditions of System III.
Fig. 5 shows the propensity for contact between specific residues of the two monomers in each simulation. In the System I simulation, the hydrophobic core (residues 23–27) of monomer A associates with residues in the hydrophobic core region of monomer B (residues 23–25 and 27). There is a tendency for the N-terminus of the B monomer to associate with the hydrophobic core region of the A monomer. The most frequent contacts between the rIAPP monomers of System II are found exclusively between the central residues, specifically residues 24–27 on monomer A and residues 23–26 on monomer B. In rIAPP, the hydrophobic C-terminus displays no tendency to associate with other residues of the peptide.
Figure 5.
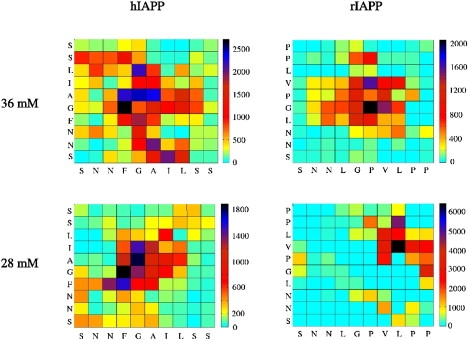
Residue-residue contact map for hIAPP and rIAPP dimers at Cp = 36 mM and Cp = 28 mM. The x axis represents the residues of the A monomer and the y axis represents residues of the B monomer. Two residues were considered to be in contact if their centers-of-mass were within 6 Å. The scale on the right gives the number of times a particular residue pair was found to be in contact, averaged over 10,000 structures.
The first two principal axes of motion for Systems I and III (Fig. S10) correspond to RCM and the parallel/antiparallel peptide alignment, respectively. In the more concentrated system, the center-of-mass distance is the second axis, whereas in the less concentrated run it is the first. In both cases there is a tendency for the antiparallel structures in close contact to be somewhat farther apart than the parallel structures in close contact. However, in System III, this trend is reversed for the configurations where the monomers are separated. In System I, there was no observable preference for monomers in close contact to be in a parallel or antiparallel alignment, whereas in system III there is a tendency for the monomers to be in a parallel alignment by a 3:2 ratio.
The residue-residue contact map of System III ((hIAPP)2; 28 mM) in Fig. 5 shows that the highest contact frequencies are exclusively found between residues of the hydrophobic core (residues 23–26). The slight preference for structures in contact to be in parallel is clearly illustrated here, as the highest frequency contact pairs are found mainly along the diagonal.
Under less concentrated conditions the rIAPP dimer forms a stable β-bridge but is unlikely to show growth beyond the dimer
Simulation of System IV = ((rIAPP)2; 28 mM) shows the formation of a β-bridge after 20 ns, initially between the C-terminal leucines (Fig. 6 a). This alignment persisted for 15 ns before the bridge residue on one monomer was shifted to the neighboring valine. This realigned bridge was still present at the end of the simulation 65-ns later (Fig. 6 a).
Figure 6.
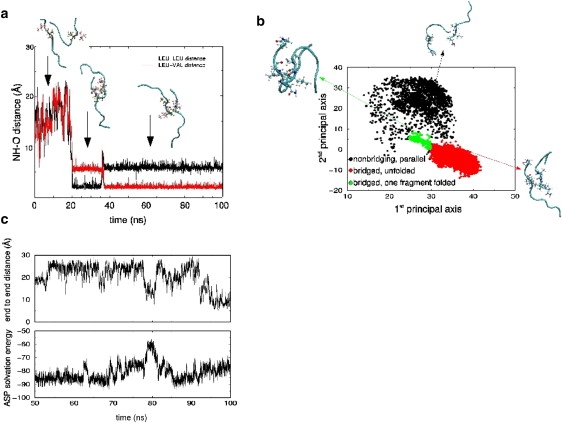
(a) Hydrogen bond distance between 1), the amide hydrogen on leucine 27 of monomer A with the carbonyl oxygen on leucine 27 of monomer B (black), and 2), the amide hydrogen on leucine 27 of monomer A with the carbonyl oxygen of valine 26 on monomer B (red). (b) Projection of the rIAPP simulation at Cp = 28 mM onto the first and second principal component axes. (c) (Bottom) Atomic Solvation Parameters solvation free energy for the two rIAPP monomers in the truncated octahedron simulation. (Top) The end-to-end distance of monomer B of rIAPP.
