

Abstract
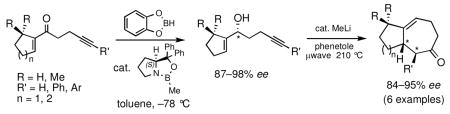
Appropriately substituted nonracemic allyl alcohols, readily prepared from the corresponding enones by application of the CBS methodology, were converted to optically active cycloheptenone derivatives with almost complete transfer of chirality via an efficient “one-pot”, cycloisomerization/Claisen rearrangement process. This methodology was directly applied to the expedient total synthesis of (−)-frondosin B.
Carbocyclic seven-membered rings are ubiquitous in nature and constitute the central structural unit in a number of polycyclic natural products, including phorbol esters,1 guanacastepenes,2 guianolides3 and the frondosins.4 Despite their prevalence and considerable medicinal relevance, the construction of cycloheptanoid systems through chemical synthesis remains challenging and is generally limited to processes other than direct intramolecular cyclization reactions. Among the most important of these are various cycloaddition strategies, such as the [5+2] and [4+3] reactions, which have proven useful for the synthesis of a number of seven-membered ring-containing natural products.5
We have recently demonstrated that a variety of cycloheptanoid fused ring systems may be conveniently accessed through a microwave-assisted tandem process that involves an oxyanionic 5-exo dig cycloisomerization reaction,6 followed by in situ Claisen rearrangement of the resulting 2-alkylidenetetrahydrofuran intermediate.7 Herein, we wish to report a new asymmetric variant of this strategy, which allows an efficient and practical construction of a number of optically active cycloheptenone derivatives. To highlight the synthetic potential of this methodology, an expedient total synthesis of (−)-frondosin B is also described.
The Claisen rearrangement reaction8 has been widely exploited for the generation of optically active products, e.g. through the use of a stereodirecting element built either within the allyl9 or the vinyl10 portion of the allyl vinyl ether moiety. Most of these processes involve enolate variants of the Claisen rearrangement, often operating under metal promoted chelate control.11 In addition, Lewis acid-promoted Claisen rearrangements, conducted in the presence of chiral ligands such as quinine and quinidine, have been employed to generate nonracemic products with varying degrees of asymmetric induction.12 However, none of these strategies have been applied to the asymmetric synthesis of substituted cycloheptenone derivatives.
Our approach to nonracemic cycloheptenones relies on the ability of a single stereogenic center installed within the allylic alcohol starting material to transfer stereochemical information to the final carbocyclic product with up to two new stereocenters through an all intramolecular tandem cycloisomerization/Claisen rearrangement sequence. Undoubtedly, the observed diastereocontrol in these reactions evolves through a preferred chair-like six-membered transition state, where the two prochiral centers of the 2-alkylidene tetrahydrofuran intermediate occupy a pseudo diequatorial relationship (Scheme 1).7h
Scheme 1.

Synthesis of optically active cycloheptenone derivatives.
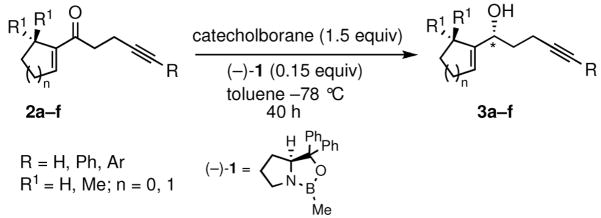
The prochiral enones employed for this investigation were prepared from the corresponding allylic alcohols7h by Swern oxidation13 or using the Bobbitt reagent.14 The enone products were then converted to the desired nonracemic homopropargylic allylic alcohols according to the Corey-Bakshi-Shibata15 (CBS) protocol employing stoichiometric catechol borane in the presence of a chiral oxazaborolidine catalyst ((−)-1, Table 1).
Table 1.