Formation of the bridge allows for a clustering of the hydrophobic residues of the C-terminus, leading to a decrease in the average van der Waals interaction energy. The change in the bridge alignment allows for better contact between the leucine 27 side chain of monomer A and the C-terminal proline 28 side chain of monomer B, leading to a net stabilization in interaction energy (Fig. S11). This point is illustrated by the residue-residue contact map (Fig. 5), in which the highest frequency contacts are observed between the C-terminal residues. The two highest frequency contact pairs involve leucine 27 of monomer A with valine 26 or proline 28 of monomer B.
Fig. 6 b shows the projection of the rIAPP trajectory of System IV onto the first and second principal component axes. The first principal axis corresponds to the extension of the two monomers, with more emphasis on the B monomer elongation. The second principal axis corresponds to the parallel/antiparallel alignment. The bridge structures are tightly clustered in the region represented by the green and red points. The small green island near the main red cluster corresponds to roughly 2 ns of simulation time, during which both the A and B monomers were folded into a U-shaped conformation. In the brief interaction, the polar N-terminal residues were buried, which led to a large spike in the solvation free energy for the dimer (Fig. 6 c). This suggests that any attempt to bury the N-terminus of the bridge would be energetically unfavorable, as would addition of other peptides to the bridge ends. These simulations show that although rIAPP can form transiently stable dimers, they are unlikely to grow and form stable higher order structures.
Discussion
The crowding and confinement of the hIAPP β-sheet dimer directly affects its solvation and has a significant effect on the mechanism by which the dimer dissociates or grows. In the most concentrated system corresponding to the most crowded conditions, the monomers continually seek conformations that occupy the least volume, as the solvation energy is insufficient to offset the cost of dissociation. In moderately concentrated systems, the greater availability of water facilitates the loss of secondary structure and dissociation. The difference in the two mechanisms illustrates the importance of hydration in determining the energetics and overall behavior of the peptides in solution. These results could also be used to interpret experimental results such as those of Mukherjee et al. (26), where the aggregation propensity of peptide is observed to increase when the level of hydration is decreased. Even under severely crowded conditions, no barrier is observed for the separation of the rIAPP β-sheet dimer.
Under moderately concentrated conditions, the propensity for hIAPP to form β-sheet structures that could serve as nucleation sites for fibril formation is significantly enhanced over the rIAPP sequence. As the hIAPP system becomes more dilute, fewer β-sheet structures are observed. Conversely, as the rIAPP system becomes more dilute, it is capable of forming a stable β-bridge structure between the two monomers. Due to the energetically unfavorable desolvation of the N-terminus, the β-bridged dimer structure is unlikely to serve as a nucleation site for fibril formation.
The apparent contrast in the behavior of the two sequences under different solvation conditions can be attributed to the effects of crowding on system stability. If the monomers of a dimer of either sequence are in close contact, the dimer will occupy the least volume when a maximum number of residues in the two monomers are aligned. In the hIAPP peptide sequence, this would lead to conformations in which the hydrophobic cores of each monomer overlap and the polar termini of the monomers are exposed to the surrounding water, a very stable alignment from the perspective of solvation. In the rIAPP peptide, an alignment of this sort also leads to an overlap of the central hydrophobic residues, with the hydrophobic residues of the C-terminus exposed to water. In terms of solvation energy, such a structure would not be as stable as that formed by hIAPP. It should be emphasized that the loss in conformational entropy due to crowding is the same for both sequences, as they have the same number of residues. The dramatically different behavior of hIAPP and rIAPP is thus due to the variation in sequence, and the effects this has on the energetics of the peptide interactions, either with itself or the surrounding environment.
Alternate alignments of the hIAPP monomers in close contact are expected to lead to conformations in which residues of the hydrophobic core overlap with polar residues, resulting in unfavorable configurations in terms of solvation, and are therefore unlikely. In the more dilute system, as the driving force provided by the crowded environment is reduced, fewer maximally compact β-sheet structures are observed. In contrast, without the restriction of a crowded environment, the rIAPP monomers are able to sample alignments that occupy larger volumes but are more favorable in terms of the solvation energy. Formation of the β-bridge structure in the rIAPP dimer requires a significant amount of free volume for reorganization. Formation of the β-bridge allows for the overlap of hydrophobic residues in the C-termini, leaving the polar N-termini exposed. The proline substitution experiments of Moriarty and Raleigh (6) demonstrate that substitution of residues 26–28 of hIAPP with proline prevents fibril formation. This result is in agreement with our finding that substitution of the C-terminal residues of hIAPP with smaller hydrophobic prolines allows for formation of the hydrophobic cluster observed in our simulation.
The sequence-dependent variations in the propensity of hIAPP and rIAPP to adopt β-sheet structures is likely to be fairly general. Our simulations suggest that, in addition to environmental factors such as crowding and cosolute effects, the context in which an amino acid is found, besides the chemical character, is an important determinant of β-content. It is therefore noteworthy to mention that the full-length hIAPP and rIAPP sequences will most likely have additional factors to consider when examining the reasons for aggregation, or lack thereof, of these species (27,28). For example, restriction of the backbone conformations in the 10-residue rIAPP sequence used for our simulations was not observed to be an impediment to its ability to dimerize, as two of the proline substitutions were located in the final residues of the C-terminus. When considering the nonamyloidogenic behavior of full-length rIAPP, this would likely produce a more pronounced effect. However, given that residues 20–29 of hIAPP have the ability to fibrilize in vitro and substitutions within this region in the full-length sequence inhibit fibril formation, our observations can still be used to identify nucleation sites, the factors contributing to their stabilization, and also suggest additional experiments to test our conclusions. For example, our simulations, which imply that the probability of adopting a β-configuration for L27 in the triad VLP of rIAPP is greater or lesser than L27 in hIAPP, depending on the level of crowding, is amenable to experimental testing by isotope edited infrared spectroscopy (29). It remains to be tested whether the formation of nucleation sites depends not only on the immediate sequence neighbors but also on residues that are further separated.
Acknowledgments
The authors thank Dr. Alan Grossfield for making the WHAM code accessible that was used in the computation of the PMF.
J.S. and D.T. gratefully acknowledge the National Institutes of Health (No. R01-NS041356) for generous support.
Supporting Material
References
- 1.Cooper G., Willis A., Clark A., Turner R., Sim R. Purification and characterization of a peptide from amyloid-rich pancreases of type-2 diabetic patients. Proc. Natl. Acad. Sci. USA. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lorenzo A., Razzaboni B., Weir G., Yanker B. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 3.Kayed R., Bernhagen J., Greenfield N., Sweimeh K., Brunner H. Conformational transitions of islet amyloid polypeptide in amyloid formation. in vitro. J. Mol. Biol. 1999;287:781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 4.Westermark P., Engstrom U., Johnson K., Westermark G., Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffiths J., Ashburn T., Auger M., Costa P., Griffin R. Rotational resonance solid-state NMR elucidates a structural model of pancreatic amyloid. J. Am. Chem. Soc. 1995;117:3539–3546. [Google Scholar]
- 6.Moriarty D., Raleigh D. Effects of sequential proline substitution on amyloid formation by human amylin(20–29) Biochemistry. 1999;38:1811–1818. doi: 10.1021/bi981658g. [DOI] [PubMed] [Google Scholar]