Preparation of optically active homopropargylic allyl alcohols via CBS reduction.
 | ||||
|---|---|---|---|---|
| entry | enone (2) | allyl alcohol (3) | yield (%)a | ee (%)b |
| 1 |
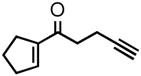 2a |
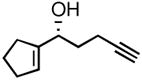 3a |
86 | 87 |
| 2 |
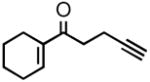 2b |
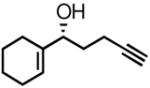 3b |
66 | 89 |
| 3 |
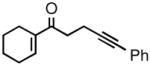 2c |
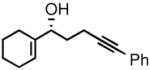 3c |
95 | 91 |
| 4 |
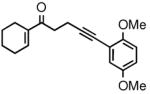 2d |
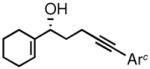 3d |
81 | 91 |
| 5 |
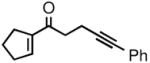 2e |
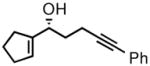 3e |
52 | 89 |
| 6 |
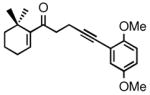 2f |
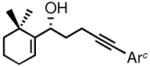 3f |
68d | 98 |
Yields of chromatographically purified products.
Enantiomer ratios were determined either by chiral GC or chiral HPLC.
Ar = 2,6-dimethoxyphenyl.
Reaction was quenched after 72 h.
In the series examined, the degree of enantioselectivity generally increased on going from cyclopentenyl to cyclohexenyl systems and was notably higher when additional substituents were placed near the reaction site (Table 1, entry 6). Thus, the more highly substituted enone 2f bearing a gem-dimethyl moiety on the cyclohexenyl ring at C6 exhibited the highest level of enantioselectivity upon CBS reduction, providing alcohol 3f in 98% ee (Table 1, entry 6). Although the absolute stereochemistry of the stereogenic center in 3f or in the other allylic alcohols prepared was not unequivocally established, ample literature precedent15 involving analogous systems and examination of the relevant transition state structures for the reduction are consistent with the formation of the R isomer as the major product in each case.
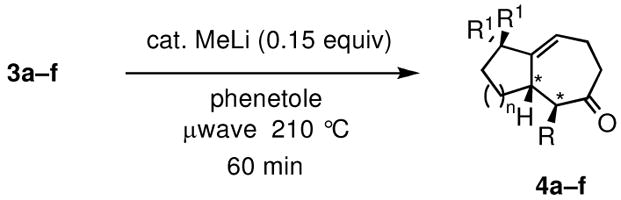
Gratifyingly, almost complete transfer of chirality was observed when each nonracemic allylic alcohol was subjected to catalytic MeLi and heat under microwave irradiation in phenetole. The expected 5–7 and 6–7 fused bicyclic ketones were produced in yields ranging from 62% to 82% and in ee’s as high as 95% (Table 2). Compounds 4c–4f represent examples where a single stereogenic center in the starting alcohol –ultimately lost in the course of the cyclization/rearrangement sequence– controls the absolute stereochemistry of two new adjacent stereocenters in the bicyclic cycloheptanoid product. It should be noted that the observed diastereomer ratios could be improved further through simple isomerization under basic conditions. For example, the initial 83:17 ratio of diastereomers in the case of 4f (Table 2, entry 6) was further enriched to a final ratio of 91:9 after stirring in a solution of MeONa/MeOH at room temperature for 24 h.
Table 2.
Synthesis of optically active bicyclic cycloheptenone derivatives via tandem 5-exocyclization/Claisen rearrangement.
 | |||||
|---|---|---|---|---|---|
| entry | allyl alcohol (3) | bicyclic ketone (4) | yield (%)a | dr | ee (%)b |
| 1 | 3a |
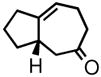 4a |
62 | 85 | |
| 2 | 3b |
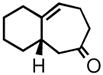 4b |
77 | 87 | |
| 3 | 3c |
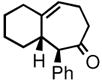 4c |
78 | 92:8 | 85 |
| 4 | 3d |
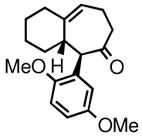 4d |
82 | 77:23 | 89 |
| 5 | 3e |
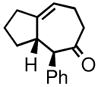 4e |
76 | 92:8 | 84 |
| 6 | 3f |
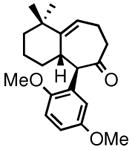 4f |
79 | 83:17(91:9)c | 95 |
Combined yield of both diastereomers.
Enantiomeric excess of the major diastereomer was determined either by chiral GC or chiral HPLC.
After 24 h of stirring in MeONa/MeOH at 25 °C.
Ketone 4f was selected as a suitable precursor for elaboration into optically active frondosin B, which is one of five structurally related natural products first isolated in 1997 from the Micronesian marine sponge dysidea frondosa (Figure 1).4 The unique structural features coupled with the therapeutic potential of this class of natural products as novel anti-inflammatory,4 anti-tumor16 and anti-HIV17 agents has sparked considerable interest in their total synthesis by several research groups,18 including our own.7f,g, 19, 20
Figure 1.
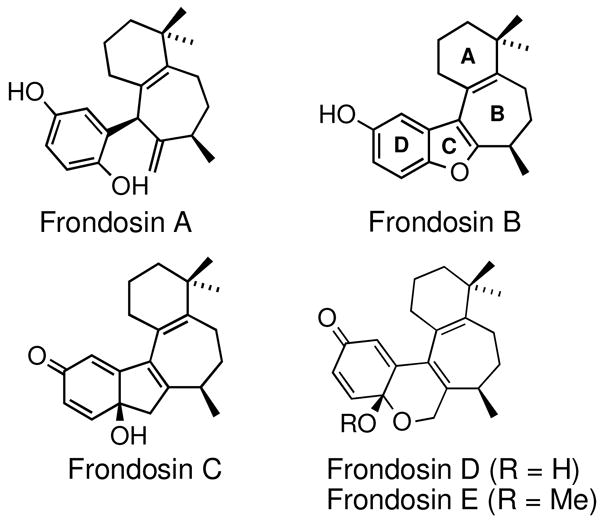
Frondosins A–E.
Thus far, the total syntheses of the naturally-occurring (+)-frondosin B and its (−)-enantiomer have been achieved by the groups of Danishefsky18b and Trauner,18c,d respectively. Although elegant in many ways, both of these approaches are somewhat lengthy, requiring multistep sequences at the early stages to prepare the requisite nonracemic starting materials. Danishefsky initially concluded that the sole stereogenic center in (+)-frondosin B is R configured; however, Trauner later challenged this assignment.18d Trauner suggested that a key nonracemic material in the Danishefsky process had undergone an inadvertent inversion of configuration early on in the synthesis, ultimately resulting in misassignment of absolute stereochemistry at C8 of the natural product.18d
Overall, our approach to (−)-frondosin B involves only two additional steps compared to that already reported for its racemic analogue (Scheme 2).20 Thus, optically active 4f (prepared from enone 2f in two steps) was methylated regio- and stereospecifically at C8, providing ketone 5 in 85% yield and 97% ee after purification. Ceric ammonium nitrate (CAN)-promoted oxidative demethylation of 5 gave the benzoquinone derivative 6, which was subsequently reduced under catalytic hydrogenation conditions. The resulting hydroquinone intermediate was directly exposed to BF3•OEt2, smoothly affording tetracycle 7, a close structural analogue of frondosin B. On treatment with catalytic p-TsOH in refluxing benzene, 7 was converted to (−)-frondosin B in 68% isolated yield ([α]21 D= −17.3, c 0.178, MeOH).21
Scheme 2.
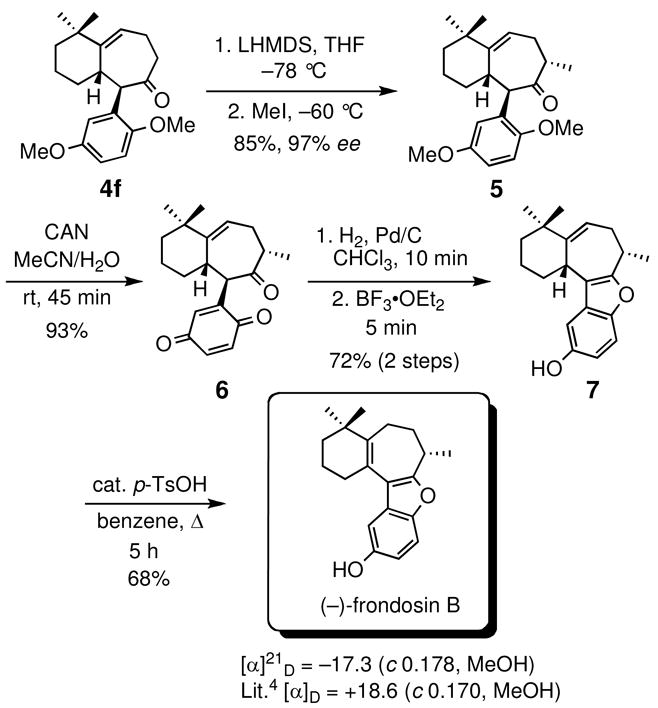
Total synthesis of (−)-frondosin B.