- 7.Abedini A., Meng F., Raleigh D. A single-point mutation converts the highly amyloidogenic human islet amyloid polypeptide into a potent fibrilization inhibitor. J. Am. Chem. Soc. 2007;129:11300–11301. doi: 10.1021/ja072157y. [DOI] [PubMed] [Google Scholar]
- 8.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 9.Hardy J., Selkoe D. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 10.Lambert M., Barlow A., Chromy B., Edwards C., Freed R. Diffusible nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsh D., Hartley D., Kusumoto Y., Fezoui Y., Condron M. Amyloid β-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 12.Straub J., Guevara J., Huo S., Lee J. Long time dynamics simulations: exploring the folding pathways of Alzheimer's amyloid β-peptide. Acc. Chem. Res. 2002;35:473–481. doi: 10.1021/ar010031e. [DOI] [PubMed] [Google Scholar]
- 13.Minton A. Implications of macromolecular crowding for protein assembly. Curr. Opin. Struct. Biol. 2000;10:34–39. doi: 10.1016/s0959-440x(99)00045-7. [DOI] [PubMed] [Google Scholar]
- 14.Cheung M., Klimov D., Thirumalai D. Molecular crowding enhances native state stability and refolding rates of globular proteins. Proc. Natl. Acad. Sci. USA. 2005;102:4753–4758. doi: 10.1073/pnas.0409630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H.-X., Rivas G., Minton A. Macromolecular crowding and confinement: biochemical, biophysical and potential physiological consequences. Annu. Rev. Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luca S., Yau W., Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacKerell A., Bashford D., Bellott M., Dunbrack R.L., Jr., Evanseck J.D. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 18.Kale L., Skeel R., Bhandarkar M., Brunner R., Gursoy A. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999;151:283–312. [Google Scholar]
- 19.Torrie G.M., Valleau J.P. Nonphysical sampling distribution in Monte Carlo free-energy estimation: umbrella sampling. J. Comput. Phys. 1977;23:187–199. [Google Scholar]
- 20.Kumar S., Bouzida D., Swendsen R.H., Kollman P.A., Rosenberg J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992;13:1011–1021. [Google Scholar]
- 21.Roux B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 1995;91:275–282. [Google Scholar]
- 22.Mascioni A., Porcelli F., Ilangovan U., Ramamoorthy A., Veglia G. Conformational preferences of amylin nucleation site in SDS micelles: an NMR study. Biopolymers. 2003;69:29–41. doi: 10.1002/bip.10305. [DOI] [PubMed] [Google Scholar]
- 23.Kitao A., Go N. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 1999;9:164–169. doi: 10.1016/S0959-440X(99)80023-2. [DOI] [PubMed] [Google Scholar]
- 24.Wiltzius J., Sievers S., Sawaya M., Cascio D., Popov D. Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin) Protein Sci. 2008;17:1467–1474. doi: 10.1110/ps.036509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Madine J., Jack E., Stockley P., Radford S., Serpell L. Structural insights into the polymorphism of amyloid-like fibrils formed by region 20–29 of amylin revealed by solid-state NMR and x-ray fiber diffraction. J. Am. Chem. Soc. 2008;130:14990–15001. doi: 10.1021/ja802483d. [DOI] [PubMed] [Google Scholar]
- 26.Mukherjee S., Chowdhury P., Gai F. Infrared study of the effect of hydration on the amide I band and aggregation properties of helical peptides. J. Phys. Chem. B. 2007;111:4596–4602. doi: 10.1021/jp0689060. [DOI] [PubMed] [Google Scholar]
- 27.Green J., Goldsbury C., Mini T., Sunderji S., Frey P. Full-length rat amylin forms fibrils following substitution of single residues from human amylin. J. Mol. Biol. 2003;326:1147–1156. doi: 10.1016/s0022-2836(02)01377-3. [DOI] [PubMed] [Google Scholar]
- 28.Abedini A., Raleigh D. Destabilization of human IAPP amyloid fibrils by proline mutations outside of the putative amyloidogenic domain: is there a critical amyloidogenic domain in human IAPP? J. Mol. Biol. 2006;355:274–281. doi: 10.1016/j.jmb.2005.10.052. [DOI] [PubMed] [Google Scholar]
- 29.Petty S., Decatur S. Experimental evidence for the reorganization of β-strands within aggregates of Aβ(16–22) peptide. J. Am. Chem. Soc. 2005;127:13488–13489. doi: 10.1021/ja054663y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.