It is worth emphasizing that the absolute configuration of the final product is ultimately derived from 3f where the R stereogenic center controls the stereochemical course of the tandem cyclization/Claisen rearrangement process, providing 4f as the 10R, 11S stereoisomer (major diastereomer). The C10 aryl substituent, in turn, directs the subsequent methylation reaction at C8, affording ketone 5. The observed trans relationship between the C10 aryl and C8 methyl substituents has been previously confirmed by X-ray crystallography involving a racemic analogue of 4f.20 Since it is unlikely that the configuration at C8 would change in the course of the remaining four steps of the reaction sequence, it is concluded that (−)-frondosin B must be S configured at C8. This result is in agreement with Danishefsky’s original assignment according to which the naturally occurring (+)-frondosin B has an R stereogenic center at C8.18b
In summary, an asymmetric variant of the oxyanionic 5-exo dig cyclization/Claisen rearrangement has been developed, allowing a straightforward synthesis of a number of optically active cycloheptanoid ring systems, including (−)-frondosin B. Further studies employing this methodology to the asymmetric synthesis of the remaining members of the frondosin family as well as other cycloheptanoid natural product targets are currently underway in our laboratories. The results from these studies will be reported in due course.
Supplementary Material
Acknowledgments
Acknowledgment This research was supported by a grant from the National Institutes of Health (NIGMS). T.V.O also gratefully acknowledges support from the Hans and Ella McCollum-Vahlteich ’21 endowment.
Supporting Information Available Full experimental details, characterization data and copies of 1H and 13C NMR spectra for all new compounds are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Evans FJ, editor. Naturally Occurring Phorbol Esters. CRC; Boca Raton, FL: 1986. [Google Scholar]
- 2.Bondi SM, Clardy J. J Am Chem Soc. 2001;123:9900–9901. doi: 10.1021/ja016176y. [DOI] [PubMed] [Google Scholar]
- 3.Wong HF, Brown GD. J Nat Prod. 2002;65:481–486. doi: 10.1021/np0103113. [DOI] [PubMed] [Google Scholar]
- 4.Patil AD, Freyer AJ, Killmer L, Offen P, Carte B, Jurewicz AJ, Johnson RK. Tetrahedron. 1997;53:5047–5060. [Google Scholar]
- 5.For a recent review, see: Battiste MA, Pelphrey PM, Wright DL. Chem Eur J. 2006;12:3438–3447. doi: 10.1002/chem.200501083.
- 6.For an early example of this process, see: Marvell EN, Titterington D. Tetrahedron Lett. 1980:2123–2134.
- 7.(a) Ovaska TV, Roark JL, Shoemaker CM. Tetrahedron Lett. 1998;39:5705–5708. [Google Scholar]; (b) Ovaska TV, Roses JB. Org Lett. 2000;2:2361–2364. doi: 10.1021/ol006139w. [DOI] [PubMed] [Google Scholar]; (c) Ovaska TV, Reisman SE, Flynn MA. Org Lett. 2001;3:115–117. doi: 10.1021/ol006823a. [DOI] [PubMed] [Google Scholar]; (d) Ovaska TV, Ravi Kumar JS, Hulford CA, O’Sullivan MF, Reisman SE. Tetrahedron Lett. 2002;43:1939–1941. [Google Scholar]; (e) McIntosh CE, Martinez I, Ovaska TV. Synlett. 2004:2579–2581. [Google Scholar]; (f) Martinez I, Alford PE, Ovaska TV. Org Lett. 2005;7:1133–1135. doi: 10.1021/ol050144o. [DOI] [PubMed] [Google Scholar]; (g) Li X, Kyne RE, Ovaska TV. Org Lett. 2006;8:5153–5156. doi: 10.1021/ol0620848. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Li X, Kyne RE, Ovaska TV. J Org Chem. 2007;72:6624–6627. doi: 10.1021/jo0710432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For reviews on the Claisen rearrangement, see: Wipf P. Comprehensive Organic Synthesis. Vol. 5. Wiley; 1991. The Claisen rearrangement; pp. 827–873.Castro AMM. Chem Rev. 2004;104:2939–3002. doi: 10.1021/cr020703u.
- 9.For representative examples, see: Katzmaier U, Schneider C. Tetrahedron Lett. 1998;39:817–818.Looper RE, Williams RM. Tetrahedron Lett. 2001;42:769–771.Samy R, Kim HY, Stein K, Toogood PL. J Org Chem. 1999;64:2711–2728. doi: 10.1021/jo982145i.Fernandez de la Pradilla R, Montero C, Tortosa M. Org Lett. 2002;4:2373–2376. doi: 10.1021/ol0261130.
- 10.For representative examples, see: Kallmerten J, Gould TJ. J Org Chem. 1986;51:1152–1155.Kazmaier U, Maier S. J Org Chem. 1999;64:4574–4575. doi: 10.1021/jo9904821.Kazmaier U, Maier S. Org Lett. 1999;1:1763–1966.
- 11.Kazmaier U, Mues H, Krebs A. Chem Eur J. 2002;8:1850–1855. doi: 10.1002/1521-3765(20020415)8:8<1850::AID-CHEM1850>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 12.For representative examples, see: Maruoka K, Banno H, Yamamoto H. J Am Chem Soc. 1990;112:7791–7793.Maruoka K, Saito S, Yamamoto H. J Am Chem Soc. 1995;117:1165–1166.Kazmaier U, Krebs A. Angew Chem Int Ed. 1995;34:2012–2013.Corey EJ, Lee DH. J Am Chem Soc. 1991;113:4026–4028.Hiersemann M, Abraham L. Eur J Org Chem. 2002:1461–1471.
- 13.Mancuso AJ, Swern D. Synthesis. 1981:165–185. [Google Scholar]
- 14.Bobbitt JM. J Org Chem. 1998;63:9367–9374. [Google Scholar]
- 15.For reviews, see: Corey EJ, Helal CJ. Angew Chem, Int Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z.Byung TC. Tetrahedron. 2006;62:7621–7643.
- 16.(a) Brat DJ, Bellail AC, Van Meir EG. Neuro-oncol. 2005;7:122–133. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhu YM, Webster SJ, Flower D, Woll PJ. Br J Cancer. 2004;91:1970–1976. doi: 10.1038/sj.bjc.6602227. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuan A, Chen JJ, Yao PL, Yang PC. Front Biosci. 2005:853–865. doi: 10.2741/1579. [DOI] [PubMed] [Google Scholar]
- 17.Hallock YF, Cardellina JH, II, Boyd MR. Nat Prod Lett. 1998;11:153–160. [Google Scholar]
- 18.(a) Inoue M, Frontier AJ, Danishefsky SJ. Angew Chem, Int Ed. 2000;39:761–764. [PubMed] [Google Scholar]; (b) Inoue M, Carson MW, Frontier AJ, Danishefsky SJ. J Am Chem Soc. 2001;123:1878–1889. doi: 10.1021/ja0021060. [DOI] [PubMed] [Google Scholar]; (c) Hughes CC, Trauner D. Angew Chem, Int Ed. 2002;41:1569–1573. doi: 10.1002/1521-3773(20020503)41:9<1569::aid-anie1569>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (d) Hughes CC, Trauner D. Tetrahedron. 2004;60:9675–9686. [Google Scholar]; (e) Kerr DJ, Willis AC, Flynn BL. Org Lett. 2004;6:457–460. doi: 10.1021/ol035822q. [DOI] [PubMed] [Google Scholar]; (f) Trost BM, Hu Y, Horne DB. J Am Chem Soc. 2007;129:11781–11790. doi: 10.1021/ja073272b. [DOI] [PubMed] [Google Scholar]; (g) Olson P, Davies HML. Org Lett. 2008;10:573–576. doi: 10.1021/ol702844g. [DOI] [PubMed] [Google Scholar]; (h) Masters K-S, Flynn BL. J Org Chem. 2008;73:8081–8084. doi: 10.1021/jo800682n. [DOI] [PubMed] [Google Scholar]; (i) Mehta G, Likhite NS. Tetrahedron Lett. 2008;49:7113–7116. [Google Scholar]
- 19.Li X, Kyne RE, Ovaska TV. Tetrahedron. 2007;63:1899–1906. [Google Scholar]
- 20.Li X, Ovaska TV. Org Lett. 2007;9:3837–3840. doi: 10.1021/ol701633z. [DOI] [PubMed] [Google Scholar]
- 21.The balance of the reaction consisted mostly of isomeric impurities, which were separated from (−)-frondosin B by column chromatography.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.